Introduction
Lung cancer is one of the most common malignant
tumors with the highest morbidity and mortality, and non-small cell
lung cancer (NSCLC) accounts for >80% of the recorded cases
(1). Epidermal growth factor receptor
(EGFR) tyrosine kinase inhibitors (TKIs), including gefitinib, are
tumor molecular-targeted agents, which have become an important
therapeutic strategy for NSCLC (2).
Icotinib hydrochloride (icotinib) was developed in China, and is a
highly selective first-generation EGFR-TKI (3). Icotinib is indicated for the treatment
of EGFR mutation-positive, advanced or metastatic NSCLC, or as a
second- or third-line treatment (3).
However, the majority of patients with NSCLC develop an acquired
resistance to EGFR-TKIs 8–10 months following the initiation of
treatment (2,4). It has been previously demonstrated that
a second point mutation on EGFR in exon 20 (T790 M) and a MET gene
amplification underlie the development of an acquired resistance to
EGFR-TKIs in patients with NSCLC (5–8). However,
the mechanisms underlying acquired resistance are not determined
for ~30% of cases (5).
Phosphatase and tensin homolog (PTEN; deleted on
chromosome 10) is a tumor-suppressor gene that has dual-specificity
phosphatase activity (9). PTEN is
mutated in various types of human cancer and regulates cellular
growth using hyperphosphorylation and dephosphorylation (10). PTEN may be involved in the
infiltration and migration of tumor cells and may affect vessel
formation (11). The tumorigenesis of
lung cancer is a complex biological process; the present study
identified that PTEN is inactivated and abnormally expressed in
lung cancer (11). It was
hypothesized that the loss of PTEN expression may contribute to the
development of EGFR-TKI resistance in EGFR mutation-positive NSCLC.
In the current study, the effect of icotinib on HCC827 cells and
the association between PTEN expression levels and cell sensitivity
to icotinib was evaluated; gene expression levels and the
biological behavior of HCC827 cells was also investigated for each
group. The aim of the present study was to elucidate the mechanisms
underlying acquired resistance to EGFR-TKIs.
Materials and methods
Cell culture and reagents
The HCC827 NSCLC cell line was provided by Xi'an
Jiaotong University (Xi'an, China); the HCC827 cells had an
acquired E746-A750 deletion mutation in the EGFR tyrosine kinase
domain. The cells were cultured in RPMI-1640 (Hyclone; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 20%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C in an atmosphere containing 5%
CO2. Icotinib was purchased from Zhejiang Beida
Pharmaceutical Co., Ltd. (Beijing, China). MTT, dimethyl sulfoxide
(DMSO), and propidium iodide (PI) were purchased from Sigma-Aldrich
(Merck Millipore, Darmstadt, Germany). Anti-PTEN (#9552; dilution,
1:2,000) was purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Horseradish peroxidase-conjugated AffiniPure
goat anti-rabbit immunoglobulin G (H+L; #AP60720; dilution,
1:2,000) was obtained from Abgent Biotech Co., Ltd. (Suzhou, China)
and the anti-β-actin antibody (#sc-130300; dilution, 1:1,500) was
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA).
PTEN-small interfering (si)RNA design,
synthesis and transfection
siRNAs corresponding to PTEN (upstream,
5′-GUAUGACAACAGCCUCAAGTT-3′; downstream,
5′-CUUGAGGCUGUUGUCAUACTT-3′) were purchased from Shanghai
GenePharma Co., Ltd. (Shanghai, China). The cells were plated in
culture plates in RPMI-1640 (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA) supplemented with 20% fetal bovine serum at 37°C in
an atmosphere containing 5% CO2. Following culturing to
70–80% confluency on 6-well plates, the cells were then transfected
with siRNA or FAM-negative siRNA using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. Following a 6-h incubation period at 37°C
with the transfection reagents, the transfection medium was
replaced with RPMI-1640 supplemented with 20% FBS. The cells were
then incubated at 37°C for 24–48 h in 5% CO2. Two wells were left
untransfected to serve as a negative control.
Drugs and growth-inhibition assay
Cells were seeded at a density of
2–4×103/100 µl in 96-well plates and incubated at 37°C
overnight in 5% CO2. Subsequently, various
concentrations (0.5 nmol/l, 1 nmol/l, 2 nmol/l, 4 nmol/l, 8 nmol/l)
of icotinib were added; following incubation times of 24, 48 and 72
h at 37°C, 20 µl MTT (5 mg/l) was added to each well and the plates
were incubated for 4 h at 37°C. Then, 150 µl DMSO was oscillated
for 10 min and the absorbance was measured at 490 nm with a 96-well
plate reader (Wako Pure Chemical Industries, Ltd., Osaka, Japan).
Each concentration was investigated in triplicate and was analyzed
by SPSS version 17.0 software (SPSS, Inc., Chicago, IL, USA). The
half maximal inhibitory concentration (IC50) was considered to be
the concentration that resulted in 50% cell growth inhibition, as
compared with the untreated control cells.
