Introduction
Hepatitis B virus (HBV) causes acute and chronic
infectious disease of the liver. Cirrhosis and liver cancer may
eventually develop in these patients (1). An estimated 350 million individuals
worldwide are currently experiencing chronic infection (2), and each year >750,000 people succumb
to infectious diseases associated with HBV.
Sustained positive expression of hepatitis B surface
antigen (HBsAg) is positively correlated with B-cell non-Hodgkin
lymphoma (NHL) (3). According to
epidemiological studies performed over the last decade, the risk of
patients infected with HBV developing NHL was 1–2-fold higher than
non-infected patients. Furthermore, HBV infection (HBsAg-negative,
HBV DNA-positive) is also linked with the development of NHL
(3). Although HBV infection may
accompany other hematological malignancies, there is less available
evidence to demonstrate the association between HBV and NHL
(3).
As HBV potentially serves an etiological function in
the development of lymphoma, it is necessary to explore whether HBV
is also able to infect and replicate in lymphoid and hematopoietic
cells. HBV is known to exist in extrahepatic sites (4). Furthermore, HBV nucleic acids have also
been demonstrated to exist in the lymph nodes, spleen, kidneys,
adrenal glands, gonads, thyroid gland and pancreas in patients with
acute HBV infection (5).
In order to investigate this, it is necessary to
determine whether the hematologic malignancy of HBV-positive
patients with NHL regresses under antiviral therapy. A direct
demonstration that HBV is involved in the development of NHL will
offer a positive answer. In addition, if HBV contributes to
lymphomagenesis, HBV vaccination will reduce the incidence of NHL,
even though this impact may only be observed following a long time
interval (3,6–8). A number
of case-control studies concerning HBV with large cohorts have been
published (9–11). In addition, a meta-analysis has
reviewed these studies, and the pooled odds ratio demonstrated the
rarity of the disorder, and was consistent with a significant
association or benefit estimation to relative risk (RR) (12).
No adequate data concerning the potential
association between major NHL subtypes and HBV is available to
assess. A previous large-scale cohort study concerning South Korean
people and their families from 1992–1995 has been performed
(7). According to a previous study,
positive expression of HBsAg resulted in a consistently increased
risk of developing NHL during the whole 14 years of follow-up
(7).
The relationship between HBV and NHL may have been
underestimated due to patients with occult hepatitis infection.
Occult HBV infection has been defined as patients who test
HBsAg-negative but HBV DNA-positive in tissues, serum or a
combination (13). HBV DNA may only
exist in tissues in certain cases; hence, HBV DNA may be
non-existent in the serum of patients with occult HBV. Although
complete HBV elimination occurs in certain cases, this is
considered to be rare at present (14,15). HBV
DNA with replication ability may exist in the lymphocytes, liver or
a combination for a long time. According to previous studies
concerning the detection of HBV DNA in the serum, occult HBV
infection further increased the relationship between NHL and
HBsAg-positive chronic lymphocytic leukaemia (16–18).
Certain individuals, who may be HBV DNA-positive in the
lymphocytes, hepatocytes or a combination but negative in the
serum, would potentially further influence the relationship between
NHL and HBV infection. The mechanism of HBV-associated lymphoma
remains indistinct, but the etiopathogenic function of HBV may help
to construct a multifactorial model for lymphomagenesis. The aim of
the present study was to answer these unresolved questions.
Materials and methods
Cell culture
The human peripheral lymphoblastoid cell line IM-9
(CCL-159) was obtained from the American Type Culture Collection
(Manassas, VA, USA). This cell line resulted from human B
lymphocytes without tumor features that were immortalized with the
Epstein-Barr virus, and are useful for genetic or functional
research as they preserve the genetic characteristics of the
lymphocyte B donor (19–21). The IM-9 cells were cultured in
RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
with 15% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid and
glutamine (Biowest, Nuaillé, France), 100 µg/ml streptomycin and
100 U/ml penicillin (Thermo Fisher Scientific, Inc.). Cells were
cultured in a humidified 5% CO2 atmosphere at 37°C.
Incubations with different concentrations of HBsAg (100, 200 and
400 µg/ml) were performed under these conditions. DMSO (0.1%) was
used as control.
