Introduction
Preeclampsia (PE) is a pregnancy-specific disorder
characterized by new-onset hypertension and proteinuria that occurs
after 20 weeks of gestation. It is a systemic disease that involves
several organs such as the brain, liver, and kidney, as well as the
coagulation system (1). This disorder
affects 2–8% of all pregnancies and continues to be a leading cause
of maternal and perinatal morbidity and mortality worldwide
(2,3).
Although the specific etiology and pathogenesis of PE remain
unknown, it is widely accepted that PE is associated with
inadequate invasion of trophocytes, and spiral artery remodeling
(4). In normal pregnancies,
extravillous cytotrophoblasts of fetal origin invade the uterine
spiral arteries of the decidua and myometrium. These invasive
cytotrophoblasts replace the endothelial layer of the maternal
spiral arteries, transforming them from small, high-resistance
vessels into large-caliber vessels. However, in the development of
preeclamptic placenta, reduced invasive ability results in
trophocytes failing to invade the deep layers of the myometrium and
thus failing to appropriately remodel the uterine spiral arteries
(5). Consequently, the transformation
of spiral arteries from high-resistance, low-flow vessels into
large-caliber vessels is suppressed. The decreased blood flow and
fetoplacental perfusion leads to placental hypoxia and ischemia,
subsequent systemic endothelial dysfunction and PE (6). However, the molecular mechanisms for the
regulation of trophoblast behavior remain largely elusive.
Transthyretin (TTR), formerly known as pre-albumin,
was first discovered in 1942, both in human cerebrospinal fluid and
blood. Subsequently, TTR synthesis has been identified in the yolk
sac, placenta, pancreas and intestine of humans. It is a 56-kDa
homotetrameric protein that binds thyroid hormone and retinol
binding protein. Dysregulated placental TTR has been found in cases
of intrauterine growth restriction and severe early onset PE
(7). While a causative role of TTR in
PE has been postulated (8), the
precise pathogenic mechanisms of PE have not yet been clarified.
Our study aimed to investigate the possible role of TTR in the
pathophysiology of PE and its function in trophoblast biology.
Materials and methods
Cell culture
The human choriocarcinoma cell line, JEG-3, was
obtained from the Cancer Hospital, Chinese Academy of Medical
Sciences (Beijing, China). JEG-3 cells were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS; Gibco-BRL,
Carlsbad, CA, USA) at 37°C in a 5% CO2 incubator. When
the cells reached 80–90% confluence, they were trypsinized and
subcultured into new culture flasks.
Construction of a recombinant plasmid
overexpressing TTR
The nucleotide sequence of the coding region of the
TTR gene was used to design amplification primers with
Primer Premier 5 software (Premier Biosoft, Palo Alto, CA, USA).
The primer sequences were: forward,
5′-GTAGAATTCGGATGGCTTCTCATCGTCTG-3′ and reverse,
5′-GTAGGTACCTCATTCCTTGGGATTGGTG-3′. Human cDNA was used as a
template to amplify the coding region of the TTR gene. The
resulting PCR product was ligated with the EcoRI/KpnI
enzyme restriction sites of the pCMV-Myc plasmid. The positive
recombinant plasmid was identified by double enzyme digestion and
the sequence was termed pCMV-Myc-TTR. The empty plasmid (pCMV-Myc)
was used as a negative control. JEG-3 cells were seeded in 6-well
plates and allowed to grow to 80–90% confluence. Transfection was
performed using the Lipofectamine 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer's
instructions. The medium was replaced with fresh culture medium 6 h
after transfection.
Western blot analysis
Protein extracts were prepared from cells at 48 h
post-transfection using ice-cold radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology, Haimen, China)
and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO,
USA). Protein concentrations were determined using a bicinchoninic
acid protein assay kit (Beyotime Institute of Biotechnology). Equal
amounts of protein were mixed with loading buffer and boiled at
100°C for 3–5 min, chilled on ice, and resolved by 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The
separated proteins were then electrophoretically transferred to
0.45-µm polyvinylidene fluoride (PVDF) membranes (Millipore,
Bedford, MA, USA) using a semi-dry western blot transfer system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) for 90 min at 200
mA. The membranes were subsequently washed with TBST (TBS
containing 0.1% Tween-20) for 1 min, after blocking with 5% non-fat
dry milk for 1 h at room temperature. The membranes were then
sequentially incubated overnight at 4°C with rabbit anti-prealbumin
monoclonal antibody (ab199074, 1:2,000 diluted), rabbit anti-matrix
metalloproteinase 2 (MMP2) monoclonal antibody (ab92536, 1:1,000
diluted) and rabbit anti-MMP9 monoclonal antibody (ab76003, 1:4,000
diluted) (all from Abcam, Cambridge, MA, USA), and anti-GAPDH (no.
