Introduction
Pyrogallol (PG; benzene-1,2,3-triol) is a polyphenol
compound that is commonly distributed in hard wood plants, and it
has anti-fungal and anti-psoriatic properties (1). PG is a reductant that is able to
generate free radicals, in particular superoxide anions
(O2•−), so has frequently been used as a
photographic developing agent and in the hair dying industry
(1). Despite the useful effects of
PG, its toxicity remains a concern for the individuals exposed to
it. Multiple studies have been performed to elucidate the
toxicological and pharmacological effects of PG (2–4). However,
the molecular mechanisms underlying the cellular effects of PG
remain only partially clarified. For example, PG induces
O2•−-mediated death of various types of cell,
including human lymphoma cells (5),
human glioma cells (6), gastric
cancer cells (7) and Calu-6 lung
cancer cells (8,9). In addition, PG triggers mutagenesis,
carcinogenesis and impairs the immune system (1).
O2•−, hydrogen peroxide
(H2O2) and hydroxyl radicals
(·•OH) are reactive oxygen species (ROS). These are
involved in various cellular events, including gene expression,
cell signaling, differentiation, cell growth and cell death. ROS
are primarily generated during mitochondrial respiration and are
specifically made by various oxidases (10). Superoxide dismutases convert
O2•− to H2O2 (11). Further metabolism yields O2
and H2O via catalase or glutathione (GSH) peroxidase
(12). Oxidative stress resulting
from either overproduction of ROS or loss of antioxidant enzymes
may initiate cellular signaling events that lead to cell death,
depending on cell type. There is evidence to suggest that ROS not
only affect extracellular signal regulated kinase 1/2 (ERK1/2) and
mitogen-activated protein kinase kinase (MEK) activation (13) but also activate c-Jun N-terminal
kinase/stress-activated protein kinase (JNK/SAPK) and p38 (14,15).
ERK1/2, JNK/SAPK and p38 are mitogen-activated protein kinases
(MAPKs), which are components of signaling pathways associated with
cell proliferation, differentiation and cell death (16). Each kinase has different upstream
activators and specific downstream substrates (17). In general, MEK-ERK signaling is
pro-survival rather than pro-apoptotic (18). JNK and p38 signaling pathways are
associated with cell death (14,15,19).
The human lung is a structurally complex organ
system (20). Fibroblast cells, which
are primarily derived from the primitive mesenchyme, synthesize
extracellular matrix components including collagen to maintain the
structural and functional integrity of the lung connective tissues.
Human pulmonary fibroblast (HPF) cells are involved in lung
inflammation, fibrosis and cancer (21). Cultured normal human cells are
frequently used in mechanistic studies of oxidative stress, being
invaluable biological models (22,23). PG
inhibits Calu-6 and A549 lung cancer cell growth via apoptosis
(8,24,25) and
depletion of GSH (24,26). In addition, MEK inhibitors, but not
JNK or p38 inhibitors, have been demonstrated to slightly attenuate
inhibition of cell growth, cell death and GSH depletion in
PG-treated Calu-6 cells (27). The
present study investigated the effect of MAPK inhibitors on
PG-treated HPF cell death, in relation to ROS and GSH levels.
Materials and methods
Cell culture
HPF cells were obtained from PromoCell GmbH
(Heidelberg, Germany) and were cultured in RPMI-1640 medium (GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) in humidified incubator containing 5% CO2
at 37°C. HPF cells were used for experiments between passages four
and eight.
Reagents
PG (Sigma-Aldrich; Merck KGaA) was dissolved in
water at 100 mM as a stock solution. The MEK inhibitor (PD98059),
JNK inhibitor (SP600125) and p38 inhibitor (SB203580) were obtained
from Calbiochem; Merck KGaA and were dissolved in dimethyl
sulfoxide (Sigma-Aldrich; Merck KGaA). Based on a previous
experiment (28), HPF cells were
pretreated with 10 µM of each MAPK inhibitor for 1 h prior to PG
treatment at 37°C.
Cell viability inhibition assays
Briefly, 5×103 HPF cells per well in 96-well
microtiter plates (Nalge Nunc International; Thermo Fisher
Scientific, Inc., Penfield, NY, USA) were exposed to 0, 50 or 100
µM PG with or without each MAPK inhibitor at 37°C for 24 h. Changes
in cell viability induced by PG and/or a given MAPK inhibitor were
determined by measuring the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich; Merck KGaA) dye absorbance as previously described
(28).
Lactate dehydrogenase (LDH)
assays
Necrosis in cells was evaluated using an LDH assay
kit (Daeil Lab Service Co., Ltd., Seoul, Korea) according to the
manufacturer's protocol. Briefly, 1×106 HPF cells in 60
mm culture plates (Nalge Nunc International; Thermo Fisher
Scientific, Inc.) were incubated with 0, 50 or 100 µM PG in the
presence or absence of each MAPK inhibitor at 37°C for 24 h. LDH
release was expressed as the percentage of extracellular LDH
activity compared with the control cells.