Regents
HBsAg (Recombinant Hepatitis B Surface Antigen, Adw;
cat. no. hbs-872; lot no. 713PADW16) was purchased from
ProSpec-Tany TechnoGene, Ltd. (Rehovot, Israel). Acetylated histone
H3 (cat. no. 8173; dilution, 1:2,000), histone 3 (cat. no. 2632;
dilution, 1:2,000), p21 (cat. no. 2947; dilution, 1:2,000), sirtuin
1 (SIRT1; cat. no. 8469; dilution, 1:3,000), nuclear factor-κB
(NF-κB; cat. no. 4882; dilution, 1:3,000), caspase-3 (cat. no.
9662; dilution, 1:2,500), caspase-8 (cat. no. 8592; dilution,
1:2,500), caspase-9 (cat. no. 9509; dilution, 1:2,500), histone
deacetylase 1 (HDAC1; cat. no. 34589; dilution, 1:2,000), B-cell
lymphoma-extra-large (Bcl-xL; cat. no. 2764; dilution, 1:2,000),
B-cell lymphoma 2 (Bcl-2; cat. no. 15071; dilution, 1:2,000) and
BCL2 associated X (Bax; cat. no. 5023; dilution, 1:2,500)
antibodies were all obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). β-actin antibodies (cat. no. SC-47778;
dilution, 1:5,000) were obtained from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Horseradish peroxidase-conjugated secondary
antibodies (goat anti-mouse immunoglobulin; cat. no. 31430;
dilution, 1:8,000) were acquired from GE Healthcare Life Sciences
(Chalfont, UK). The activator of SIRT1, SRT1720 (SRT), was
purchased from Calbiochem-Novabiochem Corporation (San Diego, CA,
USA), and the inhibitor of SIRT1, nicotinamide (NAM), was obtained
from Sigma-Aldrich (Merck KgaA, Darmstadt, Germany).
Western blotting
A lysis buffer [0.1% SDS, 50 mM Tris (pH 8), 150 mM
NaCl, 0.5% sodium deoxycholate, 0.02% NaN3 and 1% NP-40]
containing a protease inhibitor cocktail (Roche Applied Science,
Penzberg, Germany) and 1 mM phenylmethylsulfonyl fluoride was used
to extract lysates from cells. A Bradford assay with Coomassie Plus
Protein Reagent (Thermo Fisher Scientific, Inc.) was used to
measure protein concentrations. A Biolad system was used to perform
western blotting assays. Protein (50 µg) was loaded in each well of
a 12% gel for electrophoresis, then transferred to a polyvinylidene
fluoride membrane. Non-specific binding was blocked with 1X TBST
containing 5% dry skimmed milk, with agitation, at room temperature
for 1 h. The membrane was incubated with the primary antibody
solution overnight at 4°C, then with the secondary antibody, with
gentle agitation, for 1 h at room temperature. The protein bands
were detected using an enhanced chemiluminescence reagent,
purchased from GE Healthcare Life Sciences, to expose X-ray
film.
In vitro viability assay
Cells (0.5×106 cells/ml) were plated in
6-, 12-, and 24-well plates. Cell viability was assessed using the
nonradioactive MTS cell viability assay (22). Briefly, 20 µl Cell Titer 96 AQueous
One Solution reagent and 80 µl cell suspension were cultured in
96-well plates for 1 h in a 5% CO2 atmosphere at 37°C.
Formazan absorbance was then measured at 490 nm on a Quant plate
reader assembled with KC4 software (Biotek Instruments, Inc.,
Winooski, VT, USA). The measurement was repeated in triplicate and
the means were obtained.
Detection of apoptotic cells with flow
cytometry
Using Annexin V staining, fluorescence-activated
cells sorting analysis was performed following treatment of the
cells with HBsAg (100, 200 and 400 µg/ml; incubated at 37°C for 48
h) and/or SRT (10 µM; incubated at 37°C for 12 h) or NAM (300 µM;
incubated at 37°C for 1 h). DMSO was used as negative control.
Cells were harvested and stained with Annexin V and
7-aminoactinomycin D from an Annexin V-Fluorescein Isothiocyanate
Apoptosis Detection kit (BD Biosciences, Franklin Lakes, NJ, USA)
according to the manufacturer's protocol. Flow cytometry was
performed using a FACScan flow cytometer (BD Biosciences) and
FlowJo software version 10.2 (FlowJo LLC, Ashland, OR, USA).
