Introduction
Colorectal cancer (CRC) is the third cause of
cancer-associated mortalities in the world in men, and the second
in women. In 2012, ~1.36 million cases were newly diagnosed, and
694,000 patients succumbed to CRC (1). Understanding the processes that govern
this disease is key to develop novel treatment strategies. In this
regard, inflammation has been widely associated with cancer since
1863 (2). Patients diagnosed with
inflammatory bowel disease present an increased risk to develop
CRC, and the risk is higher for patients with longer disease
duration and extensive disease (3).
The link between inflammation and cancer has prompted an exhaustive
area of research where anti-inflammatory drugs have been tested for
chemoprevention and adjuvant therapy. Indomethacin, a nonsteroidal
anti-inflammatory drug (NSAID), improved survival in patients with
solid tumors from different origins (4). In addition, other NSAIDs, including
sulindac, aspirin and piroxicam, have exhibited chemopreventive
effects (5–8). Studies in the early 70s demonstrated
that aspirin and indomethacin exert their therapeutic actions
partly through the inhibition of prostaglandin (PG) synthesis
(9). These drugs inhibit the activity
of cyclooxygenase (COX) enzymes, which transform arachidonic acid
into PGH2, which is then converted into different PGs, including
PGE2, PGF2α, PGI2, PGD2 and thromboxane A2 (9). There are two functional isoforms of COX:
COX-1, whose expression is constitutive; and COX-2, which exhibits
an inducible expression and whose products have been mainly
associated with pathological processes (10). COX-1 expression has been reported to
remain stable in cancer tissues, while COX-2 expression has been
associated with advanced Dukes stage, poor long-term outcome and
lymph node metastasis in CRC patients (11–13).
In previous studies by the present authors,
increased COX-2 staining was able to predict tumor tissue content
of PGE2 in CRC patients, while COX-1 staining had an inverse
correlation with PGE2 content (14,15).
Furthermore, PGE2 levels were observed to be increased in
premalignant adenomatous polyps and colon cancer tissues, compared
with normal controls (16,17). The enzyme responsible for the
synthesis of PGE2 is microsomal human prostaglandin E synthase
(mPGES), which is overexpressed in colorectal adenomas and
carcinomas (18). Taken together, the
COX-2/mPGES/PGE2 axis appears to be important in colorectal
carcinogenesis.
Previous studies by the present authors revealed
that tumor tissues from CRC patients with high COX-2 expression
levels displayed upregulation of several transcription factors,
among which, the most overexpressed one was FBJ murine osteosarcoma
viral oncogene homolog B (FosB) (19). This transcription factor is implicated
in the activator protein-1 (AP-1)/COX-2/PGE2 axis, and may play a
relevant role in patients with high expression levels of COX-2. The
AP-1 complex consist of homo or heterodimers of members of the Jun
and Fos families, and affects proliferation, transformation,
differentiation, stress response and apoptosis, depending on the
composition of the complex, type of cell and microenvironment
(20–22). In addition, FosB overexpression has
been reported to induce malignant cell transformation in
vitro and in vivo (23).
In the present study, the role of FosB on the
regulation of COX-2 expression and PGE2 content was investigated in
a CRC cell line that overexpressed COX-2. Small interfering (si)RNA
was used to knockdown FosB expression in HCA-7 cells, and the
effects on the COX/PGE2 axis were evaluated. The present results
indicate that knocking down FosB induces an important decrease in
COX-2 expression, with no changes on PGE2 content or COX-1
expression, suggesting that COX-2 expression and PGE2 content may
be regulated by an independent mechanism.
Materials and methods
Reagents, cell lines and culture
conditions
The human colon adenocarcinoma cell line HCA-7
(catalog no. 02091238; Sigma-Aldrich, St. Lois, MO, USA), which has
exhibits high expression levels of COX-2, was routinely
sub-cultured in Dulbecco's modified Eagle's medium (catalog no.
D6546; Sigma-Aldrich) supplemented with 10% fetal bovine serum
(catalog no. F2442; Sigma-Aldrich) and 1.0% of a 200-mM L-glutamine
solution (catalog no. 091680149; MP Biomedicals, LLC, Santa Ana,
CA, USA) without antibiotics. Cells were maintained in a humidified
incubator at 37°C and 5% CO2. Early passages (4–17) were
used for the experiments.
siRNA knockdown
To evaluate the effect of FosB transcription factor
on the COX/PGE2 axis, FosB siRNA transfection was performed. A
total of 8×104 HCA-7 cells/well were seeded in a 6-well
plate and allowed to attach overnight to achieve 30% of confluence
at the time of transfection. FosB siRNA (catalog no. 4392420 ID
s230577; Ambion; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) positive control
siRNA (catalog no. 4390849; Ambion; Thermo Fisher Scientific, Inc.)
or scramble negative control siRNA (Silencer® Select
Negative Control No. 1 siRNA; catalog no. 4390843; Ambion; Thermo
Fisher Scientific, Inc.) were transfected into HCA-7 cells using
Lipofectamine® RNAiMAX Transfection Reagent (catalog no.