Annexin V/propidium iodide (PI)
staining for cell death detection
Apoptosis was determined by staining HPF cells with
Annexin V-fluorescein isothiocyanate (FITC; Ex/Em=488/519 nm;
Invitrogen; Thermo Fisher Scientific, Inc.) and PI (Ex/Em=488/617
nm; Sigma-Aldrich; Merck KGaA) as previously described (29). Briefly, 1×106 HPF cells in 60 mm
culture plates (Nalge Nunc International; Thermo Fisher Scientific,
Inc.) were incubated with 0, 50 or 100 µM PG in the presence or
absence of each MAPK inhibitor at 37°C for 24 h. Annexin V/PI
staining was analyzed using a FACStar flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA) and CellQuest Pro software
(version 5.1; BD Biosciences).
Measurement of mitochondrial membrane
potential (MMP; ΔΨm)
MMP (ΔΨm) levels were measured using a
rhodamine 123 fluorescent dye (Sigma-Aldrich; Merck KGaA;
Ex/Em=485/535 nm) as previously described (28,30,31).
Briefly, 1×106 HPF cells in 60 mm culture plates (Nalge Nunc
International; Thermo Fisher Scientific, Inc.) were incubated with
0, 50 or 100 µM PG in the presence or absence of each MAPK
inhibitor at 37°C for 24 h. Rhodamine 123 staining intensity was
determined using a FACStar flow cytometer (BD Biosciences) and
CellQuest Pro software (version 5.1; BD Biosciences). The absence
of rhodamine 123 from cells indicated the loss of MMP
(ΔΨm) in HPF cells.
Western blot analysis
Changes in apoptosis and antioxidant
system-associated protein levels were determined using western
blotting, as previously described (32). Briefly, 1×106 HPF cells in 60 mm
culture plates (Nalge Nunc International; Thermo Fisher Scientific,
Inc.) were incubated with 0, 50 or 100 µM PG in the presence or
absence of each MAPK inhibitor at 37°C for 24 h. The cells were
washed in PBS and suspended in ~100 µl of lysis buffer [20 mM
HEPES, pH 7.9, 20% glycerol, 200 mM KCl, 0.5 mM EDTA, 0.5% NP40,
0.5 mM DTT, 1% protease inhibitor cocktail (Sigma-Aldrich; Merck
KGaA)], then centrifuged at 15,900 × g at 4°C for 20 min. Samples
containing 30 µg total protein were resolved by 12.5% SDS-PAGE,
transferred to Immobilon-P polyvinylidene fluoride membranes
(Sigma-Aldrich; Merck KGaA) by electroblotting, and the membranes
were then probed with anti-poly(ADP-ribose) polymerase (PARP;
catalog no., 9542; Cell Signaling Technology, Inc., Danvers, MA,
USA; dilution, 1:5,000) and anti-GAPDH antibodies (catalog no.,
sc-25778; Santa Cruz Biotechnology, Inc., Dallas, TX, USA;
dilution, 1:5,000) at 4°C overnight without blocking. Next, the
membranes were washed with TBS with Tween-20 four times and
incubated with secondary antibody (anti-rabbit IgG; horseradish
peroxidase-linked antibody; catalog no., 7074; Cell signaling
Technology, Inc.; dilution, 1:5,000) at room temperature for 1
h.
Detection of intracellular ROS and
O2•− levels
Intracellular ROS were detected using a fluorescent
probe dye, 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCFDA; Ex/Em=495/529 nm; Invitrogen; Thermo Fisher
Scientific, Inc.), as previously described (32). Dihydroethidium (DHE; Ex/Em=518/605 nm;
Invitrogen; Thermo Fisher Scientific, Inc.) is a fluorogenic probe
that is highly selective for O2•- among ROS.
Mitochondrial O2•- levels were detected using a MitoSOX
Red mitochondrial O2•- indicator (Ex/Em=510/580 nm;
Invitrogen; Thermo Fisher Scientific, Inc.) as previously described
(30,31,33).
Briefly, 1×106 HPF cells in 60 mm culture plates (Nalge Nunc
International; Thermo Fisher Scientific, Inc.) were incubated with
0, 50 or 100 µM PG in the presence or absence of each MAPK
inhibitor for 24 h. DCF, DHE and MitoSOX Red fluorescence was
detected using a FACStar flow cytometer (BD Biosciences) and
CellQuest Pro software (version 5.1; BD Biosciences). ROS and
O2•- levels were expressed as mean fluorescence
intensity.
Detection of intracellular GSH
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMF; Ex/Em=522/595 nm;
Invitrogen; Thermo Fisher Scientific, Inc.) as previously described
(28,30,31).
Briefly, 1×106 HPF cells in 60 mm culture plates (Nalge Nunc
International; Thermo Fisher Scientific, Inc.) were incubated with
0, 50 or 100 µM PG in the presence or absence of each MAPK
inhibitor at 37°C for 24 h. CMF fluorescence intensity was
determined using a FACStar flow cytometer (BD Biosciences) and
CellQuest Pro software (version 5.1; BD Biosciences). Negative
CMF-staining (GSH-depleted) cells were expressed as a percentage of
(−) CMF cells of total cells.