ELISA
IM-9 cells were cultured, as previously described,
with 100, 200 or 400 µg/ml HBsAg for 24–72 h. DMSO (0.1%) was used
as a negative control. The cells were then centrifuged at 3,000 × g
for 10 min in room temperature and the supernatant collected. The
Human Thirty-Plex Antibody Bead kit (Invitrogen; Thermo Fisher
Scientific, Inc.) was then used according to the manufacturer's
protocol to detect C-X-C motif chemokine 10 (CXCL10, also known as
IP-10), interleukin (IL)-4, −10 and −12 and C-C motif chemokine
ligand 5 (CCL5, also known as RANTES) levels. The assay was
performed in triplicate.
Statistical analysis
Normally distributed, continuous variables were
represented as the mean ± standard deviation. Comparisons of
quantitative data between groups were performed using a paired
Student's t-tests or analysis of variance. Abnormally distributed
data were analyzed using Kruskal-Wallis tests. All statistical
analyses were conducted using SPSS software version 18.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
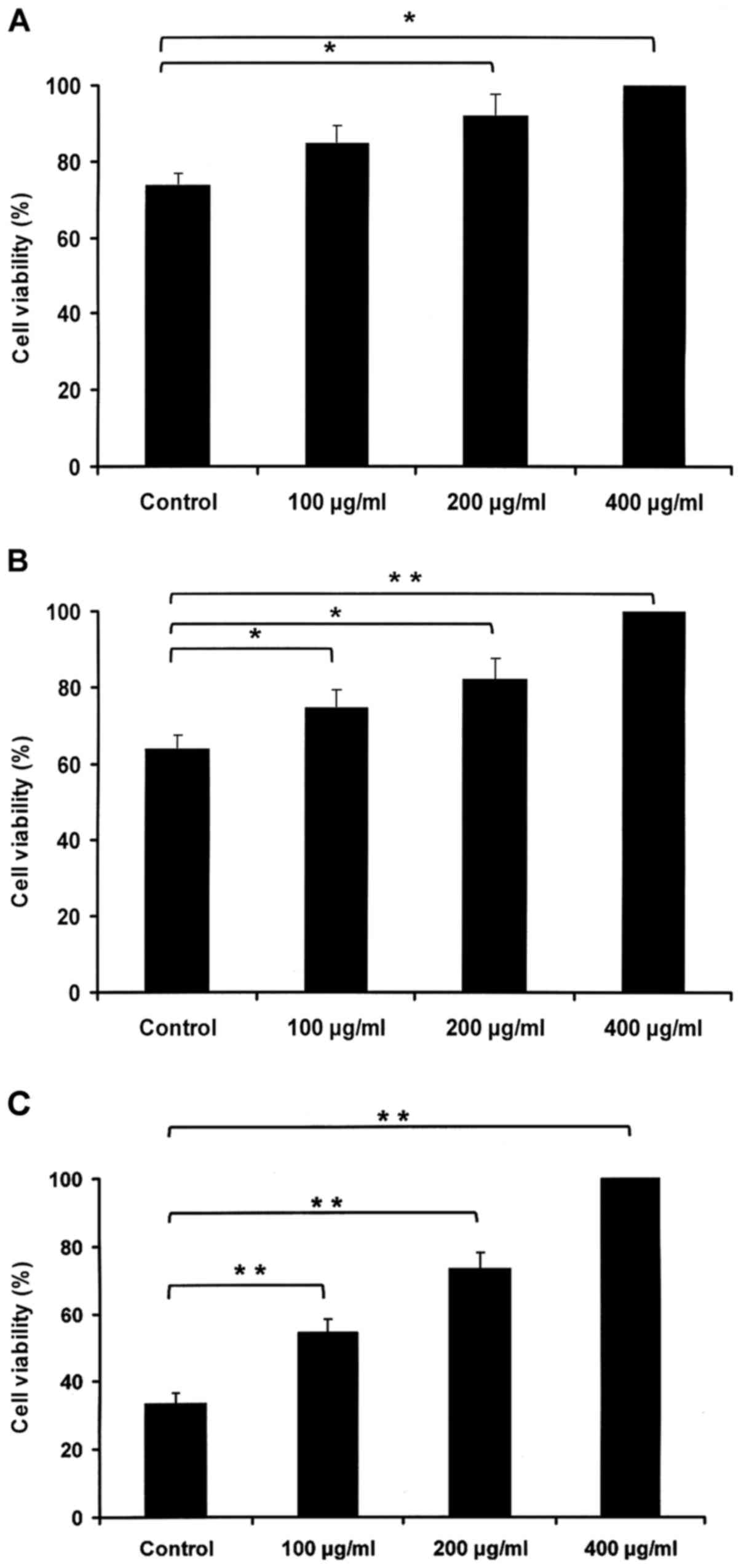
HBsAg promotes IM-9 cell viability in
a dose- and time-dependent manner
To assess the impact of HBsAg in the IM-9 cell line
in vitro, cell viability was measured using the MTS assay.