2118, 1:5,000 diluted; Cell Signaling Technology, Inc., Danvers,
MA, USA) which was used as the internal control. The following day,
the membranes were washed three times in TBST (5 min each) and
incubated with a secondary HRP-conjugated goat anti-rabbit antibody
(no. 7074, 1:10,000 diluted; Cell Signaling Technology, Inc.) for 1
h at room temperature. Finally, immunoreactive bands were detected
with an enhanced chemiluminescence kit (Millipore) and exposed to
X-ray film.
Transwell migration assay
The Transwell migration assay was performed by
determining the ability of cells to invade the 8-µm pores of
polycarbonate membranes (cat. no. 3422; Corning Costar Inc.,
Corning, NY, USA), which were placed at the bottom of Transwell
chambers in 24-well plates. JEG-3 cells were transfected with
pCMV-Myc-TTR or the empty plasmid when 80–90% confluent. After 24 h
of incubation, 1×105 JEG-3 cells were resuspended in 100
µl serum-free medium containing 1% bovine serum albumin and were
plated on the top of the chamber and incubated with 650 µl culture
medium containing 10% FBS in the bottom chamber. The cells with or
without TTR overexpression were divided into two groups, and
treated in the presence or absence of 1 µmol/l levothyroxine
(L-T4). After incubation at 37°C in an atmosphere of 5%
CO2 for 24 h, the cells on the top surface of the
Transwell membrane were wiped off with cotton swabs. Subsequently,
the cells on the bottom surface of the membrane were fixed with 95%
ethyl alcohol for 30 min and stained with 0.1% crystal violet for
3–5 min at room temperature (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China). The invaded cells on the
underside of the membrane were enumerated using an inverted
microscope in five random fields at ×50 magnification (IX-71;
Olympus, Tokyo, Japan). The results are presented as mean of
invaded cells ± standard deviation (SD) and each assay was repeated
at least 3 times.
Matrigel-based Transwell invasion
assay
A Matrigel-based Transwell invasion assay was
performed to assess the invasion ability of JEG-3 cells, 24 h after
transfection. The method was similar to the cell migration assay,
except that the membrane was pre-treated with 60 µl Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA) at a concentration of 2
mg/ml, and the results were observed after incubation for 36 h.
Experiments were performed in triplicate and the results are
presented as mean of invaded cells ± SD.
Statistical analysis
Data are presented as mean ± SD. One-way analysis of
variance (ANOVA) was conducted to compare multiple groups, followed
by the Bonferroni post hoc test for the comparisons between groups.
GraphPad Prism software, version 4.0 (GraphPad Software, Inc., San
Diego, CA, USA), was used for data analysis and plotting. P<0.05
was considered to indicate a statistically significant
difference.