13778030; Invitrogen; Thermo Fisher Scientific, Inc.). For the
transfection, 5 µl Lipofectamine® RNAiMAX Transfection
Reagent was added to 245 µl Opti-MEM I Reduced Serum Medium
(catalog no. 31985070; Thermo Fisher Scientific, Inc.), and then
mixed with 10 nM siRNA (FosB, positive control or negative control
siRNA) diluted in 250 µl Opti-MEM I Reduced Serum Medium. The mix
was incubated for 20 min at room temperature prior to be added to
the cells. Opti-MEM I Reduced Serum Medium was used to obtain a
final volume of 2 ml/well. At 72 h post-transfection, the cells and
medium were collected for RNA extraction and enzyme-linked
immunosorbent assay (ELISA). siRNA transfection was performed in
duplicate in three independent experiments.
RNA extraction and complementary
(c)DNA synthesis
Total RNA was isolated using the RNeasy Plus Micro
kit (catalog no. 74034; Qiagen GmbH, Hilden, Germany), according to
the manufacturer's protocol. RNA purity and concentration were
measured using the RNA 6000 Nano kit (catalog no. 5067-1511) on a
2100 Bioanalyzer (both Agilent Technologies, Inc., Santa Clara, CA,
USA). The sample's RNA integrity number ranged from 7 to 10. RNA
reverse transcription (RT) to cDNA was performed from 1 µg total
RNA using the Advantage RT-for-PCR kit (Clontech Laboratories,
Inc., Mountainview, CA, USA), following the manufacturer's protocol
PT1107-1 version PR023473. Oligo(dT)18 primer was used
for the synthesis of cDNA from mRNA (catalog no. 639506; Clontech
Laboratories, Inc.). Negative and positive controls were performed
alongside the samples during the RT reaction. The cycling
conditions were 30 cycles of 94°C for 45 sec, 60°C for 45 sec and
72°C for 2 min, with a final extension at 72°C for 7 min, using an
Eppendorf MasterCycler Gradient (Eppendorf-Netheler-Hinz GmbH,
Hamburg, Germany).
Quantitative polymerase chain reaction
(qPCR)
A total of 2 µl cDNA was used to analyze the
expression of FosB, COX-2, COX-1, GAPDH and 18S. The following
QuantiTect Primer Assays (Qiagen GmbH) were used: Hs_FOSB_1_SG
(FosB), Hs_PTGS2_1_SG (COX-2), Hs_PTGS1_1_SG (COX-1), Hs_GAPDH_1_SG
(GAPDH) and Hs_RRN18S_1_SG (18S). qPCR was performed in a
LightCycler 1.5 instrument (Roche Applied Science, Penzberg,
Germany) using LightCycler FastStart DNA MasterPLUS SYBR Green I
kit (catalog no. 03515885001; Roche Applied Science), according to
the manufacturer's protocol. The cycling conditions were as
follows: Activation at 95°C for 10 min, denaturation at 95°C for 10
sec and annealing at 64°C for 4 sec in 40 cycles for FosB, GAPDH
and 18S, or 45 cycles for COX-1 and COX-2. Gene expression was
analyzed using the relative standard curve method. COX-1 and COX-2
mRNA expression levels were normalized to those of 18S and GAPDH,
whereas FosB mRNA expression levels were normalized using only 18S
as a control gene, since GAPDH was used as a positive control for
FosB siRNA experiments. Data are presented as the mean ± standard
error of the mean (SEM) of the GAPDH and 18S normalized values.
Fold-changes were calculated using the negative control as a
calibrator.
ELISA
To analyze the effect of FosB knockdown on the
production of PGE2, ELISA for PGE2 was performed at 72 h post-FosB
siRNA transfection. Briefly, 500 µl medium was collected from the
FosB siRNA-transfected and control samples, and processed according
to the Biotrak EIA system protocol for PGE2 (catalog no. RPN222;
Amersham; GE Healthcare Life Sciences, Chalfont, UK). The
absorbance was measured at 630 nm in an Epoch Microplate
Spectrophotometer (Biotek Instruments, Inc., Winooski, VT, USA).
Determination of the sample concentrations (pg/well) was conducted
according to the manufacturer's protocol using a standard curve.
Each siRNA-transfected sample was analyzed in triplicate. Data are
presented as the mean ± SEM of three independent experiments
performed in duplicate, compared with the negative control
(scramble siRNA).
Cell imaging
At 72 h post-transfection with FosB siRNA, imaging
of cells was performed using an AE200 microscope with a 10X
objective (Motic Instruments, Richmond, BC, Canada).
Statistical Analysis
Data are presented as the mean ± SEM from three
independent experiments performed in duplicate or triplicate.
Statistical differences between the control group and treated cells
were evaluated using an independent t test. Statistical
analyses were performed using SPSS software version 22.0.0.0 (IBM
SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
FosB knockdown by siRNA reduces COX-2
mRNA expression but does not affect PGE2 extracellular content
To study the effects of FosB upregulation in cells
with high COX-2 expression levels, FosB expression was knocked down
on HCA-7 cells using siRNA. To standardize the siRNA treatment,
HCA-7 cells were treated with the minimum siRNA concentration
recommended by the manufacturer (10 nM) and with different
concentrations of Lipofectamine® RNAiMAX Transfection
Reagent (0.5, 3, 4 and 5 µl). The treatment was stopped at
different time points, and the RNA content was analyzed by RT-qPCR.