Statistical analysis
The results represent the mean ± standard deviation
of at least three independent experiments. The data were analyzed
using Instat software (GraphPad Prism5; GraphPad Software, Inc., La
Jolla, CA, USA). Parametric data was analyzed using the Student's
t-test (paired) or one-way analysis of variance following by post
hoc analysis with Tukey's multiple comparison test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of MAPK inhibitors on cell
viability and necrotic cell death in PG-treated HPF cells
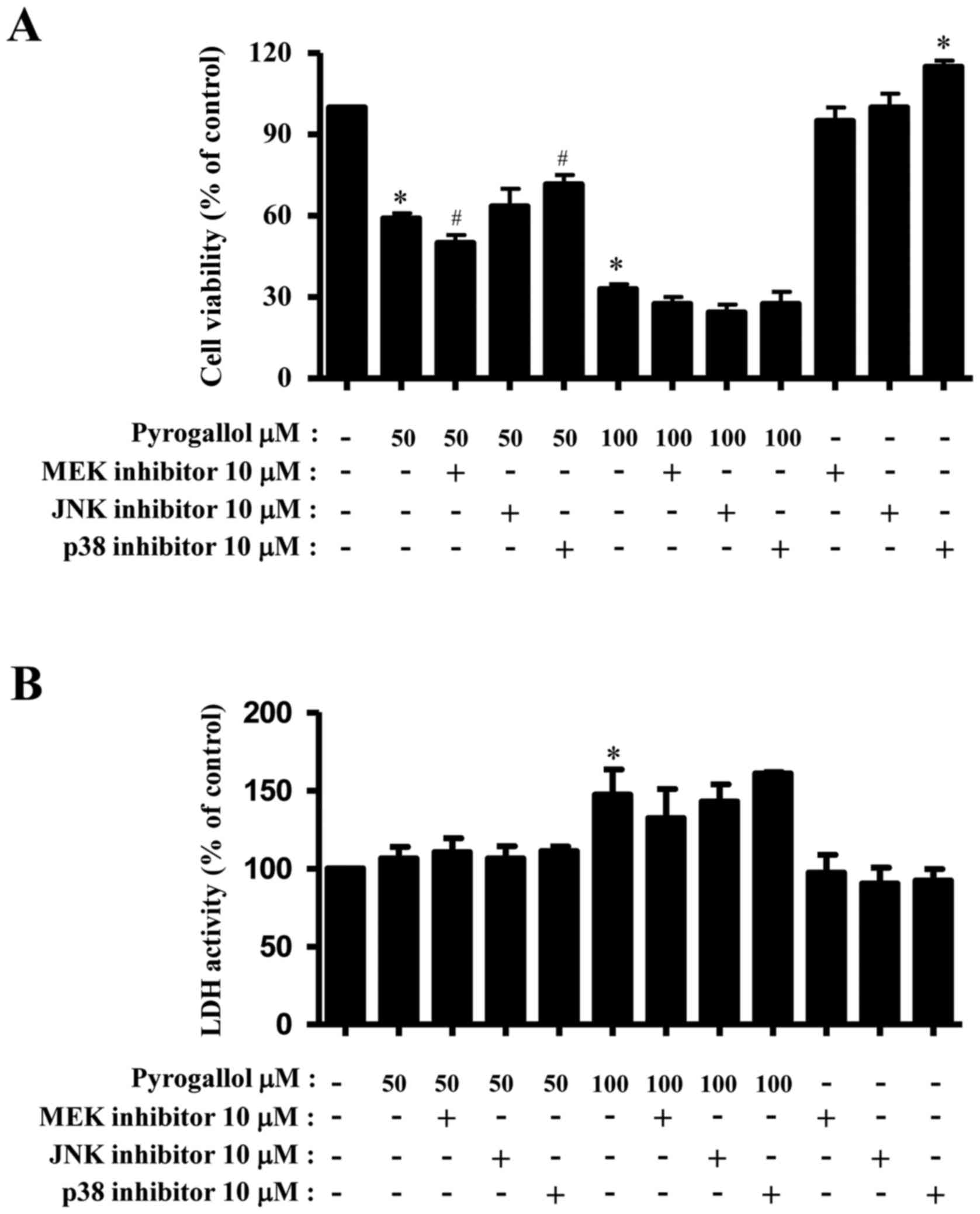
The effect of PG on HPF cell viability and necrotic
cell death was examined. For these experiments, 0, 50 or 100 µM PG
was used to differentiate the levels of cell viability inhibition
or death with or without a given MAPK inhibitor. Treatment with 50
and 100 µM PG decreased HPF viability by ~40 and 65% at 24 h,
respectively (Fig. 1A). Treatment
with an MEK inhibitor slightly enhanced the inhibition of cell
viability in 50 µM PG-treated HPF cells, whereas treatment with a
p38 inhibitor mildly attenuated the inhibition of viability
(Fig. 1A). In 100 µM PG-treated HPF
cells, all the MAPK inhibitors increased the inhibition of
viability to a certain extent (Fig.
1A), with treatment with the p38 inhibitor alone augmenting HPF
control cell viability (Fig. 1A).
Necrotic cell death was determined by measuring LDH release from
cells. While treatment with 50 µM PG did not affect LDH release
from HPF cells, 100 µM PG significantly increased LDH release
(Fig. 1B). Treatment with MAPK
inhibitors did not alter LDH activity in PG-treated and untreated
HPF cells (Fig. 1B).