IM-9 cells were cultured with PBS or HBsAg (100, 200 and 400 µg/ml)
for 24 (Fig. 1A), 48 (Fig. 1B) and 72 h (Fig. 1C). The results revealed that the
HBsAg-induced increase in viability relied on dose and time.
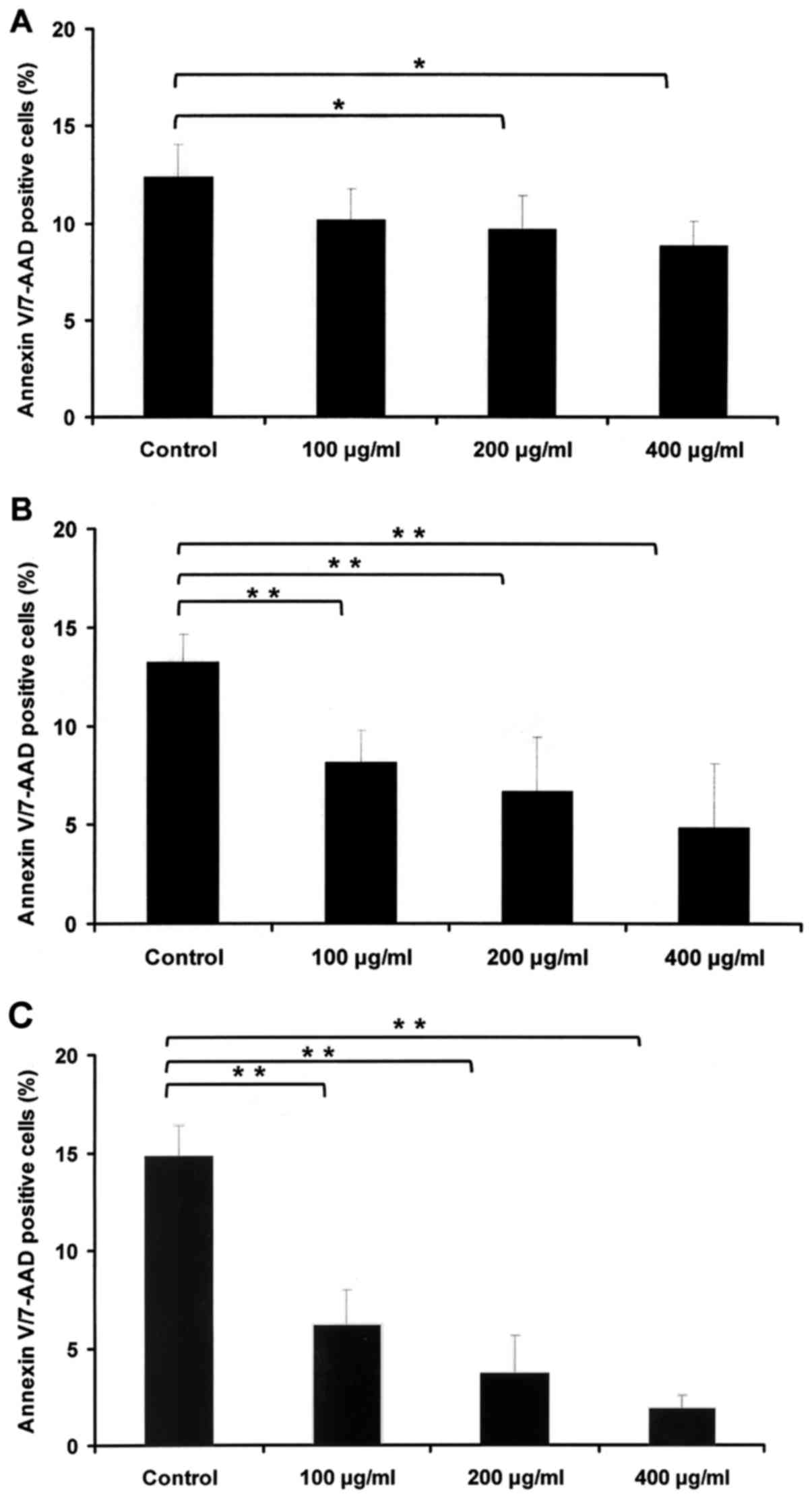
HBsAg reduces apoptosis in a dose- and
time-dependent manner
To assess the in vitro impact of HBsAg in
IM-9 cell lines, its effect on apoptosis was measured. IM-9 cells
were cultured with PBS or HBsAg (100, 200 and 400 µg/ml) for 24
(Fig. 2A), 48 (Fig. 2B) and 72 h (Fig. 2C). Cell apoptosis was subsequently
determined by Annexin V/7-aminoactinomycin D staining and
fluorescence activated cell sorting analysis, which revealed that
the HBsAg-induced decrease in apoptosis rate relied on dose and
time.
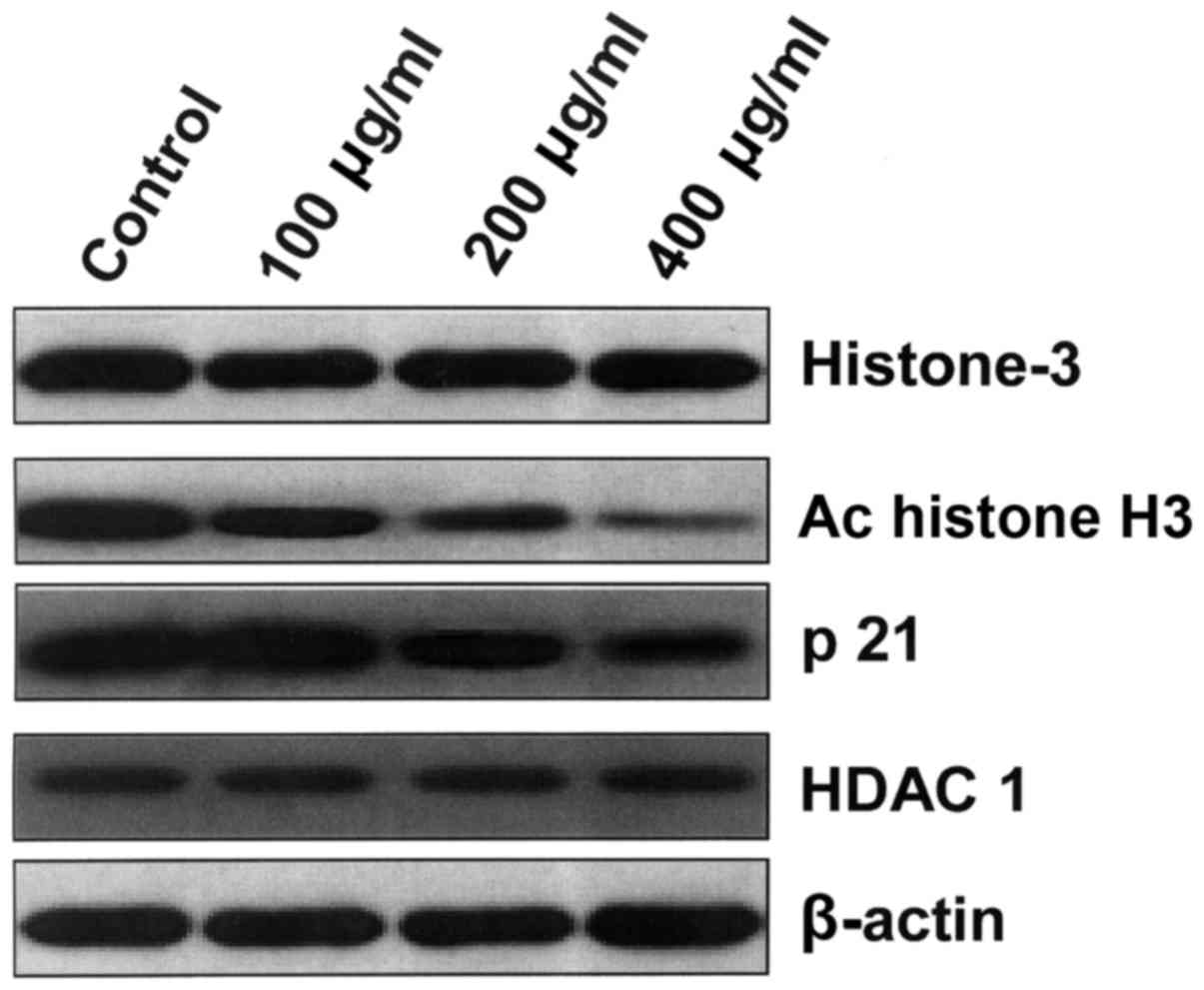
HBsAg decreases the expression of