The data was normalized using 18S as a control gene. Treatment for
72 h achieved the most significant reduction in FosB mRNA
expression in FosB siRNA-transfected cells (24 h, P=0.397; 48 h,
P=0.106; 72 h, P=0.00062; and 96 h, P=0.00533). Therefore, this
time point was selected for further experiments. There was no
significant difference between FosB mRNA expression in untreated
cells and in negative control cells (P=0.240; data not shown). The
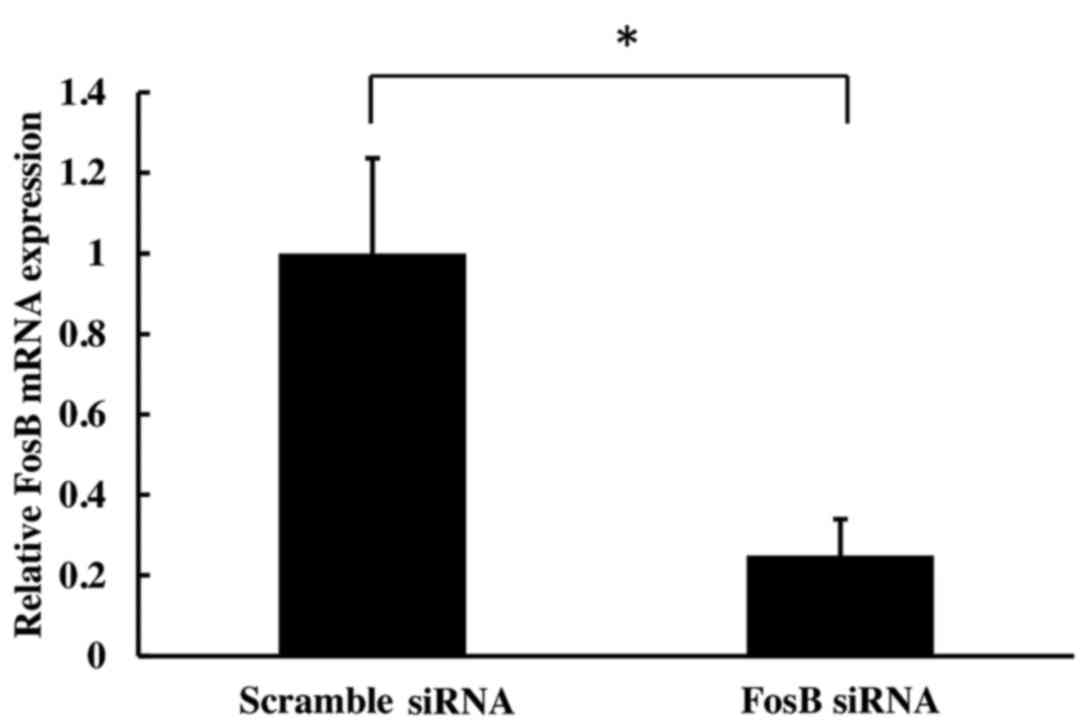
transfection of HCA-7 cells with FosB siRNA induced a significant
decrease in FosB mRNA expression levels at 72 h (75±9% reduction,
compared with the negative control (P=0.002; Fig. 1).
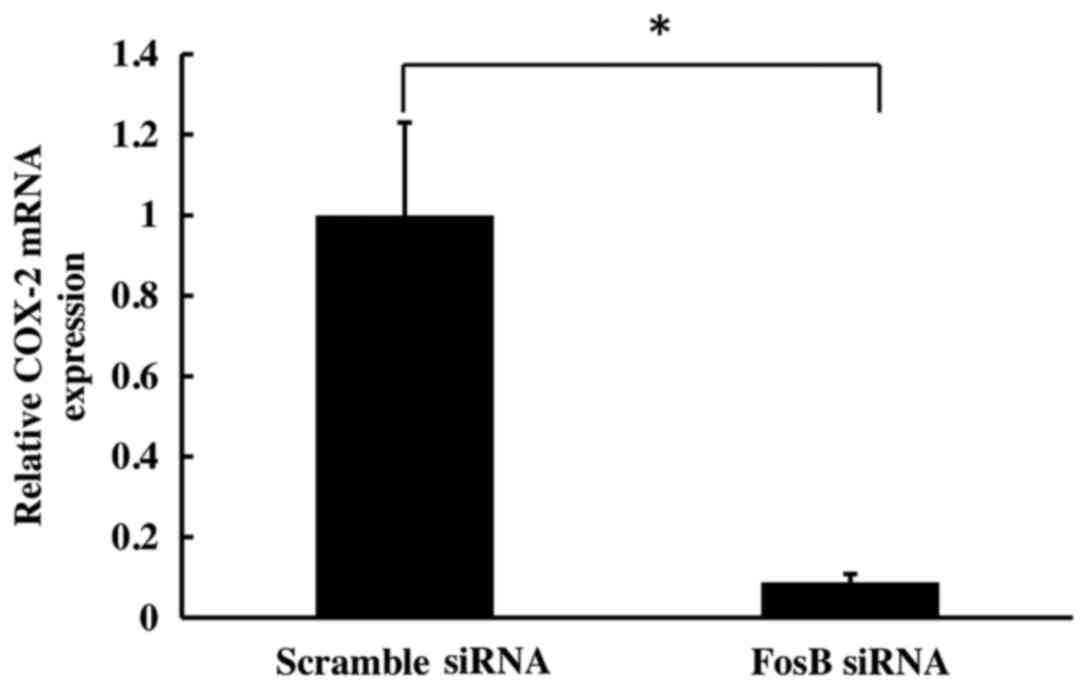
To evaluate the effect of FosB knockdown on COX-2
expression, the levels of COX-2 mRNA on cells transfected with FosB
siRNA were evaluated at 72 h using qPCR. COX-2 expression was
normalized using GAPDH and 18S as control genes. The results
revealed a significant decrease in COX-2 mRNA levels in
FosB-knocked down cells (fold-change, 0.11±0.02), compared with
negative control cells (P=0.018; Fig.