Results
TTR protein expression after
recombinant plasmid transfection
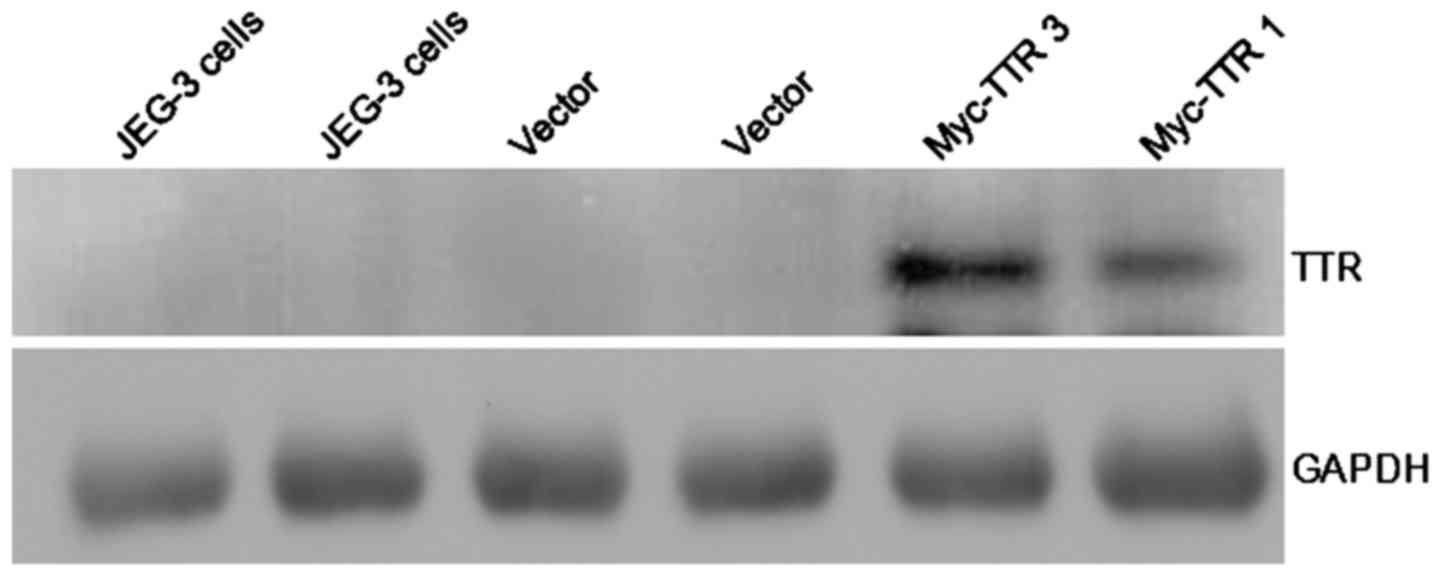
JEG-3 cells were transfected with pCMV-Myc-TTR or
empty plasmid. Myc-TTR was overexpressed in JEG-3 cells transfected
with pCMV-Myc-TTR as determined by western blot analysis, whereas
there was no expression of TTR protein in JEG-3 cells transfected
with the empty plasmid. These data indicated that JEG-3 cells
expressed extremely low levels of endogenous TTR protein (Fig. 1).
Overexpression of TTR promotes the