histone H3 acetylation and p21
IM-9 cells were cultured with HBsAg for 48 h, and
then western blotting was performed. A general characteristic of
histone deacetylases is to decrease the expression of carcinoma
suppressor genes by deacetylating histones, including p21 (23). Therefore, the impact of HBsAg on p21
expression and histone H3 acetylation was measured. Acetylation of
histone H3 and p21 expression was observed at each concentration at
48 h. HDAC1 expression was measured as a positive control. The
results revealed that HBsAg decreased the expression of p21 and
histone H3 acetylation in a dose-dependent manner (Fig. 3).
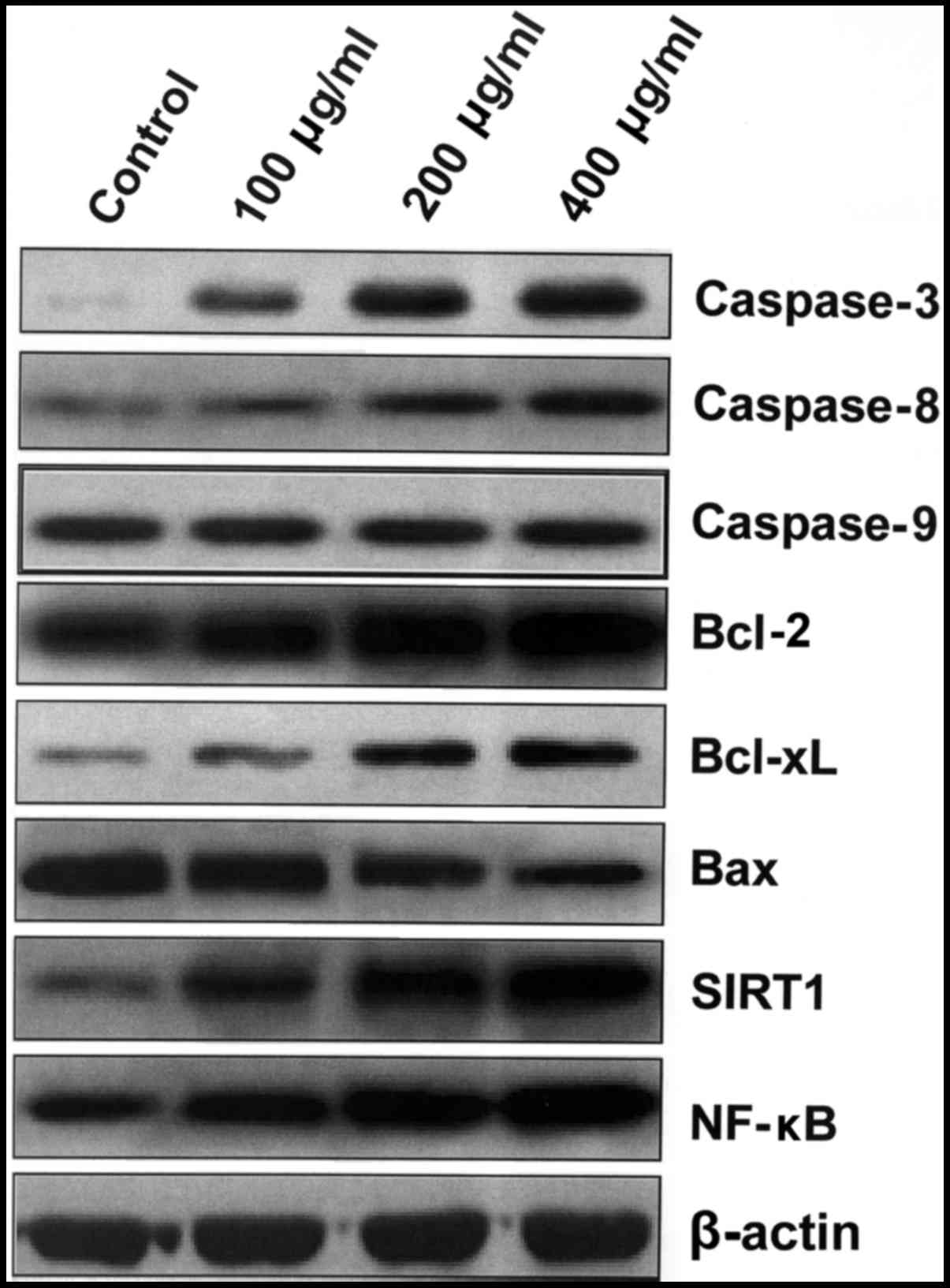
HBsAg upregulates the expression of
anti-apoptotic Bcl-2 family proteins, and inactivates the intrinsic
apoptosis pathway
Expression levels of components of the apoptosis