Cell apoptosis assay
Exponentially growing cell suspensions were seeded
into 6-well plates at a density of 2–4×103/100 µl. The
following day, various concentrations (0.5, 1, 2, 4, and 8 nmol/l)
of icotinib were added. Following incubation for 72 h at 37°C, the
cells (0.5–1×106/ml) were collected, washed with cold
PBS and 500 µl binding buffer was added; subsequently, 5 µl Annexin
V/fluorescein isothiocyanate (Shaanxi Xianfeng Biotechnology Co.,
Ltd., Xi'an, China) and 10 µl PI was added to each well, in the
absence of light, and mixed. The plates were incubated for 15 min
at 37°C and evaluated using flow cytometry (FACSCalibur; BD
Biosciences, San Jose, CA, USA) within 1 h.
Cell cycle assay
Exponentially growing cell suspensions
(0.5–1×106/ml) were seeded into 6-well plates. Following
an overnight incubation at 37°C in an atmosphere containing 5%
CO2, various concentrations of icotinib (0.5, 1, 2, 4,
and 8 nmol/l) were added. The cells were incubated for 72 h at 37°C
then collected and washed with cold PBS. The samples were frozen in
70% ethanol at −20°C and kept overnight. The cells were
subsequently washed with cold PBS, and 150 µl RNase (5 g/l;
Sigma-Aldrich; Merck Millipore) and 150 µl PI (50 µg/ml;
Sigma-Aldrich; Merck Millipore) were added. The cells were
solubilized for 30 min without light and were evaluated using flow
cytometry (FACSCalibur).
Western blot analysis
The cells were washed twice with PBS and solubilized
in a protein lysis buffer (Pierce, Rockford, IL, USA). The
supernatants, containing the whole-cell protein extracts, were
obtained following centrifugation (18,894.2 × g) for 15 min at 4°C.
The protein concentration was determined using a bicinchoninic
protein assay (Shaanxi Xianfeng Biotechnology Co., Ltd.) according
to the manufacturer's protocol. Heat-denatured protein samples (30
µg per lane) were resolved using 8% SDS-PAGE and transferred to a
polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA,
USA). The membrane was incubated for 60 min in PBS containing 0.1%
Tween-20 (Ameresco, Inc., Framingham, MA, USA) and 5% skim milk to
block non-specific binding; this was followed by incubation
overnight at 4°C with primary antibodies against PTEN and β-actisn.
The membrane was washed four times for 8 min each in PBS with 0.1%
Tween-20, then incubated for 2 h with a conjugated horseradish
peroxidase secondary antibody. The membrane was washed thoroughly
in PBS containing 0.1% Tween-20 and the bound antibody was detected
using enhanced chemiluminescence detection reagents (EMD
Millipore), according to the manufacturer's protocol. The results
of the western blotting were analyzed using ImageJ2x (National
Institutes of Health, Bethesda, MA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA (1,248.52 ng/µl) from cells was
extracted using RNA Fast200 (Sigma-Aldrich; Merck Millipore). The
PCR amplification reaction mixture (10 µl) contained the following:
5*PrimeScript buffer (2 µl), PrimeScript RT enzyme mix (0.5 µl),
oligo dT primer (0.5 µl), random hexanucleotide (0.5 µl), total RNA
(4 µl) and RNase Free (Takara Bio, Inc., Otsu Japan). The thermal
cycler conditions were as follows: 94°C for 2 min then 35 cycles
alternating between 94°C for 30 sec, 60°C for 30 sec, 72°C for 30
sec and 72°C for 2 min. The target gene expression levels were
normalized to GAPDH levels. The formula 2ss(−ΔCq)=2[Cq
(GAPDH)-Cq(target)] was used to calculate the relative gene
expression levels for each sample, reflecting the normalized target
gene expression levels. The primer sequences were as follows:
Upstream, 5′-CTATTCCCAGTCAGAGGCGCTAT-3′; downstream,
5′-TGAACTTGTCTTCCCGTCGTGT-3′.