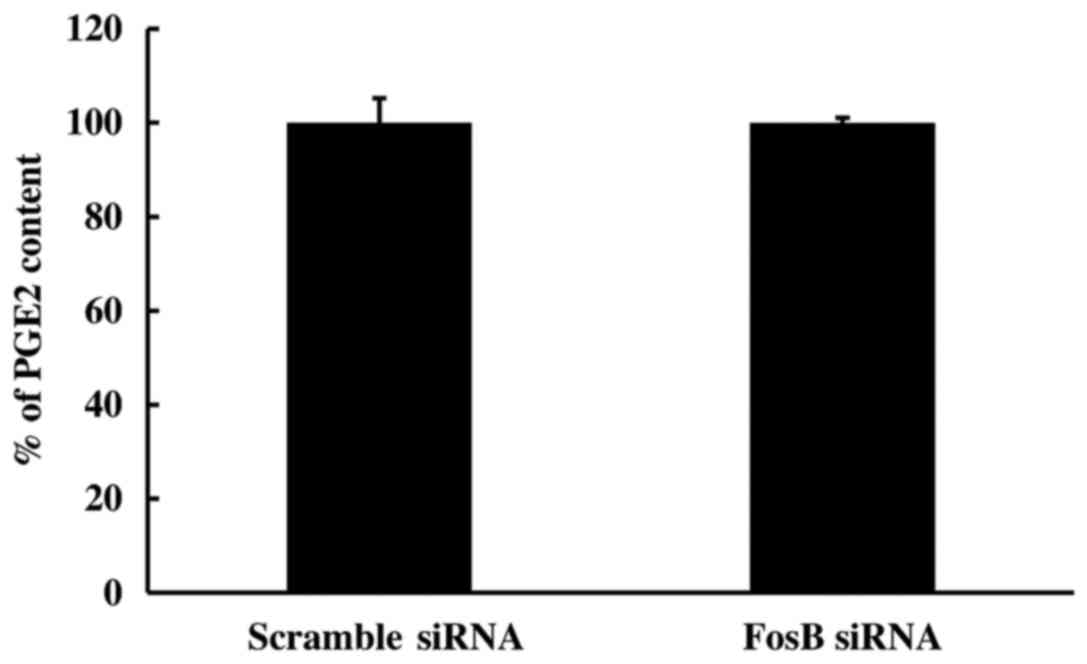
2). To further study the effect of FosB knockdown on the
COX-2/PGE2 axis, the PGE2 content in the medium of FosB-knocked
down cells was measured using ELISA. The extracellular levels of
PGE2 remained unchanged in FosB siRNA-transfected cells, compared
with those in negative control cells (P=0.980; Fig. 3).
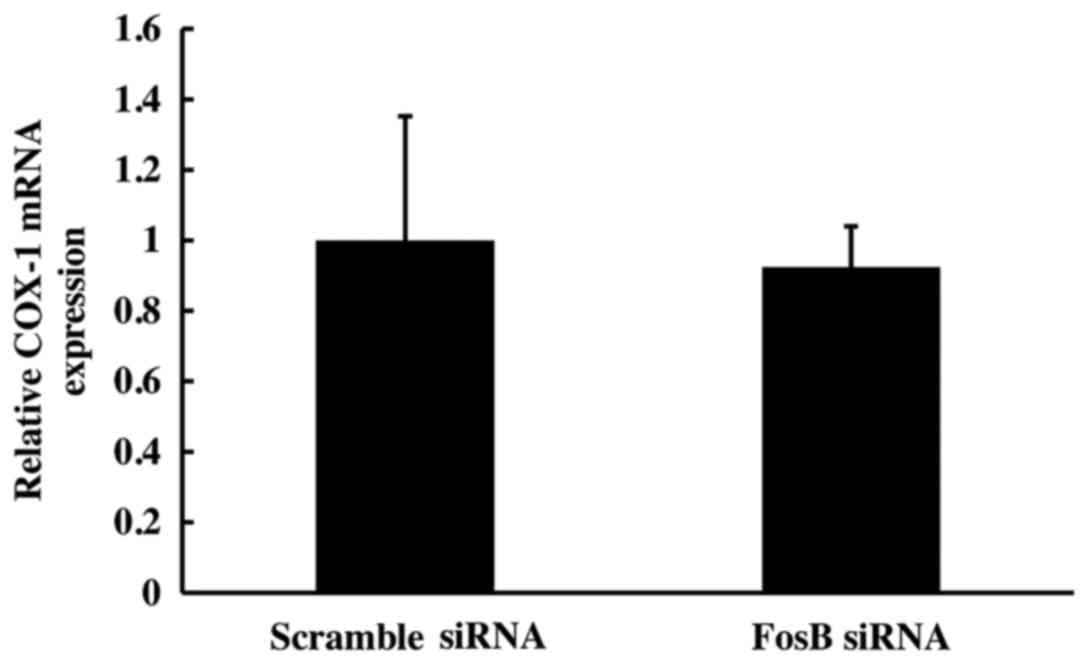
COX-1 mRNA expression is not affected
by FosB knockdown
Following 72 h of FosB siRNA transfection, there was
no significant change in COX-1 mRNA expression between FosB
siRNA-tranfected cells and negative control cells (P>0.450;
Fig. 4).
