Introduction
Protein tyrosine kinase (PTK)6, also known as breast
tumor kinase, is a non-receptor type kinase that consists of an Src
homology (SH)3 domain, an SH2 domain and a catalytic domain of
tyrosine kinase (1,2). PTK6 is overexpressed in >60% of
breast carcinoma tissue samples and in the majority of breast
cancer cell lines (3). PTK6
expression is also increased in colon, head and neck, ovary,
prostate, lung, bladder, bile duct, pancreas and gastric cancers,
and in T- and B-cell lymphomas (4,5).
Expression of PTK6 enhances the proliferation of
mammary epithelial and breast cancer cells (6). PTK6 also promotes cell migration and
invasion (7). Sublocalization of PTK6
at the plasma membrane is important for its oncogenic potential
(8). Activated PTK6 consistently
accumulates at the plasma membrane in breast cancer cell lines and
tissues (9). Although PTK6 was
detected in the nucleus and cytoplasm of normal mammary gland
epithelial cells, Tyr342 in the PTK6 activation loop was not
phosphorylated, and thus, PTK6 was not active (9).
PTK6 promotes tumorigenicity by enhancing signaling
pathways of receptor tyrosine kinases and is particularly well
known for sensitizing epidermal growth factor receptor (EGFR)
family members (10). Various
downstream substrates and interacting proteins, including signal
transducing adaptor protein-2, paxillin, Akt, p130 Crk-associated
substrate, p190Rho GTPase-activating protein-A and ArfGAP with
RhoGAP domain, ankyrin repeat and PH domain 1, contribute to the
oncogenic roles of PTK6 (11,12). Similar to other PTKs, mutations of
PTK6 identified in different cancer types increase its kinase
activity (13).
In view of its oncogenic activity and its presence
in various carcinomas such as breast cancer, PTK6 is a potentially
valuable therapeutic target for decelerating or arresting tumor
growth (14).
(E)-5-(Benzylideneamino)-1H-benzo[d] imidazol-2(3H)-one derivatives
were previously developed as novel PTK6 inhibitors that exhibited
little cytotoxicity, excellent inhibition in vitro and at
the cellular level, and selectivity for PTK6 (15). Imidazo[1,2-a]pyrazin-8-amines and
4-anilino α-carbolines were also identified as PTK6-selective
inhibitors that block its catalytic activity (16,17). In
the present study, pyrazolopyrimidine (PP)1 [4-amino-5-
(4-methylphenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine], PP2
[(4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]
pyrimidine] and a lymphocyte-specific protein tyrosine kinase (Lck)
inhibitor [4-amino-5-(4-phenoxyphenyl)-7H-pyrrolo
[3,2-d]pyrimidin-7-yl-cyclopentane] were screened as potent PTK6
inhibitors among the evaluated kinase inhibitors. The selectivity
of these compounds for PTK6 and for other PTK family members was
analyzed in HEK 293 cells, and it was then examined whether these
compounds inhibited PTK6-dependent signaling processes and the
proliferation of breast carcinoma T-47D cells.
Materials and methods
Chemicals
PP1, PP2 and the aforementioned Lck inhibitor were
purchased from Calbiochem (EMD Millipore, Billerica, MA, USA).
Genistein was purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA).
Cell culture
Human embryonic kidney (HEK) 293 cells and human
breast cancer T-47D cells (both American Type Culture Collection,
Manassas, VA, USA) were maintained in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) containing 10% fetal bovine serum (FBS; HyClone; GE Healthcare
Life Sciences, Logan, UT, USA) at 37°C in a CO2
incubator with a humidified atmosphere of 5% CO2 and 95%
air.