Statistical analysis
The data are presented as the mean ± standard
deviation. The data were analyzed using SPSS version 17.0 software
(SPSS, Inc., Chicago, IL, USA). Statistical analysis was conducted
using an independent t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Icotinib inhibited the growth of
HCC827 cells
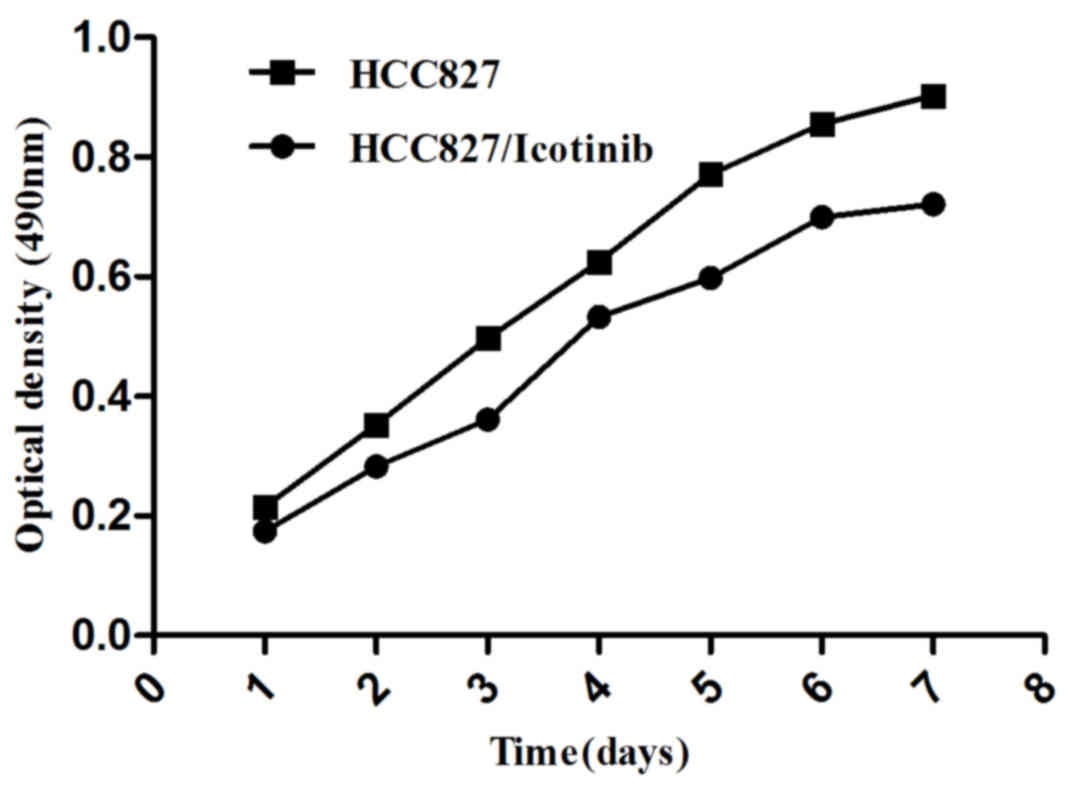
To study the effects of icotinib on HCC827 lung
cancer cells, an MTT assay was used to test cell growth and
survival (Fig. 1). The present study
identified that icotinib induces damage to HCC827 cells, which was
enhanced with increasing time and concentration (P=0.001).
Icotinib inhibits cell apoptosis and
the cell cycle of HCC827 cells
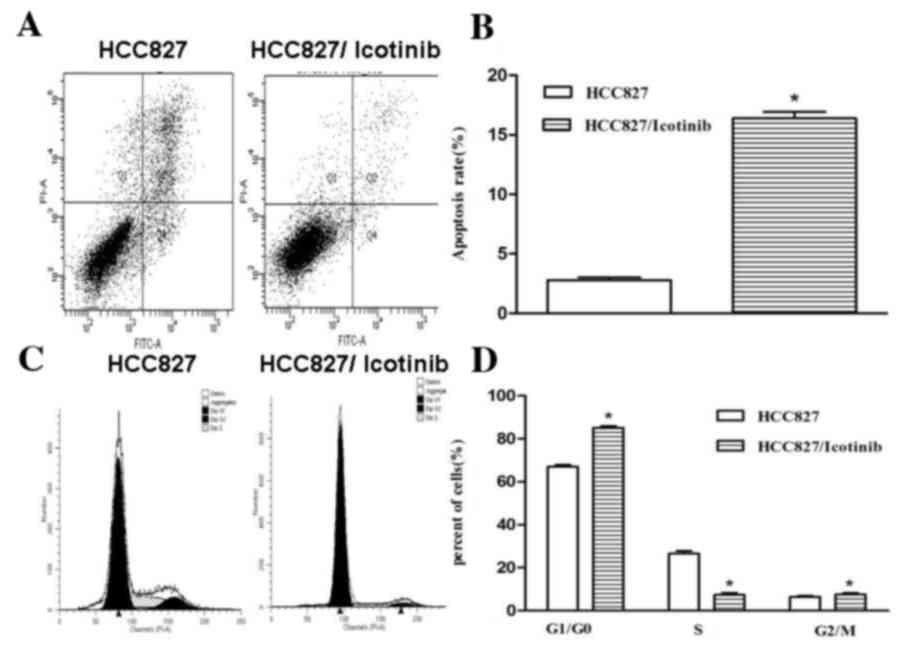
As indicated in Fig.
2, treatment with icotinib significantly inhibited the
proliferation and increased the rate of apoptosis of HCC827 cells
(P<0.001; Fig. 2A and B). The
number of cells in the G0/G1 cell cycle phase
was increased following treatment with icotinib, compared with the
control group, suggesting that icotinib significantly attenuated
the cell cycle in the G0/G1 phase
(P<0.001; Fig. 2C and D).