pathway were also analyzed. IM-9 cells were cultured with HBsAg for
48 h and western blotting was performed. Caspase-3 and −8
expression increased following treatment with HBsAg (Fig. 4). Furthermore, HBsAg treatment
resulted in the upregulation of SIRT1 and NF-κB in a dose-dependent
manner. However, the level of caspase-9 remained unchanged,
implying that the intrinsic apoptosis pathway contributed to the
inhibition of the apoptosis process induced by HBsAg (Fig. 4). HBsAg also upregulated the
expression of anti-apoptotic Bcl-xL and Bcl-2 proteins, and
inactivated the intrinsic apoptosis pathway by downregulating Bax
and increasing the expression of SIRT1 and NF-κB (Fig. 4).
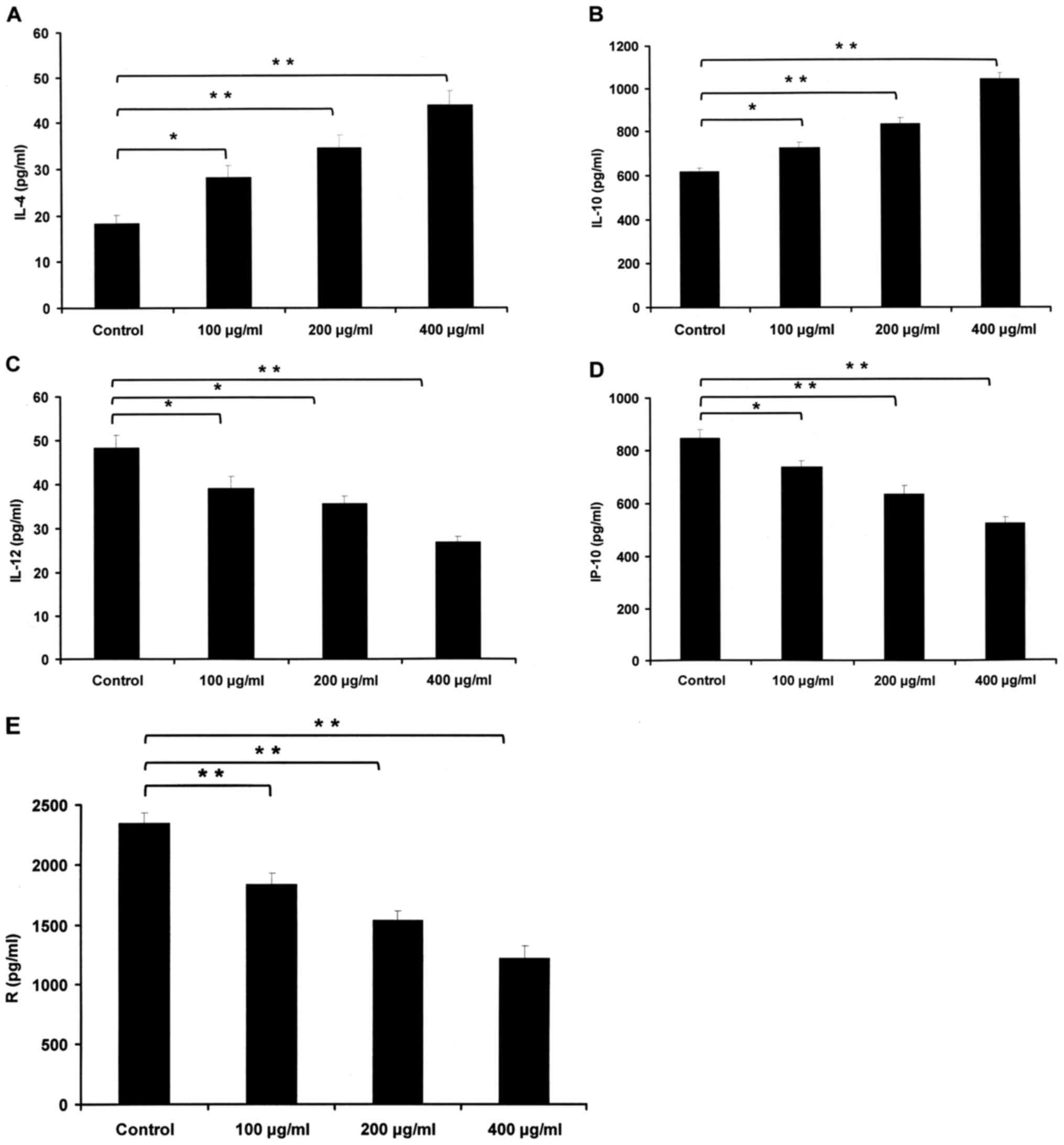
HBsAg affects the expression levels of
multiple chemokines and cytokines