Effects of MAPK inhibition on necrotic
and apoptotic cell death in PG-treated HPF cells
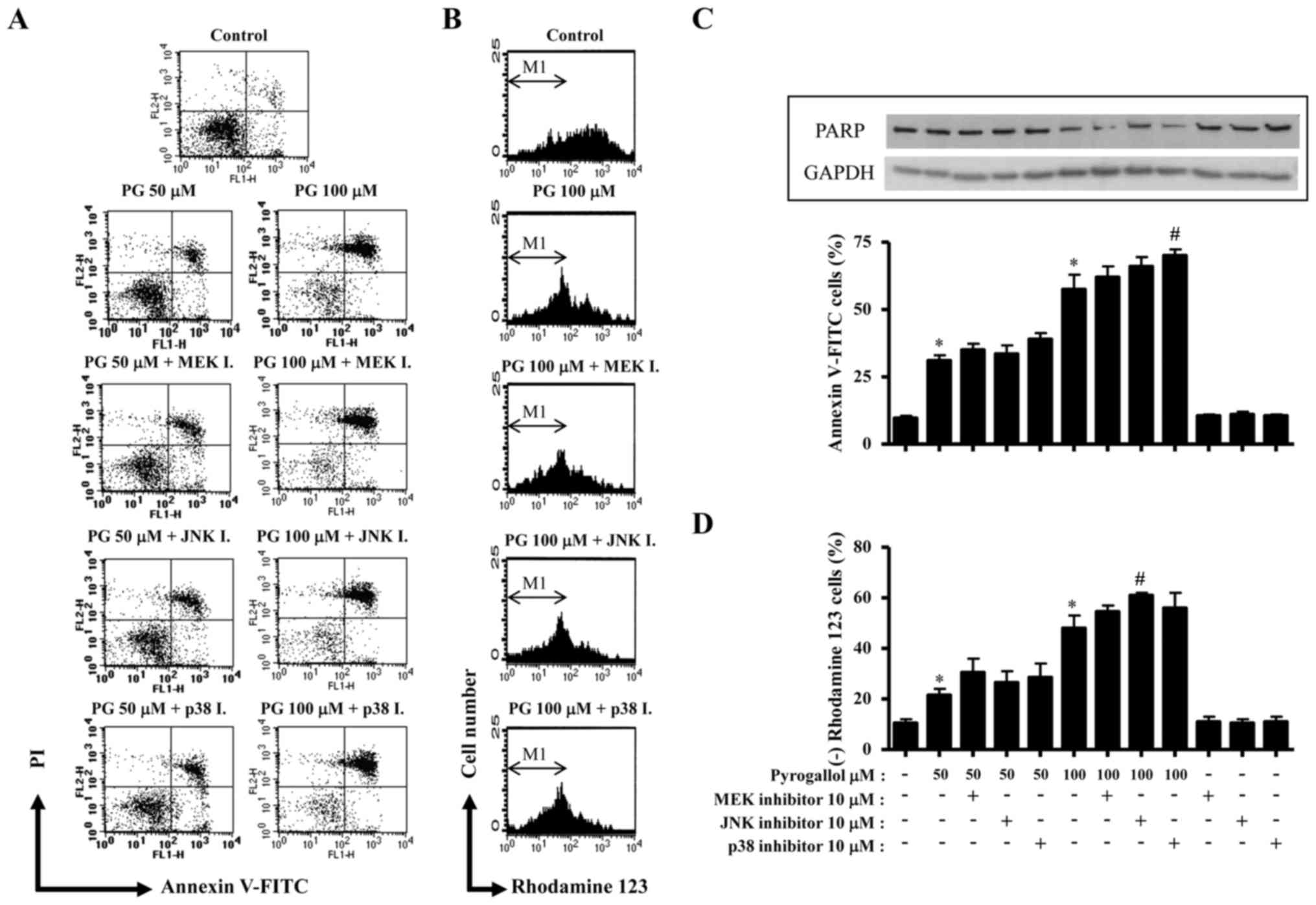
The tested doses of PG significantly increased the
rate of apoptosis in HPF cells, as evidenced by Annexin V-FITC/PI
staining (Fig. 2). In addition,
treatment with 100 µM PG increased the number of necrotic (Annexin
V- and PI+) cell death in HPF cells compared with PG-untreated
control cells (Fig. 2A). Treatment
with the p38 inhibitor slightly increased the number of Annexin V+
50 µM PG-treated HPF cells compared with only 50 µM PG-treated HPF
cells, and significantly increased the number of Annexin V+ 100 µM
PG-treated cells compared with only 100 µM PG-treated HPF cells
(Fig. 2A and C). Treatment with the
other MAPK inhibitors slightly augmented the number of Annexin V+
100 µM PG-treated HPF cells compared with only 100 µM PG-treated
HPF cells (Fig. 2A and C). PARP
protein levels were not altered in 50 µM PG-treated HPF cells,
while it was decreased in 100 µM PG-treated cells (Fig. 2C). MEK and p38 inhibitors slightly
attenuated the decrease in PARP protein levels in 100 µM PG-treated
HPF cells (Fig. 2C). Apoptotic cell
death is associated with a decrease in MMP (ΔΨm).
Treatment with 50 and 100 µM PG significantly decreased MMP
(ΔΨm) in HPF cells (Fig. 2B
and D). All the MAPK inhibitors enhanced the decrease in MMP
(ΔΨm) in PG-treated HPF cells, and treatment with the
JNK inhibitor demonstrated a significant effect on 100 µM
PG-treated HPF cells (Fig. 2B and
D).