Icotinib downregulates PTEN
expression
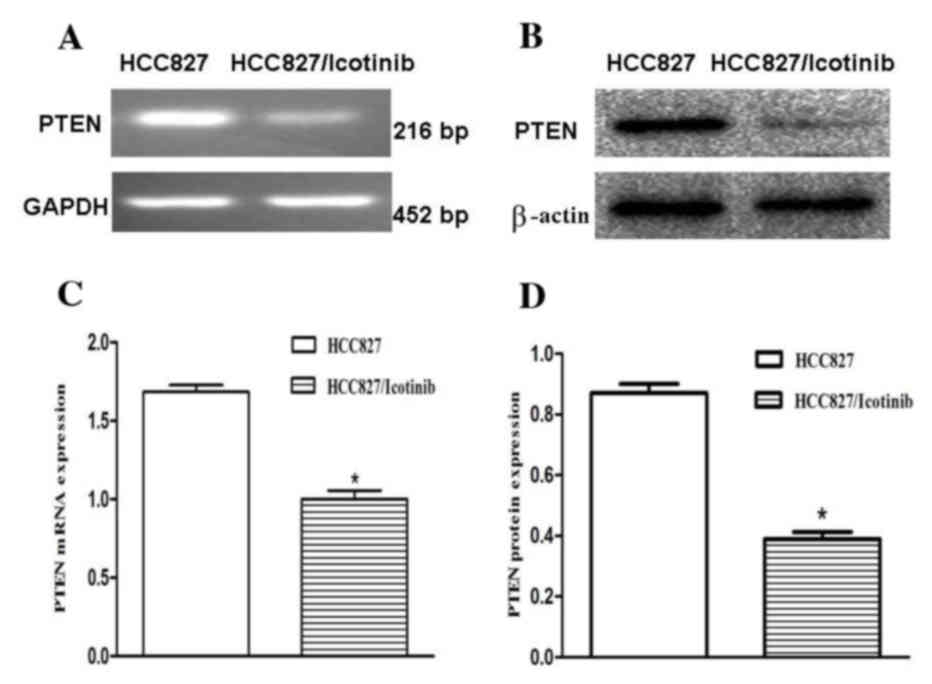
To investigate the association between icotinib and
its relevant effects, the expression levels of PTEN were examined
using RT-qPCR and western blotting (Fig.
3A and B). The RT-qPCR results revealed that PTEN mRNA
expression levels were decreased by ~34% following treatment with
icotinib (P<0.001; Fig. 3C). The
results of the western blotting demonstrated that the protein
expression levels of PTEN in the HCC827 cells were significantly
downregulated (P<0.001; Fig.
3D).
PTEN in HCC827 cells is downregulated
by transient transfection
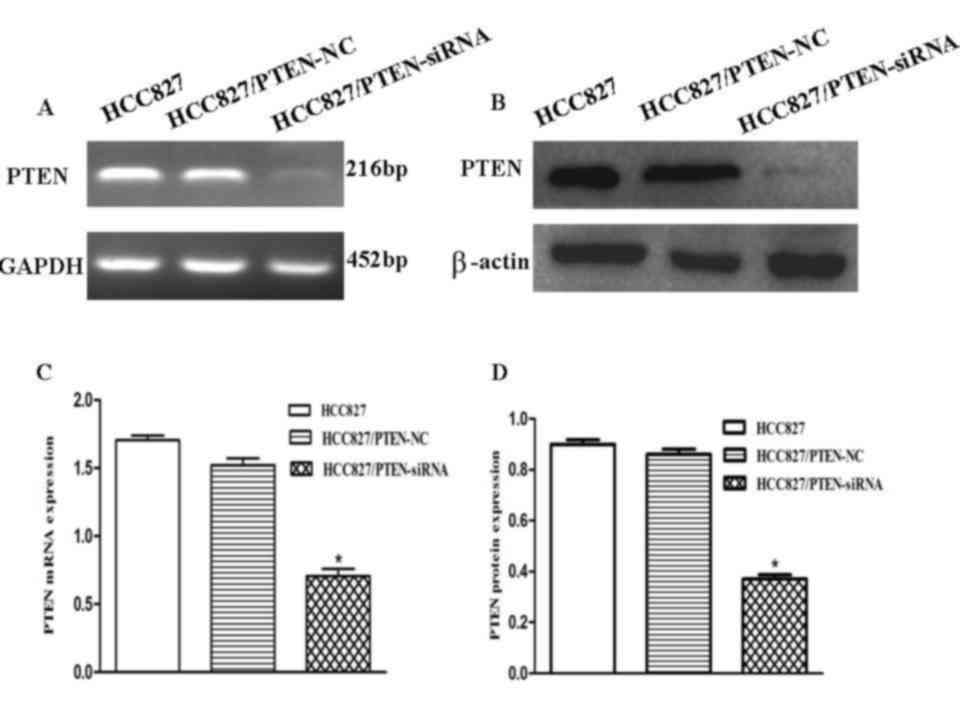
To evaluate the effects of the downregulation of
PTEN expression in HCC827 lung cancer cells, the cells were
transfected with an siRNA corresponding to PTEN. To determine
whether PTEN expression in HCC827 cells was downregulated, RT-qPCR
and western blotting were used to analyze the PTEN expression
levels in each group (Fig. 4A and B).
RT-qPCR demonstrated that PTEN expression levels decreased by ~52%
following transfection, suggesting that PTEN was significantly
downregulated by PTEN-siRNA (P<0.001; Fig. 4C). The protein expression levels of
PTEN in the HCC827 cells were significantly decreased following
transfection with the PTEN siRNA, as compared with the control
(P<0.001; Fig. 4D).