ELISA-based in vitro kinase assay for
PTK6
ELISA plates (96-well; Greiner Bio-One GmbH,
Frickenhausen, Germany) were incubated with 100 µl of 0.1 mg/ml
Poly (Glu, Tyr) (Glu:Tyr, 4:1; Sigma-Aldrich; Merck Millipore,
Darmstadt, Germany) in PBS for 16 h at 37°C, and then washed three
times with PBS. The Poly (Glu, Tyr)-coated wells were blocked with
1% bovine serum albumin in PBS for 1 h at 37°C, washed three times
with PBS, and then incubated for 30 min at room temperature with 10
nM glutathione S-transferase-fused PTK6 catalytic domain (18) in 20 µl of kinase reaction buffer [20
mM Tris-HCl (pH 7.4), 10 mM MgCl2, 1 mM MnCl2
and 50 µM Na3VO4) in the presence of the
chemical of interest (Table I)
containing a final concentration of 1% dimethyl sulfoxide. The
phosphorylation of tyrosine residues was initiated by addition of
300 µM adenosine triphosphate (ATP) to the reaction mixtures. The
wells were washed three times with PBS after incubation for 20 min
at room temperature. For the quantification of phosphorylated
tyrosines, the wells were incubated with anti-phospho-tyrosine
(4G10; 1:1,000; EMD Millipore) and horseradish
peroxidase-conjugated anti-mouse immunoglobulin G (K0211589;
1:10,000; Koma Biotech, Seoul, Korea) antibodies for 1 h each at
room temperature. The optical density was measured with a
3,3′,5,5′-tetramethylbenzidine solution (Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol.
 | Table I.PTK6 inhibitory activities of protein
kinase inhibitors screened from a targeted kinase inhibitor. |
Table I.
PTK6 inhibitory activities of protein
kinase inhibitors screened from a targeted kinase inhibitor.
| Chemical name | PubChem compound
identifier | Molecular weight
(Da) | Known target | In vitro
IC50 (nM)a |
|---|
| Genistein | 5280961 | 270.2 | PTKs | >10,000 |
| PP1 | 1400 | 281.4 | Src, Fyn | <300 |
| PP2 | 4878 | 301.8 | Lck, Fyn, Hck | <300 |
| Lck inhibitor | 6603792 | 370.5 | Lck, Src | <300 |
| LFM-A13 | 54676905 | 360.0 | Btk | >10,000 |
| JAK inhibitor I | 5494425 | 309.3 | JAK1- 3 | >10,000 |
| Syk inhibitor | 6419747 | 353.4 | Syk | >3,000 |
| Emodin | 3220 | 270.2 | Lck, ErbB2 | >10,000 |
| Erbstatin analog | 5353609 | 194.2 | EGFR, Abl | >300 |
| Lavendustin C | 3896 | 275.3 | EGFR, Src,
CaMKII | >300 |
| Daphnetin | 5280569 | 178.1 | EGFR, PKA, PKC | >3,000 |
| PD 157432 | 10085225 | 283.3 | EGFR, ErbB2 | >10,000 |
| Flt-3 inhibitor | 1048845 | 360.4 | Flt-3 | >10,000 |
| cFMS inhibitor | 11617559 | 366.4 | FMS | >10,000 |
| GTP-14564 | 3385203 | 234.3 | FMS, Kit, Flt-3,
PDGFRB | >10,000 |
| PDGFR inhibitor
I | 5330535 | 276.3 |
PDGFRB | >10,000 |
| Oxindole I | 5908088 | 210.2 | VEGFR1, PDGFRB | >10,000 |
| VEGFR2 inhibitor
II | 23301538 | 343.2 | VEGFR2, PDGFRB | >3,000 |
| TrkA inhibitor | 5413390 | 315.4 | TrkA | >10,000 |
|
Picropodophyllin | 72435 | 414.4 | IGF1R | >10,000 |
| JNK inhibitor
II | 8515 | 220.2 | JNK1-3 | >10,000 |
Western blot analysis
HEK 293 cells expressing hyperactive PTK6
(Flag-PTK6-3PA/Y447F) (19), Src,
Fyn, Lck, bone marrow tyrosine kinase gene on chromosome X (Bmx) or
EGFR were treated with the indicated concentrations of compounds
for 2 days. Western blot analysis and immunoprecipitation were
performed as previously described (15). Immunoreactive proteins were visualized
using anti-phospho-tyrosine (4G10; 1:1,000), anti-PTK6 (sc-1188;
1:2,000; Santa Cruz Biotechnology, Inc.), anti-phospho-signal
transducer and activator of transcription (STAT)3 (sc-8059;
1:2,000; Santa Cruz Biotechnology, Inc.), anti-STAT3 (sc-7179;
1:2,000; Santa Cruz Biotechnology, Inc.) and anti-GAPDH (AbC-2003,
1:2,000; AbClon, Inc., Seoul, Korea) primary antibodies overnight
at 4°C, followed by incubation with a horseradish
peroxidase-conjugated secondary antibody (K0211589 or K0211708;
1:10,000; Koma Biotech, Seoul, Republic of Korea) for 1 h at room
temperature and an enhanced chemiluminescence detection kit (EMD
Millipore). For the quantification of the phosphorylation levels in
the cell lysates, chemiluminescence was detected using a LAS-3000
imaging system (Fujifilm, Tokyo, Japan) and analyzed using Multi
Gauge version 2.2 software (Fujifilm). The half maximal inhibitory
concentration (IC50) at the cellular level was
determined by quantifying the phosphorylation levels in the HEK 293
cell system. The reference level was the phosphorylation level of
the chemical-free control, which was set at 100%.