Effects of MAPK inhibitors on ROS
levels in PG-treated HPF cells
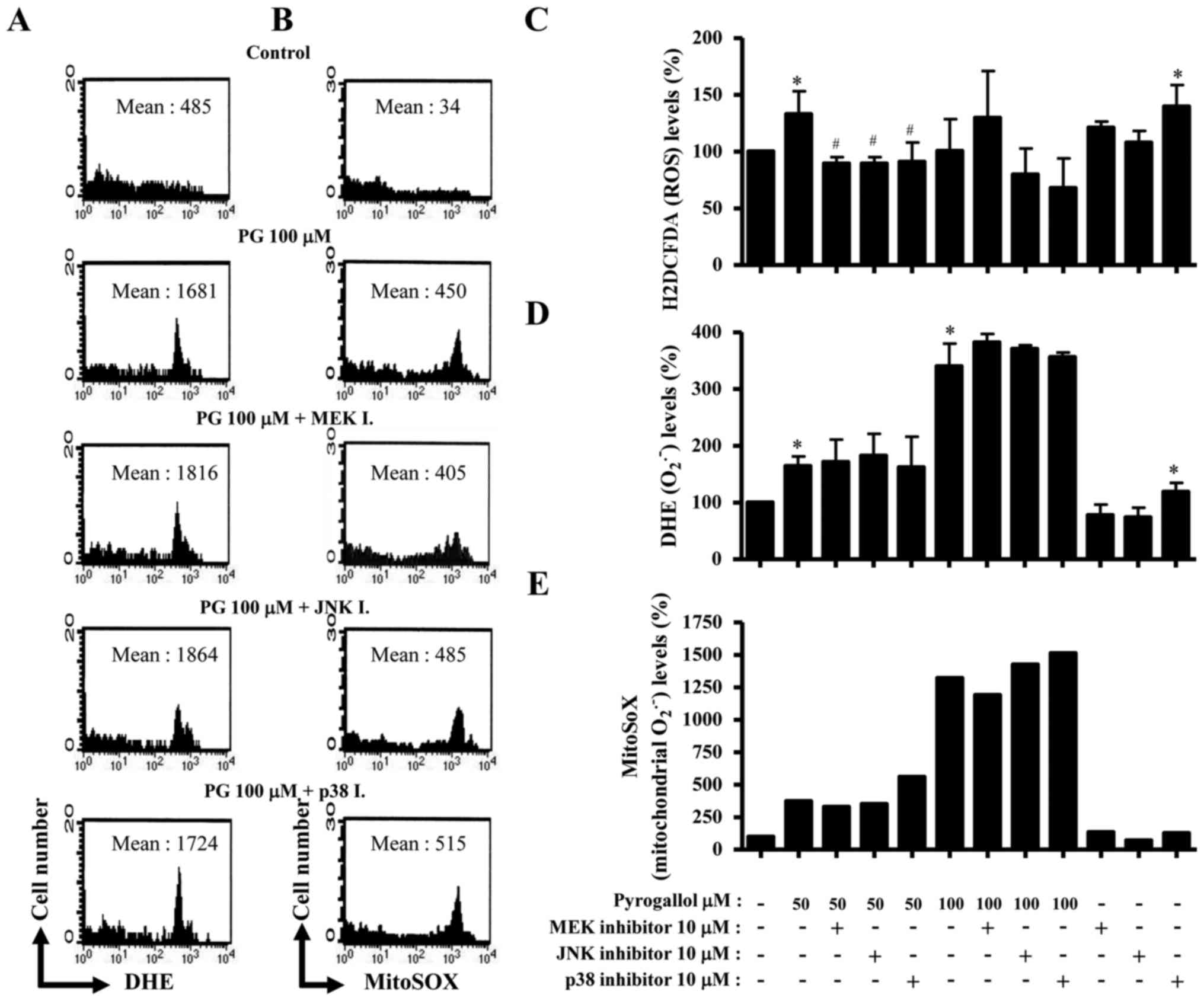
To assess the level of intracellular ROS in HPF
cells treated with PG, DHE, H2DCFDA and MitoSOX Red
fluorescent dyes were used (Fig. 3).
The level of DHE fluorescent dye, which reflects the accumulation
of O2•- in cells, increased in HPF cells treated with PG
(Fig. 3A and D). All the MAPK
inhibitors were likely to increase O2•- level in 100 µM
PG-treated HPF cells (Fig. 3A and D).
ROS (DCF) level in HPF cells was increased by 50 µM PG treatment
but not 100 µM PG treatment (Fig.
3C). All the MAPK inhibitors decreased ROS levels in HPF cells
treated with 50 µM PG, and treatment with p38 and JNK inhibitors
also decreased the level of ROS in 100 µM PG-treated HPF cells
(Fig. 3C). Furthermore, MitoSOX Red
fluorescence levels, indicating the presence of mitochondrial
O2•-, were markedly increased in PG-treated HPF cells
(Fig. 3B and E). Treatment with a p38
inhibitor increased mitochondrial O2•- levels in
PG-treated HPF cells, whereas treatment with an MEK inhibitor
slightly decreased mitochondrial O2•- levels (Fig. 3B and E). Treatment with a JNK
inhibitor reduced the mitochondrial O2•- level in HPF
control cells (Fig. 3E).