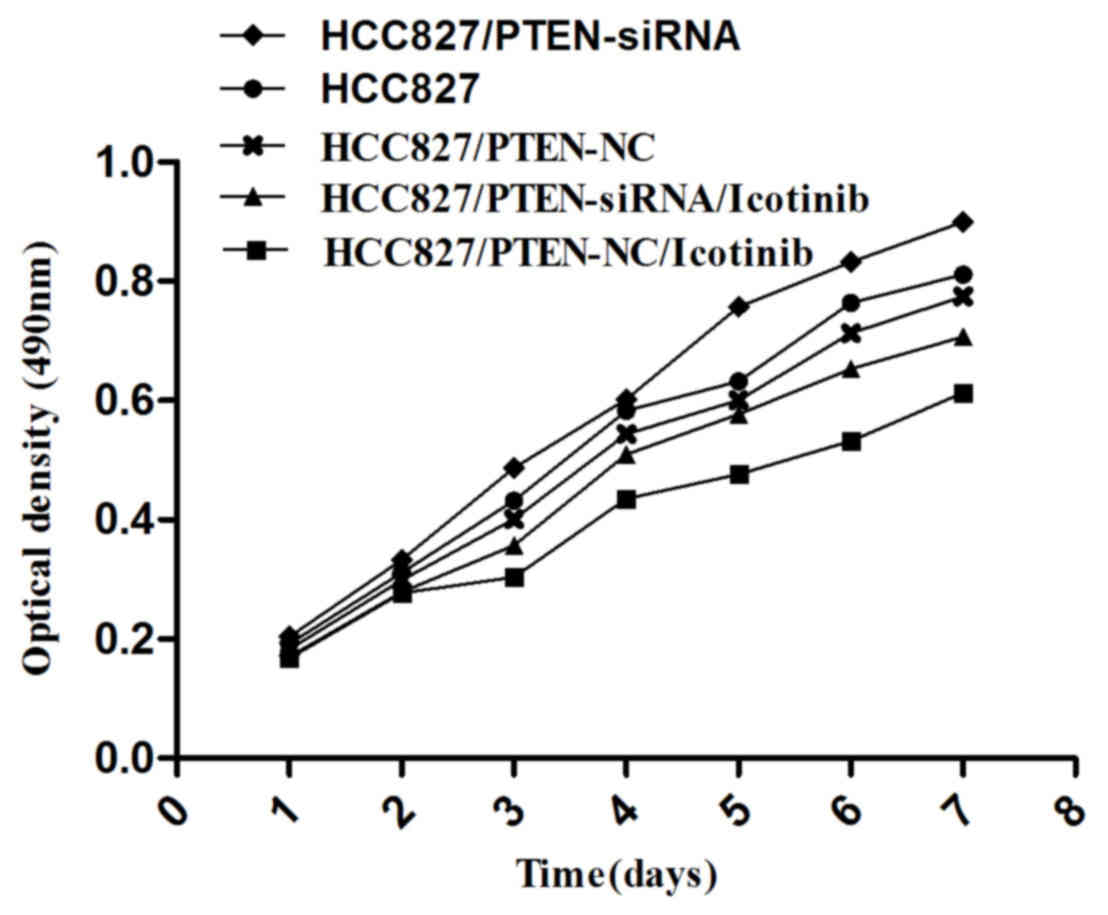
Silencing PTEN expression promotes the
growth of HCC827 cells
An MTT assay was used to examine cell growth in each
group, as indicated in Fig. 5; the
growth of HCC827 cells was observed to be inhibited by icotinib
treatment (P=0.0042). However, the growth of HCC827 cells was
promoted if the PTEN gene was silenced (P=0.0117).
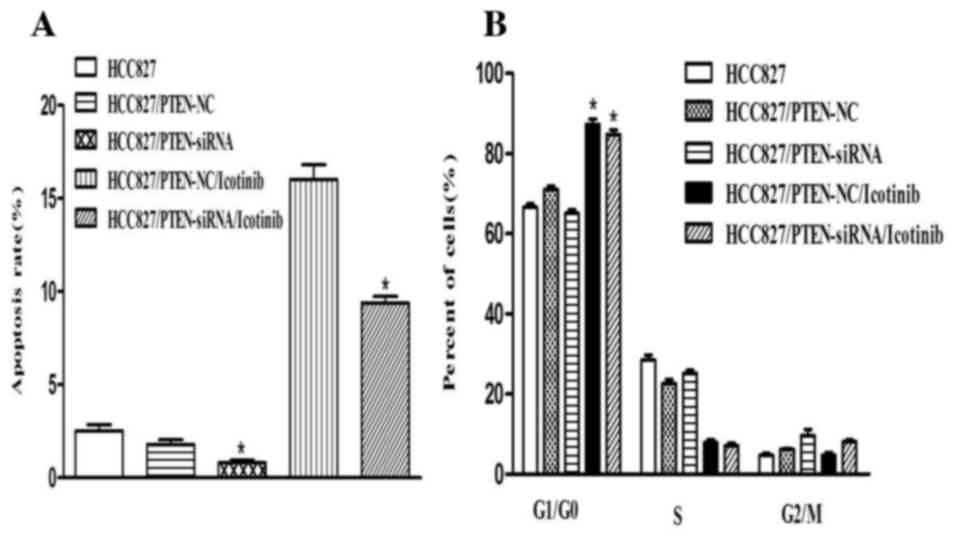
Silencing PTEN expression decreases
the apoptosis of HCC827 cells
Transfection with PTEN siRNA decreased the rate of
apoptosis of HCC827 cells, compared with the control group
(P<0.001). Concordant with the previous results, the number of
cells in the G0/G1 phase was increased
following icotinib treatment (P<0.001) and no significant
difference was observed between the control and PTEN-siRNA groups
(Fig. 6).