migration and invasion of JEG-3 cells
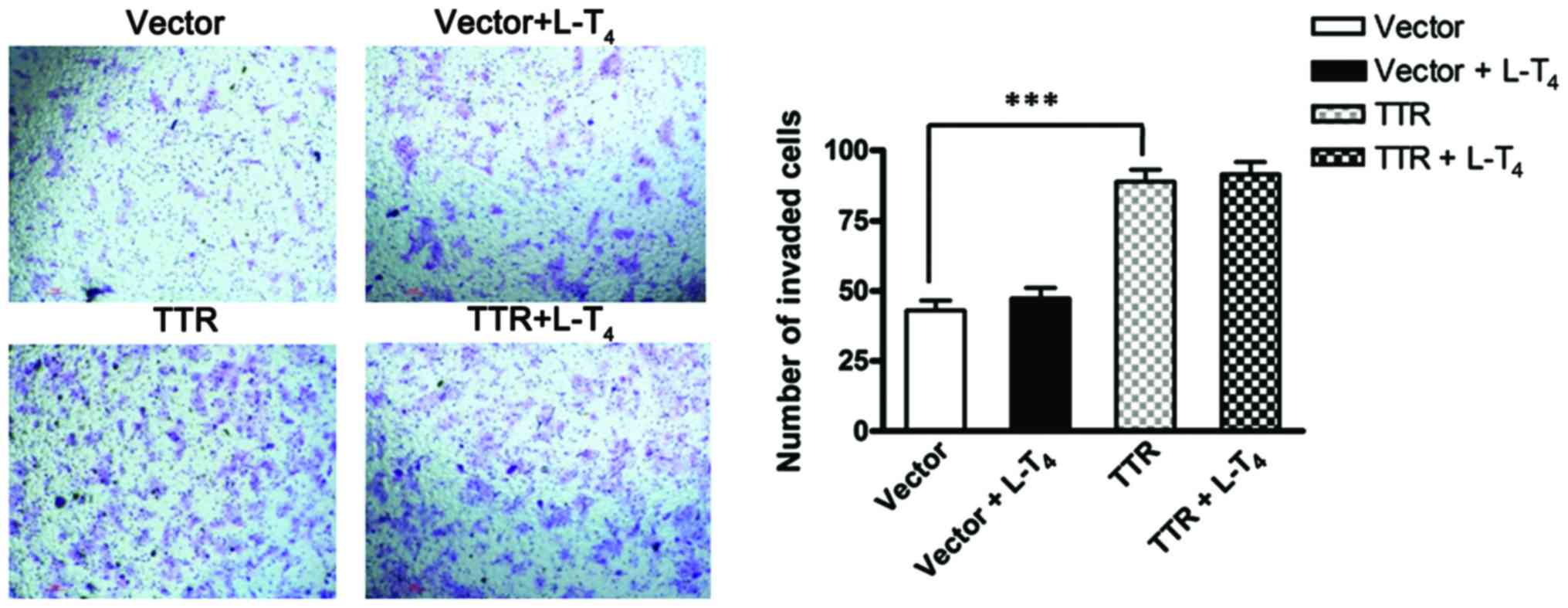
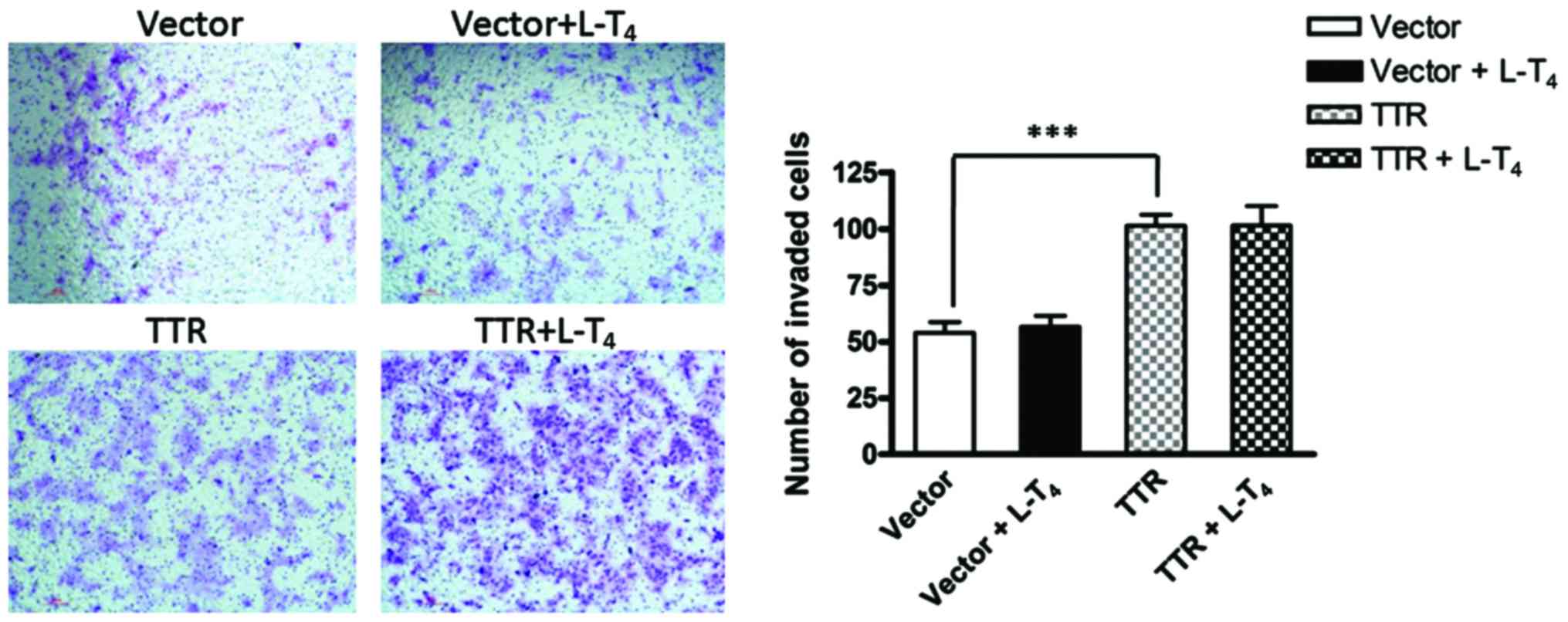
The migration and invasion ability of JEG-3 cells
transfected with the TTR-expressing construct or empty plasmid were
assessed by a Transwell migration assay and Matrigel-based invasion
assay, respectively. In the 24-h migration assay, the number of
invaded cells increased significantly when JEG-3 cells were
transfected with pCMV-Myc-TTR compared with the empty plasmid.