Effects of MAPK inhibitors on GSH
levels in PG-treated HPF cells
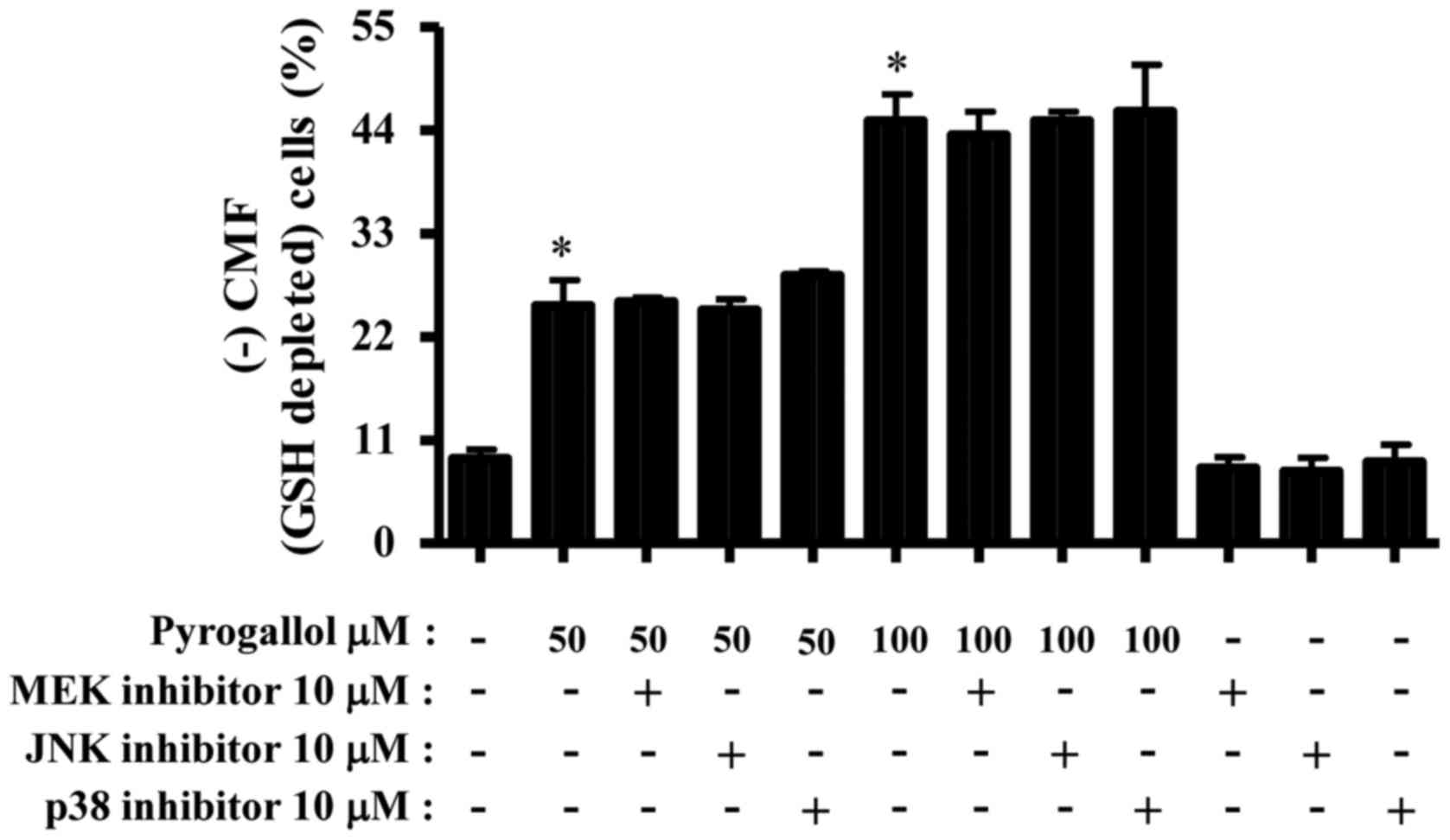
Changes in intracellular GSH levels in HPF cells
treated with PG and/or each MAPK inhibitor were assessed using a
CMFDA dye. Treatment with 50 or 100 µM PG significantly increased
the number of GSH-depleted cells in HPF cells compared with the
negative control (Fig. 4). None of
the MAPK inhibitors significantly altered GSH depletion in
PG-treated or untreated HPF cells (Fig.
4).
Discussion
PG is known to trigger the collapse of MMP
(ΔΨm) and O2•−-mediated cell death
via apoptosis in various types of cancer cell (7,8,24,25,34). In
the present study, PG increased O2•− levels,
particularly mitochondrial O2•− levels, in
HPF cells and induced decreased MMP (ΔΨm). The high
production of mitochondrial O2•− in
PG-treated HPF cells resulted in cell death. In particular,
treatment with 100 µM PG induced apoptosis as well as necrosis in
HPF cells.