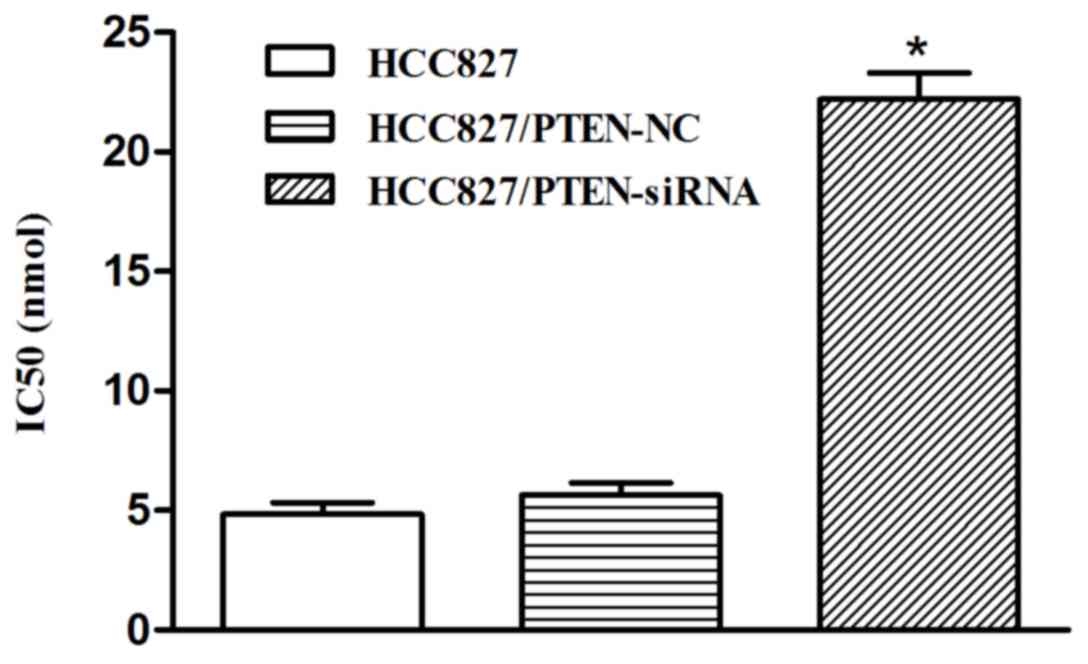
Silencing PTEN expression decreases
the sensitivity of HCC827 cells to icotinib
In the HCC827 cells transfected with PTEN siRNA, the
inhibition rate of icotinib was significantly decreased, compared
with the control group, and the IC50 was significantly increased
(22.52 nmol) following transfection (P<0.001; Fig. 7).
Discussion
Icotinib hydrochloride is an EGFR-TKI that targets
the mutated EGFR gene (3). If ligands
including epidermal growth factor, transforming growth factor or
amphiregulin are combined with the extracellular domain of EGFR,
the intracellular tyrosine kinase may be activated and combine with
adenosine triphosphate (ATP) (12).
The phosphorylation of ATP activates downstream signal transduction
pathways, including the rat sarcoma (RAS)/mitogen-activated protein
kinases (MAPK), phosphatidylinositol-4,5-bisphosphate 3-kinase
(PI3K)/protein kinase B (Akt) and proto-oncogene tyrosine-protein
kinase Src signaling pathway, which may induce tumor cell
proliferation, anti-apoptosis, invasion and metastasis (5).
It has previously been reported that EGFR-TKIs may
suppress tumor growth by binding to the intracellular domain
(magnesium-ATP) of EGFR, which may inhibit the activity and
phosphorylation of the tyrosine kinase and block downstream signal
transduction pathways (12). However,
the majority of patients with NSCLC develop an acquired resistance
to EGFR-TKIs within 8–10 months (13). A previous study on TKI-sensitive NSCLC
indicated that a mutation in the EGFR tyrosine kinase domain is
responsible for activating various anti-apoptotic signaling
pathways (14). An EGFR mutation may
be detected in 43–89% of patients with lung cancer; mutations on
exons 19 and 21 are the most common (15). These mutations have been observed to
confer increased cell sensitivity to TKIs, including to icotinib
(3). In addition to a second point
mutation in EGFR on exon 20 (T790 M) and a MET gene amplification,
Kirsten-RAS, PIK3, insulin-like growth factor-1 and
epithelial-mesenchymal transition target gene amplification
mutations are also associated with acquired resistance to EGFR-TKIs
(13,14,16).