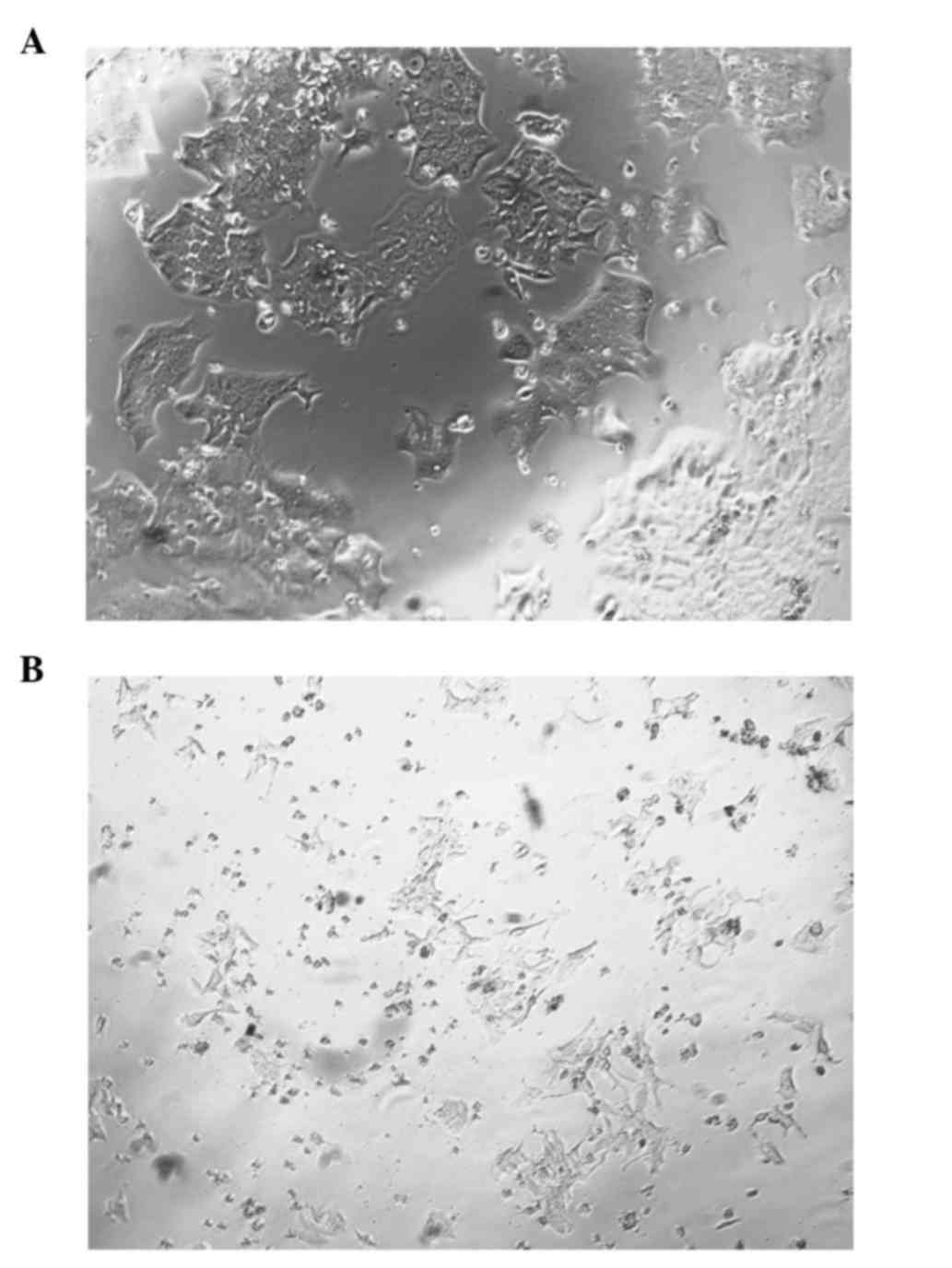
Morphological changes following FosB
knockdown
During the course of the FosB siRNA transfection
experiments, differences in the morphology of FosB-knocked down
cells vs. control cells were noticed. Fig. 5A represents the FosB siRNA-transfected
cells, while Fig. 5B represents the
negative control cells.