MTT assay
Subconfluent empty vector-transfected and
PTK6-knockdown T-47D cells were incubated for 4 days in DMEM-10%
FBS containing various concentrations of the chemicals. Viable
cells were measured using MTT assay, as previously described
(15). The viability of
chemical-free, vector-transfected T-47D cells was set at 100%.
Statistical analysis
All data were expressed as the mean ± standard
deviation of three independent experiments. Statistical analysis
was performed using Microsoft Excel (version, 2007; Microsoft
Corporation, Redmond, WA, USA). The significant differences between
the groups were assessed using a Student's t-test. P>0.05 was
considered to indicate a statistically significant difference.
Results
PP1, PP2 and Lck inhibitor inhibit the
catalytic activity of PTK6 in vitro
Protein kinase inhibitors were analyzed for the
inhibition of the PTK6 catalytic activity using an ELISA-based
in vitro kinase assay system for PTK6. Among the tested
kinase inhibitors, PP1, PP2 and the Lck inhibitor exhibited strong
inhibition of PTK6 (Table I). The
IC50 values for PP1, PP2 and the Lck inhibitor were
230.0, 50.0 and 60.0 nM, respectively (Table II).
| Table II.Inhibition of the catalytic activity
of purified PTK6 by selected kinase inhibitors in vitro. |
PP1, PP2 and Lck inhibitor are highly
selective for PTK6 at the cellular level
PP1, PP2 and the aforementioned Lck inhibitor were
developed as Src family kinase (SFK) inhibitors (20,21). The
present study analyzed the selectivity of each inhibitor for the
inhibition of several PTKs, including PTK6, SFK members (Src, Fyn
and Lck), a non-receptor type PTK (Bmx) and a receptor type PTK
(EGFR). HEK 293 cells expressing hyperactive PTK6, Src, Fyn, Lck,
Bmx or EGFR were incubated in the presence of various
concentrations of these inhibitors. Inhibition of each PTK activity
at the cellular level was assessed for each inhibitor by measuring
the decrease in tyrosine phosphorylation levels of cellular
proteins via western blot analysis using an anti-phospho-tyrosine
antibody. As expected, PP1, PP2 and the Lck inhibitor exhibited
strong inhibition of SFK members. The IC50 value of PP1
to Lck was 1.76 µM; the IC50 value of PP2 to Lck was
4.36 µM; and the IC50 values of the Lck inhibitor to
Lck, Fyn and Src were 0.37, 1.22 and 3.46 µM, respectively
(Table III). Unexpectedly, PP1, PP2
and the Lck inhibitor inhibited PTK6 to a greater degree than SFK
members. The IC50 values of PP1, PP2 and the Lck
inhibitor to PTK6 were 2.5, 13.0 and 53.0 nM, respectively
(Table III and Fig. 1, top panel). This result suggested
that PP1, PP2 and the Lck inhibitor were highly selective for PTK6
compared with other PTKs, including the SFK family members, at the
cellular level.