However, in TTR-overexpressing cells, there was no significant
difference in cell invasion in the presence or absence of
L-T4. Similarly, the invasion ability of JEG-3 cells
increased significantly after TTR overexpression, compared with the
control cells in the 36-h invasion assay. However, there was no
difference in cell invasion between TTR-overexpressing cells in the
presence or absence of L-T4 (Figs. 2 and 3).
Overexpression of TTR upregulates MMP2
and MMP9
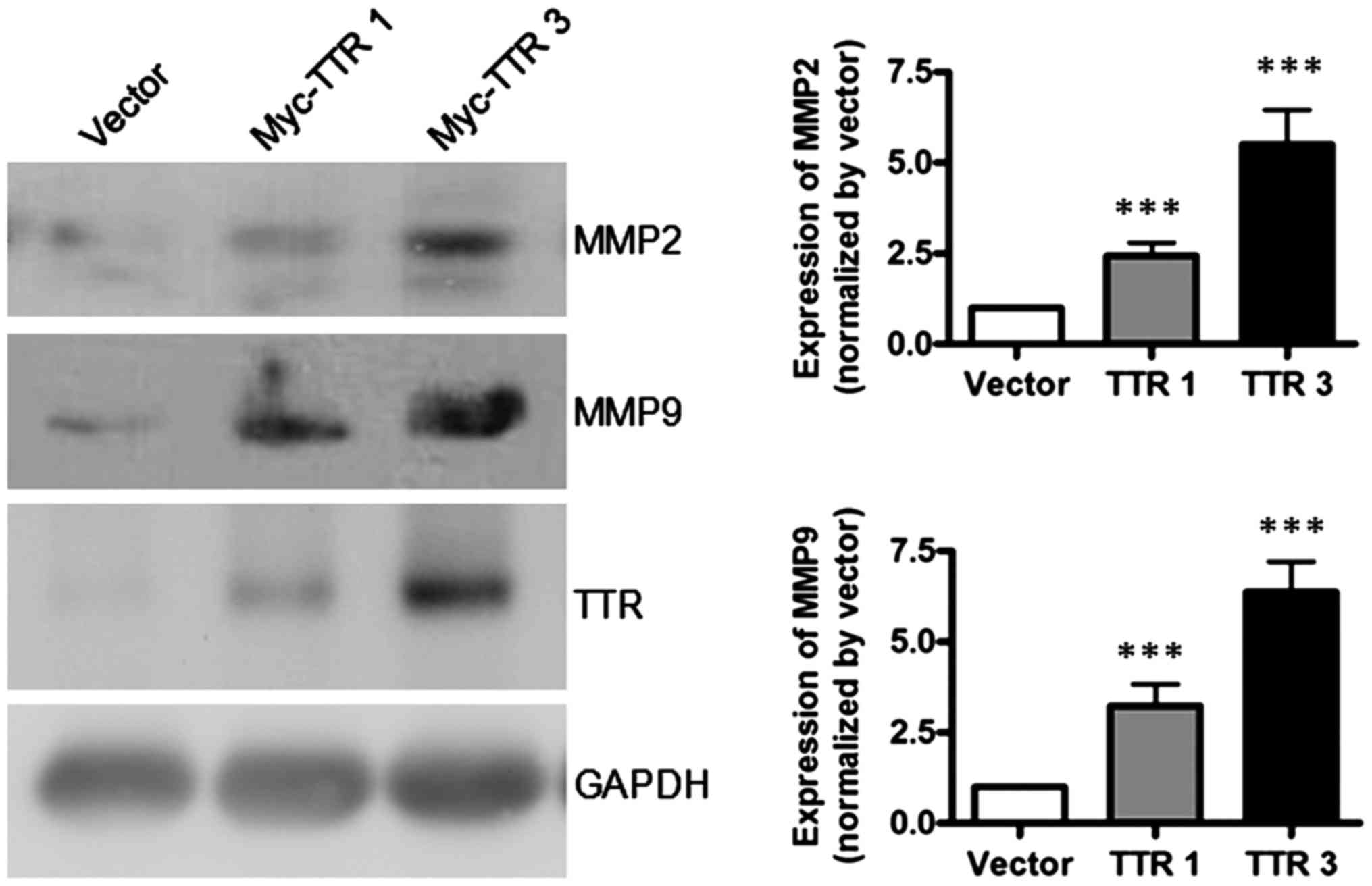
MMPs are a family of proteolytic enzymes that have
been implicated in extracellular matrix remodeling in the process
of trophocyte invasion. To investigate the molecular mechanisms of
TTR in mediating trophoblast invasion, we used western blot
analysis to analyze the protein levels of MMP2 and MMP9 at 48 h
after transfection. The results showed that overexpression of TTR
significantly increased the expression levels of MMP2 and MMP9
(Fig. 4), indicating that TTR likely
regulates the invasion ability of these cells through MMP-mediated
remodeling of extracellular matrix.
Discussion
JEG-3 is a line of choriocarcinoma cells that
originated from choriocarcinoma explants. They are phenotypically
similar to extravillous trophoblasts (EVTs). JEG-3 cells express
human choriogonadotropin; human leukocyte antigen-G, integrin α
(ITGA)1, 5 and 6; and MMP2 and MMP9; and have been extensively used
as an in vitro model to study the properties of trophoblast
migration and invasion (9,10).
The process of EVT cell invasion of the maternal
spiral arteries is critical for the establishment of a successful
pregnancy and is very similar to the invasion of tumor cells
(11,12). Failure of the process has been
recognized as a common pathologic feature of PE (13,14). In
addition, this process is regulated by many cytokines and
chemokines (15–17). In the placentas of patients with PE,
trophoblast dysfunction reduces the invasion and migration
capacities of EVTs. When the invasion of trophocytes into the
endometrium at the maternal-fetal interface of PE patients is very