Discussion
Inflammation has an important role on CRC
carcinogenesis. Inflammatory molecules, including COX-2 and PGE2,
are increased in tumor tissues of CRC patients, and their high
expression has been associated with poor outcomes (11,12,14,16,17).
Drugs capable of inhibiting COX enzymes such as NSAIDs have
demonstrated chemopreventive and antitumoral potential in animal
models and humans (4–8). However, the therapeutic effects of
NSAIDs varied among different CRC patients. These discrepancies may
be due to the variations in the expression levels of COX-2 and gene
mutational status across patients (24–26), since
those with high COX-2 expression levels and mutations in
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha exhibited improved survival with NSAID treatment (24).
Despite the important role of COX-2 and PGE2 in CRC,
the mechanisms that regulate the expression of these two molecules
are still not well understood. Previous studies by the present
authors correlated the expression of COX-2, but not that of COX-1,
to the PGE2 content in tissues from CRC patients (14), thus suggesting a common regulatory
mechanism for COX-2 expression and PGE2 production. Since PGE2 is a
downstream product of COX-2, it is possible to assume that the
expression of COX-2 may affect the levels of PGE2. A previous study
by the present authors proposed FosB as an important transcription
factor in CRC patients with high COX-2 tumor tissue expression,
since FosB was overexpressed in these patients, comparing with
patients who exhibited low COX-2 expression (19). FosB is a member of the AP-1 complex,
which has been linked to malignant transformation in several types
of cancer, and its components, including FosB, have important roles
on cell proliferation (among other functions), which suggests their
involvement in cancer development (27–30). In
the present study, the role of FosB on the COX-2/PGE2 axis was
investigated. The results revealed that FosB knockdown induced an
important decrease on COX-2 expression, while the PGE2 content was
not changed. These results suggest that the levels of PGE2 may be
regulated by mechanisms independent of COX-2 expression, regardless
of the fact that COX-2 expression is able to predict the content of
PGE2 in CRC tissues. Since the extracellular levels of PGE2 did not
change in cells with FosB knockdown, despite a marked decrease on
COX-2 mRNA expression in these cells, the present findings imply
that there are other important players in the production of PGE2.
As PGE2 could also be synthesized by COX-1, the expression levels
of this enzyme were also measured in the present study, and it was
observed that the knockdown of FosB did not affect COX-1
expression, which is in agreement with a constitutive expression of
COX-1 in the tested cells. Since FosB knockdown is able to decrease
COX-2 expression, but neither COX-1 expression nor PGE2 content, it
is possible to suggest that FosB transcription factor is important
in the regulation of COX-2 expression, but is not involved in the
regulation of enzymes catalyzing the direct synthesis of PGE2 or in
the regulation of COX-1 expression. In the present study, important
changes in cell morphology were observed subsequent to FosB
knockdown, which may be explained by the role of FosB in processes
such as stress response, proliferation and apoptosis (20).
Previous reports support the present finding that
PGE2 content is not changed upon decreasing COX-2 expression
(31–35). A number of authors have demonstrated
that in the
Vhl∆IE/Apcmin/+
intestinal tumor model, the production of PGE2 is not dependent on
the levels of COX-2, since despite the blockage of COX-2 activity
by nimesulide, the PGE2 content remained elevated, suggesting that
the high PGE2 levels observed were due to an increase in mPGES1
expression (31,32). The high expression of COX-2 and mPGES1
in their studies was explained by the direct activation of the
promoter region of these two genes by hypoxia-inducible factor 2α
(31), which associates hypoxia with
the COX-2/mPGES1/PGE2 axis. In other report, PGs were able to
induce mPGES1 expression through the activation of early growth
response 1, implying a positive regulatory feedback between these
components (33). In addition, mPGES1
has been observed to be increased in CRC tumor tissues and to
correlate with a worse prognosis (34). Almendingen et al (35) reported that treatment with rofecoxib,
a COX-2 selective inhibitor, did not affect the PGE2 levels in
plasma of patients with familial adenomatous polyposis. Altogether,
those findings offer an explanation for the present results. Thus,
it is possible that high expression of mPGES1 contributes to high
PGE2 levels independently of a reduced COX-2 expression.
Considering that the levels of COX-1 and PGE2 remain unchanged
following FosB knockdown, it is also possible that basal COX-1
expression maintains the PGE2 levels. Further studies are required
to investigate in more detail the regulation of COX-2 expression
and PGE2 content, as well as the role of FosB as a regulatory
factor in COX-2 expression.
The present study aids to explain the regulatory
mechanisms behind the expression of COX-2. As NSAIDs have been
regarded as potential chemopreventive and/or adjuvant agents for
cancer treatment, particularly in CRC patients (19), due to their inhibitory effect on COX
enzymes, it is important to understand the mechanisms by which
these enzymes exert their actions in the process of carcinogenesis.