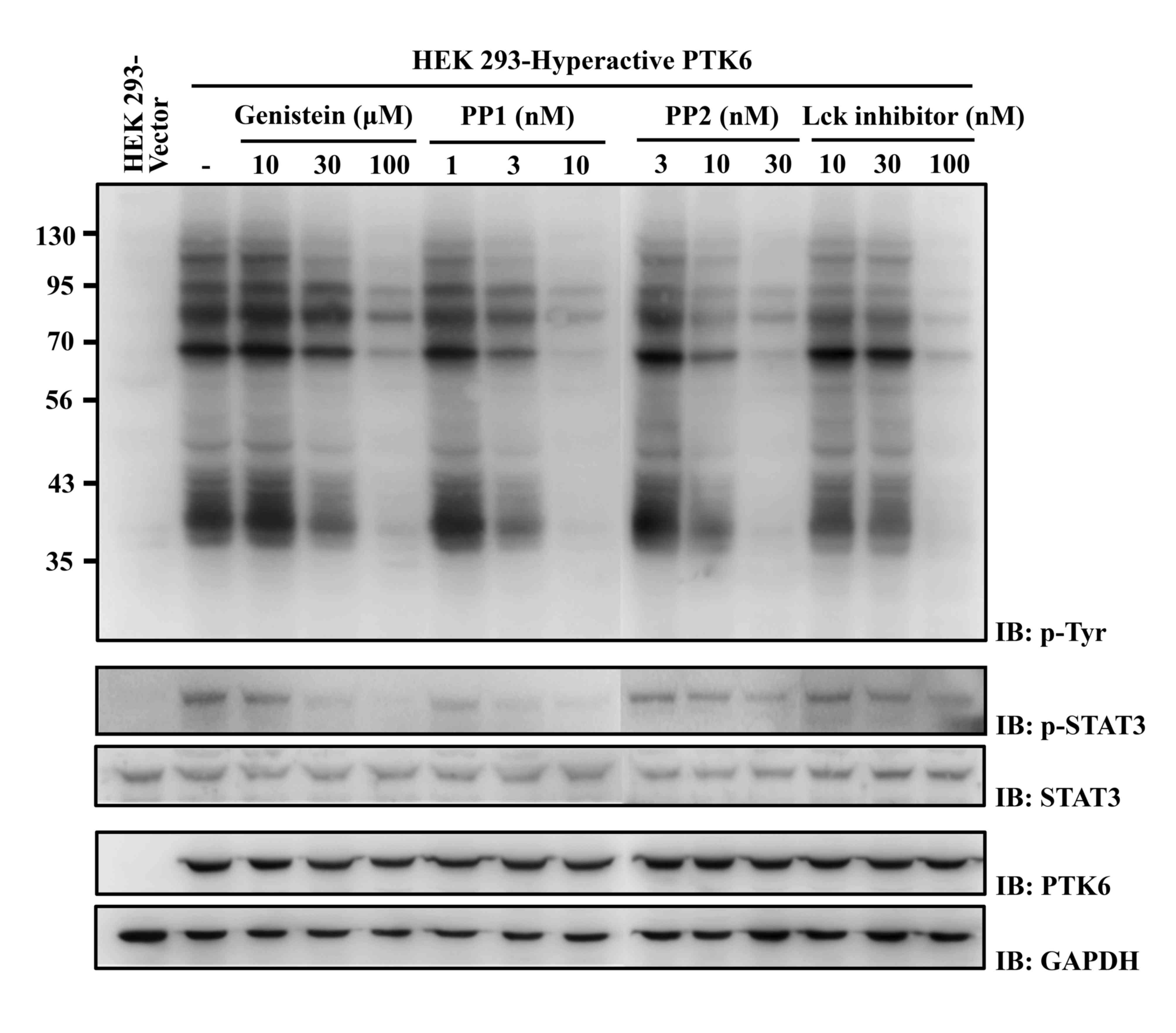 | Figure 1.Effects of selected chemicals on the
phosphorylation of cellular proteins and a specific PTK6 substrate,
STAT3, in HEK 293 cells. HEK 293 cells stably expressing
hyperactive PTK6 were incubated with the indicated concentrations
of chemicals for 48 h. Cell lysates were analyzed by western
blotting using anti-phospho-tyrosine (4G10), anti-phospho-STAT3,
anti-STAT3, anti-PTK6 and anti-GAPDH antibodies. The numbers on the
left of the top panel indicate apparent molecular weights in kDa.
HEK, human embryonic kidney; PP, pyrazolopyrimidine; Lck,
lymphocyte-specific protein tyrosine kinase; PTK, protein tyrosine
kinase; STAT, signal transducer and activator of transcription; p-,
phosphorylated. |
| Table III.Selectivity of genistein, PP1, PP2
and a Lck inhibitor for various PTKs in the HEK 293 cell
system. |
Table III.