shallow, and the vascular endothelium has not been replaced by
trophocytes, this will result in placental hypoperfusion, hypoxia,
or ischemia and thus induce the occurrence and deterioration of PE
(18).
It is widely believed that the pathophysiological
changes of PE may result from the abnormal expression of certain
proteins. A previous study of PE using surface-enhanced laser
desorption ionization time-of-flight mass spectrometry
(SELDI-TOF-MS) detected 10 significantly different protein peaks
between patients with hypertensive disorders of pregnancy and
healthy controls (18). Liu et
al (19) used peptide ligand
library affinity chromatography combined with 1D gel-LC-MS/MS
analysis to identify proteins that were differentially expressed in
sera of preeclamptic patients compared with sera from healthy
pregnant women, and found that TTR was significantly downregulated
in preeclamptic patients. Kalkunte et al (20) used SELDI-TOF-MS combined with
two-dimensional gel electrophoresis to test sera collected from
patients with PE or subjects with normal pregnancy and found
reduced levels of TTR in PE serum. This study also demonstrated
that TTR aggregates to form deposits in preeclamptic placental
tissue and causes apoptosis. Furthermore, native TTR inhibited all
PE-like features in the humanized mouse model, including new-onset
proteinuria, increased blood pressure, glomerular endotheliosis and
production of anti-angiogenic factors. Chen et al (8) assumed that TTR may cause a disorder of
maternal vascular function and contribute to the pathology of PE by
deposition of TTR amyloid fibrils in the vascular system, which are
produced by variant TTR proteins, resulting in organ ischemia. The
study by Zhu et al (21) using
ELISA and western blot analysis revealed that TTR levels were
markedly decreased in early onset severe PE cases compared with
controls, and TTR levels were lower in the early onset PE patients
than in the late onset group, indicating that changes in TTR levels
may correlate with the severity of PE.