The development of more effective therapies using combinations of
NSAIDs and other inhibitors of the PG pathways depend on a deeper
knowledge in this matter.
In conclusion, the present study has demonstrated
that FosB partly regulates COX-2 expression in HCA-7 cells with no
apparent participation in the regulation of PGE2 production.
Acknowledgements
D. L. C-M was a PhD student in the Doctoral Program
in Biomedical Sciences of the National Autonomous University of
Mexico (Mexico City, Mexico), and received a scholarship from the
National Council of Science and Technology (Mexico City, Mexico;
grant no. 245314), the Program of Support for Postgraduate Studies
and the Mobility Program of International Students of the General
Direction of Postgraduate Studies of the National Autonomous
University of Mexico (Mexico City, Mexico). The present study was
partly supported by grants from the Swedish Cancer Society
(Stockholm, Sweden; grant nos. 2014 and 4261), the Swedish Research
Council (Stockholm, Sweden; grant nos. 08712, 13268 and 11611),
Assar Gabrielsson Foundation (AB Volvo, Gothenburg, Sweden), the
Magnus Bergvall Foundation (Stockholm, Sweden), the Sahlgrenska
University Hospital Foundation (Gothenburg, Sweden), the Wilhelm
and Martina Lundgren Foundation (Stockholm, Sweden) and the Tore
Nilsson Foundation (Stockholm, Sweden).
References
|
1
|
International Agency for Research on
Cancer, . GLOBOCAN 2012: Estimated cancer incidence, mortality and
prevalence worldwide in 2012. http://globocan.iarc.fr/Pages/fact_sheets_population.aspxAccessed
January 5, 2016.
|
|
2
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lutgens MW, van Oijen MG, van der Heijden
GJ, Vleggaar FP, Siersema PD and Oldenburg B: Declining risk of
colorectal cancer in inflammatory bowel disease: An updated
meta-analysis of population-based cohort studies. Inflamm Bowel
Dis. 19:789–799. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lundholm K, Gelin J, Hyltander A, Lönnroth
C, Sandström R, Svaninger G, Körner U, Gülich M, Kärrefors I and
Norli B: Anti-inflammatory treatment may prolong survival in
undernourished patients with metastatic solid tumors. Cancer Res.
54:5602–5606. 1994.PubMed/NCBI
|
|
5
|
Thun MJ, Namboodiri MM and Heath CW Jr:
Aspirin use and reduced risk of fatal colon cancer. N Engl J Med.
325:1593–1596. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Narisawa T, Sato M, Tani M, Kudo T,
Takahashi T and Goto A: Inhibition of development of
methylnitrosourea-induced rat colon tumors by indomethacin
treatment. Cancer Res. 41:1954–1957. 1981.PubMed/NCBI
|
|
7
|
Rao CV, Tokumo K, Rigotty J, Zang E,
Kelloff G and Reddy BS: Chemoprevention of colon carcinogenesis by
dietary administration of piroxicam, alpha-difluoromethylornithine,
16 alpha-fluoro-5-androsten-17-one, and ellagic acid individually
and in combination. Cancer Res. 51:4528–4534. 1991.PubMed/NCBI
|
|
8
|
Moorghen M, Ince P, Finney KJ, Sunter JP,
Appleton DR and Watson AJ: A protective effect of sulindac against
chemically-induced primary colonic tumours in mice. J Pathol.
156:341–347. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vane JR: Inhibition of prostaglandin
synthesis as a mechanism of action for aspirin-like drugs. Nat New
Biol. 231:232–235. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rouzer CA and Marnett LJ: Cyclooxygenases:
Structural and functional insights. J Lipid Res. 50:Suppl. S29–S34.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sano H, Kawahito Y, Wilder RL, Hashiramoto
A, Mukai S, Asai K, Kimura S, Kato H, Kondo M and Hla T: Expression
of cyclooxygenase-1 and −2 in human colorectal cancer. Cancer Res.
55:3785–3789. 1995.PubMed/NCBI
|
|
12
|
Sheehan KM, Sheahan K, O'Donoghue DP,
MacSweeney F, Conroy RM, Fitzgerald DJ and Murray FE: The
relationship between cyclooxygenase-2 expression and colorectal
cancer. JAMA. 282:1254–1258. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kunzmann AT, Murray LJ, Cardwell CR,
McShane CM, McMenamin UC and Cantwell MM: PTGS2 (cyclooxigenase-2)
expression and survival among colorectal cancer patients: A
systematic review. Cancer Epidemiol Biomarkers Prev. 22:1490–1497.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cahlin C, Lönnroth C, Arvidsson A,
Nordgren S and Lundholm K: Growth associated proteins in tumor
cells and stroma related to disease progression of colon cancer
accounting for tumor tissue PGE2 content. Int J Oncol. 32:909–918.
2008.PubMed/NCBI
|
|
15
|
Wang D and DuBois RN: An inflammatory
mediator, prostaglandin E2, in colorectal cancer. Cancer J.
19:502–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rigas B, Goldman IS and Levine L: Altered
eicosanoid levels in human colon cancer. J Lab Clin Med.