Similar to other EGFR-TKIs, icotinib may induce cell
proliferation inhibition, cell apoptosis and cell cycle arrest by
inhibiting the activity and phosphorylation of the tyrosine kinase
domain (3). The current study
identified that icotinib may damage HCC827 cells in a time- and
concentration-dependent manner. Treatment with icotinib
significantly inhibited the proliferation and increased the rate of
apoptosis of HCC827 cells, in which the mRNA and protein expression
levels of PTEN were significantly downregulated. The PTEN gene is a
tumor suppressor gene located on chromosome 10q23.3 (10). PTEN is frequently mutated in a variety
of types of human cancer, including glioblastoma, melanoma, breast,
prostate, renal and endometrial carcinoma (11). PTEN exerts its tumor-suppressor
effects through its phosphatase domain and C2 domain (17). It may inhibit phosphatidylinositol
(3,4,5)-trisphosphate phosphorylation by
specifically antagonizing the PI3K/Akt signaling pathway and
suppressing Akt expression, which may induce the apoptosis of
Akt-dependent cells (17–21).
A previous study has reported that alterations in
PTEN in NSCLC cell lines are associated with a loss of
heterozygosity, or with gene deletion (22). Loss of PTEN is involved in the
development of EGFR inhibitor resistance in certain tumor cell
lines (23,24) and in patients with glioblastoma
(25). The underlying mechanisms may
include promoter hypermethylation, post-translational modifications
or the alternative splicing of the pre-mRNA (26).
PTEN expression patterns in NSCLC require further
study, and the role of PTEN in icotinib treatment in NSCLC has yet
to be fully elucidated. Yamamoto et al (22) constructed a PC-9/GR EGFR-TKI resistant
cell line, and identified that its PTEN expression levels were
significantly decreased, and phosphorylated Akt levels were
significantly increased. In the present study, it was identified
that PTEN mRNA and protein expression levels were significantly
downregulated following icotinib treatment. Silencing PTEN
expression may promote cell proliferation, decrease the rate of
apoptosis of HCC827 cells and reduce the sensitivity of HCC827
cells to icotinib. The current study hypothesized that the
underlying mechanisms may involve the loss of PTEN with an
increasing PIP-3 concentration, the hyperactivation of Akt and the
subsequent release of cytochrome c and the inactivation of
forkhead, caspase-9 and B-cell-lymphoma-2 associated agonist of
cell death (26). PTEN may influence
cell proliferation and apoptosis by regulating the MAPK signaling
pathway and the extracellular signal-regulated protein kinase cell
survival pathway (22,27,28).
In conclusion, the present study identified that
PTEN expression levels affected the sensitivity of HCC827 cells to
icotinib treatment, indicating that PTEN may be involved in
regulating the icotinib-induced cytotoxicity of HCC827 cells. These
results suggest that PTEN may serve as a novel target for
monitoring the sensitivity of NSCLC cells to EGFR-TKIs, including
icotinib in patients with lung cancer.
References
|
1
|
Walker S: Updates in non-small cell lung
cancer. Clin J Oncol Nurs. 12:587–596. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu Q, Wang Y, Liu H, Meng S, Zhou S, Xu J,
Schmid-Bindert G and Zhou C: Treatment outcome for patients with
primary NSCLC and synchronous solitary metastasis. Clin Transl
Oncol. 15:802–809. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tan F, Shi Y, Wang Y, Ding L, Yuan X and
Sun Y: Icotinib, a selective EGF receptor tyrosine kinase
inhibitor, for the treatment of non-small-cell lung cancer. Future
Oncol. 11:385–397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kosaka T, Yatabe Y, Endoh H, Yoshida K,
Hida T, Tsuboi M, Tada H, Kuwano H and Mitsudomi T: Analysis of
epidermal growth factor receptor gene mutation in patients with
non-small cell lung cancer and acquired resistance to gefitinib.
Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Onitsuka T, Uramoto H, Nose N, Takenoyama
M, Hanagiri T, Sugio K and Yasumoto K: Acquired resistance to
gefitinib: The contribution of mechanisms other than the T790M, MET
and HGF status. Lung Cancer. 68:198–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sos ML, Rode HB, Heynck S, Peifer M,
Fischer F, Klüter S, Pawar VG, Reuter C, Heuckmann JM, Weiss J, et
al: Chemogenomic profiling provides insights into the limited
activity of irreversible EGFR Inhibitors in tumor cells expressing
the T790M EGFR resistance mutation. Cancer Res. 70:868–874. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ju L and Zhou C: Association of integrin
beta1 and c-MET in mediating EGFR TKI gefitinib resistance in
non-small cell lung cancer. Cancer Cell Int. 13:152013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bean J, Brennan C, Shih JY, Riely G, Viale
A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eng C: PTEN: One gene, many syndromes. Hum
Mutat. 22:183–198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ortega-Molina A and Serrano M: PTEN in
cancer, metabolism, and aging. Trends Endocrinol Metab. 24:184–189.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soria JC, Lee HY, Lee JI, Wang L, Issa JP,
Kemp BL, Liu DD, Kurie JM, Mao L and Khuri FR: Lack of PTEN
expression in non-small cell lung cancer could be related to
promoter methylationc. Clin Cancer Res. 8:1178–1184.