Selectivity of genistein, PP1, PP2
and a Lck inhibitor for various PTKs in the HEK 293 cell
system.
|
| IC50 at
the cellular level (nM)a |
|---|
|
|
|
|---|
| PTKs | Genistein | PP1 | PP2 | Lck inhibitor |
|---|
| PTK6 | 43,180±1,340 | 2.5±0.3 | 13±2 | 53±7 |
| Src | >100,000 | 70,300±7,320 | 53,060±8,230 | 3,460±850 |
| Fyn | 70,640±8,520 | 13,630±3,160 | 35,980±1,030 | 1,220±50 |
| Lck | >100,000 | 1,760±250 | 4,360±730 | 370±40 |
| Bmx | 69,550±7,460 | 2,390±430 | 16,460±2,780 | 15,980±3,680 |
| EGFR | 24,830±1,590 | 21,840±1,140 | 56,800±3,920 | >10,000 |
PP1, PP2 and Lck inhibitor inhibit the
phosphorylation of PTK6 substrate proteins in HEK 293 cells
To analyze whether PP1, PP2 and the Lck inhibitor
block the PTK6-mediated signaling pathway, HEK 293 cells expressing
hyperactive PTK6 were treated with these chemicals at
concentrations i) lower than, ii) approximately the same as, and
iii) higher than their IC50 values. Phosphorylation of
STAT3, which is a known specific substrate of PTK6 (22), was inhibited at concentrations equal
or greater than the IC50 value of each chemical
(Fig. 1, mid panel).
PP1 and PP2 inhibit the PTK6-mediated
proliferation of T-47D cells
PTK6 is often expressed in breast cancer cell lines
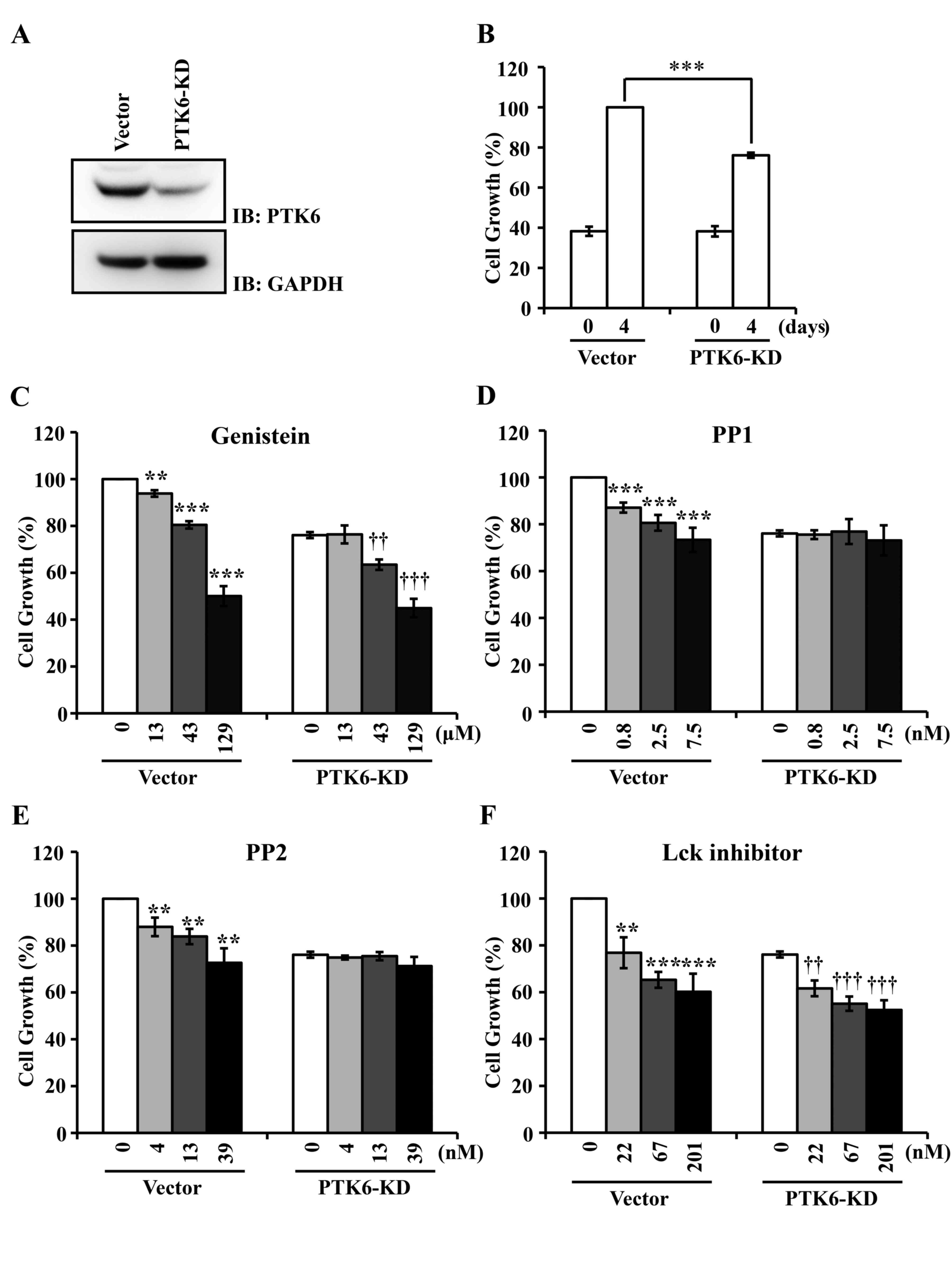
(3). Knockdown of PTK6 decreases the
proliferation of breast cancer cells (14). Consistent with this observation, the
silencing of PTK6 in breast carcinoma T-47D cells using a small
hairpin RNA vector suppressed ~24% of cell proliferation, compared
with vector transfection (Fig. 2A and
B). PP1, PP2, the Lck inhibitor and genistein (used as a
control) were applied to T-47D cells at concentrations of 0.33-, 1-
and 3-fold their IC50 values. PP1 and PP2 inhibited the
proliferation of vector-transfected T-47D cells in a dose-dependent
manner, but did not affect the proliferation of PTK6-knockdown
T-47D cells at values of ≤3-fold their IC50 values
(Fig. 2D and E). However, the Lck
inhibitor and genistein suppressed the proliferation of both
vector-transfected and PTK6-knockdown T-47D cells (Fig. 2C and F). These results suggest that
the Lck inhibitor is not specific for PTK6, which is similar to the
general PTK inhibitor genistein (23).
Discussion
In our study, PP1, PP2 and a Lck inhibitor were