Previous studies suggested that decreased TTR
expression may be responsible for the occurrence of PE (20,21). TTR
levels may change prior to the onset of PE and may represent a
candidate biomarker to predict PE. However, it remains unclear how
TTR is involved in the pathogenesis of PE. The present study showed
that the number of invaded cells increased significantly when JEG-3
cells were transfected with Myc-TTR compared with the empty
plasmid, in both the Transwell migration assay and Matrigel-based
invasion assay. This suggested that TTR can promote the migration
and invasion ability of trophoblasts. Additionally, a decreased TTR
expression leads to inhibition of the migration and invasion of
trophoblasts in PE patients. Shallow trophoblast invasion in turn,
results in placental hypoxia and ischemia, which eventually leads
to the occurrence of PE. However, the present results also showed
that there was no significant difference in the number of invaded
TTR-overexpressing cells in the presence or absence of
L-T4. It is known that TTR is a carrier protein. Its
main function is the transport of T4 (19). However, we showed that the migration
and invasion ability of JEG-3 cells was not enhanced by the
addition of L-T4, indicating that TTR may not exert its
roles through the TTR-T4 complex to promote the
migration and invasion of trophoblasts. Our study further confirmed
the role of TTR in the pathogenesis of PE, although further study
is required to clarify the molecular mechanisms.
MMPs are a family of zinc-containing endopeptidases
capable of degrading a wide range of extracellular matrix
components (22). Of these, MMP2 and
MMP9, also known as gelatinase A and B, respectively, are the most
frequently investigated MMPs and are thought to play important
roles in trophoblast invasion. It has been demonstrated that
patients with PE exhibit low levels of placental MMP2 and MMP9
(23,24). It has also been shown that MMPs are
involved in the remodeling of uterine spiral arteries, and reduced
MMP activity may affect trophoblastic invasion, which subsequently
induces abnormal placental development and onset of PE (25). The present findings showed that
overexpression of TTR significantly increased the protein levels of
MMP2 and MMP9, indicating that TTR likely regulates the invasion
ability of cells through MMP-mediated remodeling of extracellular
matrix. More specifically, TTR likely promotes the migration and
invasion ability of trophoblasts by increasing the secretion of
MMP2 and MMP9. The reduced levels of TTR may decrease the secretion
of MMP2 and MMP9 in PE patients and subsequently inhibit the
migration and invasion ability of trophoblasts. Shallow trophoblast
invasion contributes to placental hypoxia and ischemia, which
eventually leads to the occurrence of PE.
In conclusion, we have demonstrated that
overexpression of TTR effectively promotes the migration and
invasion ability of JEG-3 cells, which was associated with
increased protein levels of MMPs. Our findings support an important
role for TTR in regulating trophoblast invasion and migration,
representing a possible underlying pathological and molecular
mechanism of PE.
Acknowledgements
The present study was supported by the Sino-RUS
Cooperation Funds (no. 2015DFR31070) and by the National Natural
Science Funds (no.81571455).
References
|
1
|
ACOG Committee on Obstetric Practice;
American College of Obstetricians and Gynecologists, . ACOG
practice bulletin. Diagnosis and management of preeclampsia and
eclampsia. Number 33, January 2002. Int J Gynaecol Obstet.
77:67–75. 2002.PubMed/NCBI
|
|
2
|
Duley L, Meher S and Abalos E: Management
of pre-eclampsia. BMJ. 332:463–468. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Poon LC and Nicolaides KH: Early
prediction of preeclampsia. Obstet Gynecol Int. 2014:2973972014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tal R: The role of hypoxia and
hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol
Reprod. 87:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goldman-Wohl D and Yagel S: Regulation of
trophoblast invasion: from normal implantation to pre-eclampsia.
Mol Cell Endocrinol. 187:233–238. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lam C, Lim KH and Karumanchi SA:
Circulating angiogenic factors in the pathogenesis and prediction
of preeclampsia. Hypertension. 46:1077–1085. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fruscalzo A, Schmitz R, Klockenbusch W,
Köhler G, Londero AP, Siwetz M and Huppertz B: Human placental
transthyretin in fetal growth restriction in combination with
preeclampsia and the HELLP syndrome. Histochem Cell Biol.
138:925–932. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Y and Zhang Z: Does transthyretin
function as one of contributors for preeclampsia? Med Hypotheses.