2002.PubMed/NCBI
|
|
12
|
Mezzapelle R, Miglio U, Rena O, Paganotti
A, Allegrini S, Antona J, Molinari F, Frattini M, Monga G, Alabiso
O and Boldorini R: Mutation analysis of the EGFR gene and
downstream signalling pathway in histologic samples of malignant
pleural mesothelioma. Br J Cancer. 108:1743–1749. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu H, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jones HE, Goddard L, Gee JM, Hiscox S,
Rubini M, Barrow D, Knowlden JM, Williams S, Wakeling AE and
Nicholson RI: Insulin-like growth factor-I receptor signalling and
acquired resistance to gefitinib (ZD1839; Iressa) in human breast
and prostate cancer cells. Endocr Relat Cancer. 11:793–814. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boch C, Kollmeier J, Roth A,
Stephan-Falkenau S, Misch D, Grüning W, Bauer TT and Mairinger T:
The frequency of EGFR and KRAS mutations in non-small cell lung
cancer (NSCLC): Routine screening data for central Europe from a
cohort study. BMJ Open. 3:pii. e0025602013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma C, Wei S and Song Y: T790M and acquired
resistance of EGFR TKI: A literature review of clinical reports. J
Thorac Dis. 3:10–18. 2011.PubMed/NCBI
|
|
17
|
Tian T, Nan KJ, Guo H, Wang WJ, Ruan ZP,
Wang SH, Liang X and Lu CX: PTEN inhibits the migration and
invasion of HepG2 cells by coordinately decreasing MMP expression
via the PI3K/Akt pathway. Oncol Rep. 23:1593–1600. 2010.PubMed/NCBI
|
|
18
|
Chalhoub N and Baker SJ: PTEN and the
PI3-kinase pathway in cancer. Annu Rev Pathol. 4:127–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Leslie NR and Downes CP: PTEN function:
How normal cells control it and tumour cells lose it. Biochem J.
382:1–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moon SH, Kim DK, Cha Y, Jeon I, Song J and
Park KS: PI3K/Akt and Stat3 signaling regulated by PTEN control of
the cancer stem cell population, proliferation and senescence in a
glioblastoma cell line. Int J Oncol. 42:921–928. 2013.PubMed/NCBI
|
|
21
|
Fidler MJ, Morrison LE, Basu S, Buckingham
L, Walters K, Batus M, Jacobson KK, Jewell SS, Coon J IV and Bonomi
PD: PTEN and PIK3CA gene copy numbers and poor outcomes in
non-small cell lung cancer patients with gefitinib therapy. Br J
Cancer. 105:1920–1926. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yamamoto C, Basaki Y, Kawahara A,
Nakashima K, Kage M, Izumi H, Kohno K, Uramoto H, Yasumoto K,
Kuwano M and Ono M: Loss of PTEN expression by blocking nuclear
translocation of EGR1 in gefitinib-resistant lung cancer cells
harboring epidermal growth factor receptor-activating mutations.
Cancer Res. 70:8715–8725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamasaki F, Johansen MJ, Zhang D,
Krishnamurthy S, Felix E, Bartholomeusz C, Aguilar RJ, Kurisu K,
Mills GB, Hortobagyi GN and Ueno NT: Acquired resistance to
erlotinib in A-431 epidermoid cancer cells requires down-regulation
of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res.
67:5779–5788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
She QB, Solit DB, Ye Q, O'Reilly KE, Lobo
J and Rosen N: The BAD protein integrates survival signaling by
EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor
cells. Cancer Cell. 8:287–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mellinghoff IK, Wang MY, Vivanco I,
Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH and
Chute DJ: Molecular determinants of the response of glioblastomas
to EGFR kinase inhibitors. N Engl J Med. 353:2012–2024. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang YS, Wang YH, Xia HP, Zhou SW,
Schmid-Bindert G and Zhou CC: MicroRNA-214 Regulates the Acquired
Resistance to Gefitinib via the PTEN/AKT pathway in EGFR-mutant
cell lines. Asian Pac J Cancer Prev. 13:255–260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sos ML, Koker M, Weir BA, Heynck S,
Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P,
et al: PTEN loss contributes to erlotinib resistance in EGFR-mutant
lung cancer by activation of Akt and EGFR. Cancer Res.
69:3256–3261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ludovini V, Bianconi F, Pistola L, Chiari
R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A,
Flacco A, et al: Phosphoinositide-3-kinase catalytic alpha and KRAS
mutations are important predictors of resistance to therapy with
epidermal growth factor receptor tyrosine kinase inhibitors in
patients with advanced non-small cell lung cancer. J Thorac Oncol.
6:707–715. 2011. View Article : Google Scholar : PubMed/NCBI
|