screened as potential inhibitors for PTK6 using an in vitro
kinase assay. These chemicals were initially developed as
ATP-competitive inhibitors of SFKs (21,22). PP1
inhibits Lck, Fyn and Src with IC50 values of 5, 6 and
170 nM, respectively, as assessed with an in vitro kinase
assay (20). PP2 inhibits Lck and Fyn
with IC50 values of 5 and 6 nM, respectively (20). The Lck inhibitor was developed as a
derivative of PP1 and PP2, and inhibits Lck and Src with
IC50 values of <1 and 70 nM, respectively (21). These chemicals were widely used to
investigate the physiological roles for SFKs, but were not used in
human clinical trials. PTK6 is evolutionarily distinct from, but
still closely associated with, SFKs (2). Thus, it is expected that PP1, PP2 and
the Lck inhibitor inhibit PTK6.
Although PP1 and PP2 are more selective for SFKs
than the previous generation of PTK inhibitors (including
herbimycin A and genistein), they can inhibit off-target kinases
(including C-terminal Src kinase, ephrin type-A receptor 2,
platelet-derived growth factor receptor, fibroblast growth factor
receptor 1, p21 activated kinase, receptor-interacting protein 2,
p38 and casein kinase 1δ) with sufficient potency (24–27). When
the selectivity of these chemicals for various PTKs was analyzed in
HEK 293 cells expressing one of the PTKs, they displayed high
selectivity for PTK6 over various SFK members, including Src, Fyn,
Lck, and other PTK family members such as Bmx and EGFR. In
particular, PP1 and PP2 inhibited PTK6 activity at IC50
values of 2.5 and 13.0 nM, respectively. Although they also
inhibited the catalytic activities of other PTKs, including SFKs,
their IC50 values were mostly at micro-molar
concentrations. Incubation of T-47D cells with 0.8–7.5 nM PP1 or
4.0–39.0 nM PP2 reduced the PTK6-dependent proliferation of T-47D
cells without a decrease in the proliferation of PTK6-knockdown
cells. The Lck inhibitor also exhibited inhibitory selectivity for
PTK6, but reduced the proliferation of T-47D cells in a
PTK6-independent manner. These results demonstrate that PP1 and PP2
reduce the PTK6-mediated signaling pathways and cell proliferation
in PTK6-positive cells. Thus, it can be suggested that PP1 and PP2
could be applied as therapeutic agents in PTK6-positive malignant
diseases.