In general, the activation of ERK is pro-survival
rather than pro-apoptotic (18). The
results of the present study demonstrated that treatment with an
MEK inhibitor, which resulted in decreased ERK activity, enhanced
the inhibition of cell viability, cell death and MMP
(ΔΨm) loss in PG-treated HPF cells, suggesting that ERK
signaling in PG-treated HPF cells is involved in HPF cell survival.
In addition, treatment with an MEK inhibitor increased ROS levels
in 100 µM PG-treated HPF cells. However, this inhibitor decreased
ROS levels in 50 µM PG-treated HPF cells and decreased the
mitochondrial O2•− level in PG-treated 50 and
100 µM HPF cells. These results suggested that the different
effects of MEK inhibition on ROS levels in HPF cells are dependent
on the incubation doses of PG.
The JNK and p38 signaling pathways have been
suggested to be associated with cell death (14,19,35).
Previous data have also demonstrated that JNK and p38 inhibitors
significantly prevent the inhibition of cell growth, cell death and
MMP (ΔΨm) loss in PG-treated As4.1 juxtaglomerular cells
(36). In contrast, treatment with a
JNK inhibitor enhanced the inhibition of cell growth, cell death
and MMP (ΔΨm) loss in PG-treated calf pulmonary arterial
endothelial cells, whereas treatment with a p38 inhibitor
significantly attenuated these effects in the same cells (28). According to the results of the present
study, treatment with JNK and p38 inhibitors increased the
inhibition of HPF cell viability, cell death and MMP
(ΔΨm) loss following treatment with 100 µM PG, implying
that the JNK and p38 signaling pathways in PG-treated HPF cells are
pro-survival rather than pro-apoptotic. In addition, treatment with
JNK and p38 inhibitors slightly increased
O2•− levels in 100 µM PG-treated HPF cells,
whereas these same inhibitors decreased ROS (DCF) levels in 50 and
100 µM PG-treated HPF cells. These results suggested that it was
the JNK or p38 MEK inhibitor-induced altered
O2•− levels rather than ROS (DCF) levels that
influenced PG-induced cell death. Furthermore, treatment with a p38
inhibitor partially attenuated the inhibition of 50 µM PG-treated
HPF cell viability, and this inhibitor alone significantly
increased cell viability and ROS levels, including
O2•−, in HPF control cells without the
induction of cell death. Treatment with a JNK inhibitor alone also
specifically affected mitochondrial O2•−
levels independent to HPF cell viability and death. These results
indicated that JNK and p38 inhibition differently influences ROS
levels, cell viability and cell death in HPF cells, which are
altered depending on the concentration of PG.
PG induces GSH depletion in a variety of cells
(9,26,28,37). The
results of the present study also demonstrated that PG treatment
increased the number of GSH-depleted HPF cells in a dose-dependent
manner. However, none of the MAPK inhibitors, which demonstrated a
partial effect on HPF cell death, altered the number of
GSH-depleted cells following treatment with 50 and 100 µM PG.
Therefore, the intracellular GSH content was at least partially
associated with PG-induced HPF cell death.
In conclusion, PG induced apoptosis as well as
necrosis in HPF cells. MAPK inhibitors slightly promoted cell death
in PG-treated HPF cells. HPF cell death following treatment with PG
and/or MAPK inhibitors was partially associated with the
O2•− level and changes in GSH content. The
results of the present study enhance understanding of PG-induced
cell death on normal lung cells in association with MAPK signaling
pathways and ROS levels.
Acknowledgements
The present study was supported by a grant from the
National Research Foundation of Korea, funded by the Korean
government (Ministry of Science, ICT and Future Planning; grant
nos., 20080062279 and 2016R1A2B4007773).
Glossary
Abbreviations
Abbreviations:
|
HPF
|
human pulmonary fibroblast
|
|
PG
|
pyrogallol
|
|
ROS
|
reactive oxygen species
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential
|
|
GSH
|
glutathione
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
mitogen-activated protein kinase
kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LDH
|
lactate dehydrogenase
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
CMF
|
5-chloromethylfluorescein
diacetate
|
References
|
1
|
Upadhyay G, Gupta SP, Prakash O and Singh
MP: Pyrogallol-mediated toxicity and natural antioxidants: Triumphs
and pitfalls of preclinical findings and their translational
limitations. Chem Biol Interact. 183:333–340. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Upadhyay G, Tiwari MN, Prakash O, Jyoti A,
Shanker R and Singh MP: Involvement of multiple molecular events in
pyrogallol-induced hepatotoxicity and silymarin-mediated
protection: Evidence from gene expression profiles. Food Chem
Toxicol. 48:1660–1670. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gupta YK, Sharma M and Chaudhary G:
Pyrogallol-induced hepatotoxicity in rats: A model to evaluate
antioxidant hepatoprotective agents. Methods Find Exp Clin
Pharmacol. 24:497–500. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharma M, Rai K, Sharma SS and Gupta YK:
Effect of antioxidants on pyrogallol-induced delay in gastric
emptying in rats. Pharmacology. 60:90–96. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saeki K, Hayakawa S, Isemura M and Miyase
T: Importance of a pyrogallol-type structure in catechin compounds
for apoptosis-inducing activity. Phytochemistry. 53:391–394. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sawada M, Nakashima S, Kiyono T, Nakagawa
M, Yamada J, Yamakawa H, Banno Y, Shinoda J, Nishimura Y, Nozawa Y
and Sakai N: p53 regulates ceramide formation by neutral
sphingomyelinase through reactive oxygen species in human glioma
cells. Oncogene. 20:1368–1378. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park WH, Park MN, Han YH and Kim SW:
Pyrogallol inhibits the growth of gastric cancer SNU-484 cells via
induction of apoptosis. Int J Mol Med. 22:263–268. 2008.PubMed/NCBI
|
|
8
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han YH, Kim SZ, Kim SH and Park WH:
Apoptosis in pyrogallol-treated Calu-6 cells is correlated with the
changes of intracellular GSH levels rather than ROS levels. Lung
Cancer. 59:301–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: An update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: A comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wilcox CS: Reactive oxygen species: Roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2. Role in cell survival following oxidant injury. J Biol Chem.