122:518–523. 1993.PubMed/NCBI
|
|
17
|
Pugh S and Thomas GA: Patients with
adenomatous polyps and carcinomas have increased colonic mucosal
prostaglandin E2. Gut. 35:675–678. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoshimatsu K, Golijanin D, Paty PB, Soslow
RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K and Dannenberg AJ:
Inducible microsomal prostaglandin E synthase is overexpressed in
colorectal adenomas and cancer. Clin Cancer Res. 7:3971–3976.
2001.PubMed/NCBI
|
|
19
|
Asting AG, Carén H, Andersson M, Lönnroth
C, Lagerstedt K and Lundholm K: COX-2 gene expression in colon
cancer tissue related to regulating factors and promoter
methylation status. BMC Cancer. 11:2382011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ashida R, Tominaga K, Sasaki E, Watanabe
T, Fujiwara Y, Oshitani N, Higuchi K, Mitsuyama S, Iwao H and
Arakawa T: AP-1 and colorectal cancer. Inflammopharmacology.
13:113–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang F, Nam S, Zhao R, Tian Y, Liu L,
Horne DA and Jove R: A novel synthetic derivative of the natural
product berbamine inhibits cell viability and induces apoptosis of
human osteosarcoma cells, associated with activation of JNK/AP-1
signaling. Cancer Biol Ther. 14:1024–1031. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shaulian E and Karin M: AP-1 in cell
proliferation and survival. Oncogene. 20:2390–2400. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kovary K, Rizzo CA, Ryseck RP, Noguchi T,
Raynoschek C, Pelosin JM and Bravo R: Constitutive expression of
FosB and its short form, FosB/SF, induces malignant cell
transformation in rat-A1 cells. New Biol. 3:870–879.
1991.PubMed/NCBI
|
|
24
|
Liao X, Lochhead P, Nishihara R, Morikawa
T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, et
al: Aspirin use, tumor PIK3CA mutation, and colorectal-cancer
survival. N Engl J Med. 367:1596–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chan AT, Ogino S and Fuchs CS: Aspirin use
and survival after diagnosis of colorectal cancer. JAMA.
302:649–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chan AT, Ogino S and Fuchs CS: Aspirin and
the risk of colorectal cancer in relation to the expression of
COX-2. N Engl J Med. 356:2131–2142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Babu RL, Kumar M Naveen, Patil RH,
Devaraju KS, Ramesh GT and Sharma SC: Effect of estrogen and
tamoxifen on the expression pattern of AP-1 factors in MCF-7 cells:
Role of c-Jun, c-Fos, and Fra-1 in cell cycle regulation. Mol Cell
Biochem. 380:143–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harwood FG, Kasibhatla S, Petak I, Vernes
R, Green DR and Houghton JA: Regulation of FasL by NF-kappaB and
AP-1 in Fas-dependent thymineless death of human colon carcinoma
cells. J Biol Chem. 275:10023–10029. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim YJ, Yoon SY, Kim JT, Choi SC, Lim JS,
Kim JH, Song EY, Lee HG, Choi I and Kim JW: NDRG2 suppresses cell
proliferation through down-regulation of AP-1 activity in human
colon carcinoma cells. Int J Cancer. 124:7–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rivat C, Le Floch N, Sabbah M, Teyrol I,
Redeuilh G, Bruyneel E, Mareel M, Matrisian LM, Crawford HC,
Gespach C and Attoub S: Synergistic cooperation between the AP-1
and LEF-1 transcription factors in the activation of the matrilysin
promoter by the src oncogene: Implications in cellular invasion.
FASEB J. 17:1721–1723. 2003.PubMed/NCBI
|
|
31
|
Xue X and Shah YM: Hypoxia-inducible
factor-2α is essential in activating the COX-2/mPGES-1/PGE2
signaling axis in colon cancer. Carcinogenesis. 34:163–169. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xue X, Taylor M, Anderson E, Hao C, Qu A,
Greenson JK, Zimmermann EM, Gonzalez FJ and Shah YM:
Hypoxia-inducible factor-2α activation promotes colorectal cancer
progression by dysregulating iron homeostasis. Cancer Res.
72:2285–2293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stamatakis K, Jimenez-Martinez M,
Jimenez-Segovia A, Chico-Calero I, Conde E, Galán-Martínez J, Ruiz
J, Pascual A, Barrocal B, López-Pérez R, et al: Prostaglandins
induce early growth response 1 transcription factor mediated
microsomal prostaglandin E2 synthase up-regulation for colorectal
cancer progression. Oncotarget. 6:39941–39959. 2015.PubMed/NCBI
|
|
34
|
Seo T, Tatsuguchi A, Shinji S, Yonezawa M,
Mitsui K, Tanaka S, Fujimori S, Gudis K, Fukuda Y and Sakamoto C:
Microsomal prostaglandin E synthase protein levels correlate with
prognosis in colorectal cancer patients. Virchows Arch.
454:667–676. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Almendingen K, Larsen LN, Fausa O, Bratlie
J, Høstmark AT and Aabakken L: Selective COX-2 inhibition affects
fatty acids, but not COX mRNA expression in patients with FAP. Fam
Cancer. 9:571–580. 2010. View Article : Google Scholar : PubMed/NCBI
|