76:8–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huppertz B, Kertschanska S, Demir AY,
Frank HG and Kaufmann P: Immunohistochemistry of matrix
metalloproteinases (MMP), their substrates, and their inhibitors
(TIMP) during trophoblast invasion in the human placenta. Cell
Tissue Res. 291:133–148. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hannan NJ, Paiva P, Dimitriadis E and
Salamonsen LA: Models for study of human embryo implantation:
choice of cell lines? Biol Reprod. 82:235–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harris LK: Review: trophoblast-vascular
cell interactions in early pregnancy: how to remodel a vessel.
Placenta. 31(Suppl): S93–S98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hiden U, Bilban M, Knöfler M and Desoye G:
Kisspeptins and the placenta: regulation of trophoblast invasion.
Rev Endocr Metab Disord. 8:31–39. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pennington KA, Schlitt JM, Jackson DL,
Schulz LC and Schust DJ: Preeclampsia: multiple approaches for a
multifactorial disease. Dis Model Mech. 5:9–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kaitu'u-Lino TJ, Ye L, Tuohey L,
Dimitriadis E, Bulmer J, Rogers P, Menkhorst E, Van Sinderen M,
Girling JE, Hannan N, et al: Corin, an enzyme with a putative role
in spiral artery remodeling, is up-regulated in late secretory
endometrium and first trimester decidua. Hum Reprod. 28:1172–1180.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ren L, Liu YQ, Zhou WH and Zhang YZ:
Trophoblast-derived chemokine CXCL12 promotes CXCR4 expression and
invasion of human first-trimester decidual stromal cells. Hum
Reprod. 27:366–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim YJ: Pathogenesis and promising
non-invasive markers for preeclampsia. Obstet Gynecol Sci. 56:2–7.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu X, Dai LI and Zhou R: Association
between preeclampsia and the CXC chemokine family (Review). Exp
Ther Med. 9:1572–1576. 2015.PubMed/NCBI
|
|
18
|
Gong LY, Zhang ZY, Zheng YH and Zhang JZ:
Study of a serum protein fingerprint diagnostic model in patients
with hypertensive disorder complicating pregnancy. Zhonghua Fu Chan
Ke Za Zhi. 42:822–825. 2007.(In Chinese). PubMed/NCBI
|
|
19
|
Liu C, Zhang N, Yu H, Chen Y, Liang Y,
Deng H and Zhang Z: Proteomic analysis of human serum for finding
pathogenic factors and potential biomarkers in preeclampsia.
Placenta. 32:168–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kalkunte SS, Neubeck S, Norris WE, Cheng
SB, Kostadinov S, Vu Hoang D, Ahmed A, von Eggeling F, Shaikh Z,
Padbury J, et al: Transthyretin is dysregulated in preeclampsia,
and its native form prevents the onset of disease in a preclinical
mouse model. Am J Pathol. 183:1425–1436. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhu L, Chen Y, Liu C, Deng H, Zhang N,
Wang S and Zhang Z: Transthyretin as a novel candidate biomarker
for preeclampsia. Exp Ther Med. 7:1332–1336. 2014.PubMed/NCBI
|
|
22
|
Heissig B, Hattori K, Friedrich M, Rafii S
and Werb Z: Angiogenesis: vascular remodeling of the extracellular
matrix involves metalloproteinases. Curr Opin Hematol. 10:136–141.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Omran OM, Shokry M, Ismail H, Omar G and
Rezk M: Expression of matrix metalloproteinases 2 and 9 in human
trophoblasts of normal and preeclamptic placentas. Int J Health Sci
(Qassim) 5 (Suppl 1). 21–23. 2011.
|
|
24
|
Yang Y, Zhang J, Gong Y, Liu X, Bai Y, Xu
W and Zhou R: Increased expression of prostasin contributes to
early-onset severe preeclampsia through inhibiting trophoblast
invasion. J Perinatol. 35:16–22. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cockle JV, Gopichandran N, Walker JJ,
Levene MI and Orsi NM: Matrix metalloproteinases and their tissue
inhibitors in preterm perinatal complications. Reprod Sci.
14:629–645. 2007. View Article : Google Scholar : PubMed/NCBI
|