271:4138–4142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gomez-Lazaro M, Galindo MF,
Melero-Fernandez de Mera RM, Fernandez-Gómez FJ, Concannon CG,
Segura MF, Comella JX, Prehn JH and Jordan J: Reactive oxygen
species and p38 mitogen-activated protein kinase activate Bax to
induce mitochondrial cytochrome c release and apoptosis in response
to malonate. Mol Pharmacol. 71:736–743. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blenis J: Signal transduction via the MAP
kinases: Proceed at your own RSK. Proc Natl Acad Sci USA. 90:pp.
5889–5892. 1993; View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kusuhara M, Takahashi E, Peterson TE, Abe
J, Ishida M, Han J, Ulevitch R and Berk BC: p38 Kinase is a
negative regulator of angiotensin II signal transduction in
vascular smooth muscle cells: Effects on Na+/H+ exchange and
ERK1/2. Circ Res. 83:824–831. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: Implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang YH and Lee SJ: The role of p38 MAPK
and JNK in Arsenic trioxide-induced mitochondrial cell death in
human cervical cancer cells. J Cell Physiol. 217:23–33. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsia CC, Hyde DM and Weibel ER: Lung
structure and the intrinsic challenges of gas exchange. Compr
Physiol. 6:827–895. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kalluri R: The biology and function of
fibroblasts in cancer. Nat Rev Cancer. 16:582–598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
You BR and Park WH: Arsenic trioxide
induces human pulmonary fibroblast cell death via increasing ROS
levels and GSH depletion. Oncol Rep. 28:749–757. 2012.PubMed/NCBI
|
|
23
|
You BR and Park WH: Gallic acid-induced
human pulmonary fibroblast cell death is accompanied by increases
in ROS level and GSH depletion. Drug Chem Toxicol. 34:38–44. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Han YH, Kim SH, Kim SZ and Park WH:
Pyrogallol inhibits the growth of human pulmonary adenocarcinoma
A549 cells by arresting cell cycle and triggering apoptosis. J
Biochem Mol Toxicol. 23:36–42. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of human lung cancer Calu-6 cells
via arresting the cell cycle arrest. Toxicol in vitro.
22:1605–1609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol as a glutathione depletor induces apoptosis in HeLa
cells. Int J Mol Med. 21:721–730. 2008.PubMed/NCBI
|
|
27
|
Han YH, Moon HJ, You BR and Park WH: The
effects of MAPK inhibitors on pyrogallol-treated Calu-6 lung cancer
cells in relation to cell growth, reactive oxygen species and
glutathione. Food Chem Toxicol. 48:271–276. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han YH, Moon HJ, You BR, Kim SZ, Kim SH
and Park WH: JNK and p38 inhibitors increase and decrease
apoptosis, respectively, in pyrogallol-treated calf pulmonary
arterial endothelial cells. Int J Mol Med. 24:717–722.
2009.PubMed/NCBI
|
|
29
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*-) generator induces apoptosis via the depletion of
intracellular GSH contents in Calu-6 cells. Lung Cancer.
63:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park WH and You BR: Antimycin A induces
death of the human pulmonary fibroblast cells via ROS increase and
GSH depletion. Int J Oncol. 48:813–820. 2016.PubMed/NCBI
|
|
32
|
You BR, Shin HR, Han BR and Park WH: PX-12
induces apoptosis in Calu-6 cells in an oxidative stress-dependent
manner. Tumour Biol. 36:2087–2095. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
34
|
Han YH, Moon HJ, You BR and Park WH: The
anti-apoptotic effects of caspase inhibitors on propyl
gallate-treated HeLa cells in relation to reactive oxygen species
and glutathione levels. Arch Toxicol. 83:825–833. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hsin YH, Chen CF, Huang S, Shih TS, Lai PS
and Chueh PJ: The apoptotic effect of nanosilver is mediated by a
ROS- and JNK-dependent mechanism involving the mitochondrial
pathway in NIH3T3 cells. Toxicol Lett. 179:130–139. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han YH and Park WH: Pyrogallol-induced
As4.1 juxtaglomerular cell death is attenuated by MAPK inhibitors
via preventing GSH depletion. Arch Toxicol. 84:631–640. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Park WH, Han YW, Kim SH and Kim SZ: A
superoxide anion generator, pyrogallol induces apoptosis in As4.1
cells through the depletion of intracellular GSH content. Mutat
Res. 619:81–92. 2007. View Article : Google Scholar : PubMed/NCBI
|