Resistance to chemotherapy and molecularly targeted
therapies is a major problem confronting current cancer research.
The results of a recent study indicated that PTK6 confers
resistance of breast cancer SUM102 cells to cetuximab, an
EGFR-blocking antibody that is approved for the treatment of
several types of human solid tumors (28). Knockdown of PTK6 sensitized the cells
to cetuximab by inducing apoptosis. Assuming that PTK6 catalytic
activity is essential for drug resistance (28), PTK6 inhibitors such as PP1 and PP2 may
be useful for the treatment of chemotherapy-resistant cancer
cells.
Acknowledgements
The present study was supported by a grant from the
National Research Foundation of Korea funded by the Ministry of
Science, ICT & Future Planning (grant no.
2014M3C9A2064536).
References
|
1
|
Mitchell PJ, Barker KT, Martindale JE,
Kamalati T, Lowe PN, Page MJ, Gusterson BA and Crompton MR: Cloning
and characterisation of cDNAs encoding a novel non-receptor
tyrosine kinase, brk, expressed in human breast tumours. Oncogene.
9:2383–2390. 1994.PubMed/NCBI
|
|
2
|
Lee H, Kim M, Lee KH, Kang KN and Lee ST:
Exon-intron structure of the human PTK6 gene demonstrates that PTK6
constitutes a distinct family of non-receptor tyrosine kinase. Mol
Cells. 8:401–407. 1998.PubMed/NCBI
|
|
3
|
Barker KT, Jackson LE and Crompton MR: BRK
tyrosine kinase expression in a high proportion of human breast
carcinomas. Oncogene. 15:799–805. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brauer PM and Tyner AL: Building a better
understanding of the intracellular tyrosine kinase PTK6-BRK by BRK.
Biochim Biophys Acta. 1806:66–73. 2010.PubMed/NCBI
|
|
5
|
Mizuguchi Y, Specht S, Isse K, Sasatomi E,
Lunz JG III, Takizawa T and Demetris AJ: Breast tumor
kinase/protein tyrosine kinase 6 (Brk/PTK6) activity in normal and
neoplastic biliary epithelia. J Hepatol. 63:399–407. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kamalati T, Jolin HE, Mitchell PJ, Barker
KT, Jackson LE, Dean CJ, Page MJ, Gusterson BA and Crompton MR:
Brk, a breast tumor-derived non-receptor protein-tyrosine kinase,
sensitizes mammary epithelial cells to epidermal growth factor. J
Biol Chem. 271:30956–30963. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen HY, Shen CH, Tsai YT, Lin FC, Huang
YP and Chen RH: Brk activates rac1 and promotes cell migration and
invasion by phosphorylating paxillin. Mol Cell Biol.
24:10558–10572. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ie Kim H and Lee ST: Oncogenic functions
of PTK6 are enhanced by its targeting to plasma membrane but
abolished by its targeting to nucleus. J Biochem. 146:133–139.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peng M, Emmadi R, Wang Z, Wiley EL, Gann
PH, Khan SA, Banerji N, McDonald W, Asztalos S, Pham TN, et al:
PTK6/BRK is expressed in the normal mammary gland and activated at
the plasma membrane in breast tumors. Oncotarget. 5:6038–6048.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ostrander JH, Daniel AR and Lange CA:
Brk/PTK6 signaling in normal and cancer cell models. Curr Opin
Pharmacol. 10:662–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goel RK and Lukong KE: Tracing the
footprints of the breast cancer oncogene BRK-Past till present.
Biochim Biophys Acta. 1856:39–54. 2015.PubMed/NCBI
|
|
12
|
Kang SA, Lee ES, Yoon HY, Randazzo PA and
Lee ST: PTK6 inhibits down-regulation of EGF receptor through
phosphorylation of ARAP1. J Biol Chem. 285:26013–26021. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsui T and Miller WT: Cancer-associated
mutations in breast tumor kinase/PTK6 differentially affect enzyme
activity and substrate recognition. Biochemistry. 54:3173–3182.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harvey AJ and Crompton MR: The Brk protein
tyrosine kinase as a therapeutic target in cancer: Opportunities
and challenges. Anticancer Drugs. 15:107–111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shim HJ, Yang HR, Kim HIe, Kang SA, No KT,
Jung YH and Lee ST: Discovery of
(E)-5-(benzylideneamino)-1H-benzo[d]imidazol-2(3H)-one derivatives
as inhibitors for PTK6. Bioorg Med Chem Lett. 24:4659–4663. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zeng H, Belanger DB, Curran PJ, Shipps GW
Jr, Miao H, Bracken JB, Siddiqui M Arshad, Malkowski M and Wang Y:
Discovery of novel imidazo[1,2-a]pyrazin-8-amines as Brk/PTK6
inhibitors. Bioorg Med Chem Lett. 21:5870–5875. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mahmoud KA, Krug M, Wersig T, Slynko I,
Schächtele C, Totzke F, Sippl W and Hilgeroth A: Discovery of
4-anilino α-carbolines as novel Brk inhibitors. Bioorg Med Chem
Lett. 24:1948–1951. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim HIe and Lee ST: An intramolecular
interaction between SH2-kinase linker and kinase domain is
essential for the catalytic activity of protein-tyrosine kinase-6.
J Biol Chem. 280:28973–28980. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang SA, Cho HS, Yoon JB, Chung IK and Lee
ST: Hsp90 rescues PTK6 from proteasomal degradation in breast
cancer cells. Biochem J. 447:313–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hanke JH, Gardner JP, Dow RL, Changelian
PS, Brissette WH, Weringer EJ, Pollok BA and Connelly PA: Discovery
of a novel, potent and Src family-selective tyrosine kinase
inhibitor. Study of Lck- and FynT-dependent T cell activation. J
Biol Chem. 271:695–701. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arnold LD, Calderwood DJ, Dixon RW,
Johnston ND, Kamens JS, Munschauer R, Rafferty P and Ratnofsky SE:
Pyrrolo[2,3-d]pyrimidines containing an extended 5-substituent as
potent and selective inhibitors of lck I. Bioorg Med Chem Lett.
10:2167–2170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu L, Gao Y, Qiu H, Miller WT, Poli V and
Reich NC: Identification of STAT3 as a specific substrate of breast
tumor kinase. Oncogene. 25:4904–4912. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smal C, Cardoen S, Bertrand L, Delacauw A,
Ferrant A, Van den Berghe G, Van Den Neste E and Bontemps F:
Activation of deoxycytidine kinase by protein kinase inhibitors and
okadaic acid in leukemic cells. Biochem Phamacol. 68:95–103. 2004.
View Article : Google Scholar
|
|
24
|
Summy JM, Trevino JG, Lesslie DP, Baker
CH, Shakespeare WC, Wang Y, Sundaramoorthi R, Metcalf CA III, Keats
JA, Sawyer TK and Gallick GE: AP23846, a novel and highly potent
Src family kinase inhibitor, reduces vascular endothelial growth
factor and interleukin-8 expression in human solid tumor cell lines
and abrogates downstream angiogenic processes. Mol Cancer Ther.
4:1900–1911. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma QL, Yang F, Frautschy SA and Cole GM:
PAK in Alzheimer disease, Huntington disease and X-linked mental
retardation. Cell Logist. 2:117–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bain J, McLauchlan H, Elliott M and Cohen
P: The specificities of protein kinase inhibitors: An update.
Biochem J. 371:199–204. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bain J, Plater L, Elliott M, Shpiro N,
Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR and
Cohen P: The selectivity of protein kinase inhibitors: A further
update. Biochem J. 408:297–315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li X, Lu Y, Liang K, Hsu JM, Albarracin C,
Mills GB, Hung MC and Fan Z: Brk/PTK6 sustains activated EGFR
signaling through inhibiting EGFR degradation and transactivating
EGFR. Oncogene. 31:4372–4383. 2012. View Article : Google Scholar : PubMed/NCBI
|