NHL with dysregulated cytokines may cause the immune
system to fail to respond to carcinoma cells (24). Thus, the expression levels of multiple
chemokines and cytokines were detected with a Human Thirty-Plex
Antibody Bead kit in IM-9 cells treated with or without 100, 200 or
400 µg/ml HBsAg for 48 h to examine whether HBsAg influenced
production of these cytokines. The expression levels of cytokine
IL-12, chemokines IP-10 and RANTES, which contribute to Th1 cell
differentiation and recruitment, were all significantly decreased
following treatment with HBsAg. IP-10 is a ligand for C-X-C motif
chemokine receptor 2 and RANTES is a ligand for C-C motif chemokine
receptor (CCR)5 and CCR3. IL-10, which is a pivotal growth factor
of NHL cancer cells, increased significantly, as did IL-4, which is
necessary for Th2 cell differentiation. HBsAg elevated the levels
of IL-10, and IL-4, but reduced the levels of IP-10, IL-12, and
RANTES (Fig. 5).
Inhibition of SIRT1 suppresses the
effects induced by HBsAg
IM-9 cells were cultured with PBS or HBsAg (200
µg/ml) for 48 h, and then treated with SRT (10 µM) or NAM (10 mM)
for a further 48 h (25). Cell
viability was determined, and the HBsAg-induced increase of cell
viability was demonstrated to be promoted by SRT and inhibited by
NAM (Fig. 6A). The rate of cell
apoptosis was then determined, which revealed that the
anti-apoptosis effect of HBsAg was promoted by SRT and inhibited by
NAM (Fig. 6B). The downregulation of
histone H3 acetylation and p21 induced by HBsAg was increased by
SRT and inhibited by NAM (Fig. 6C).
Furthermore, the upregulation of SIRT1 and NF-κB induced by HBsAg
was increased by SRT and inhibited by NAM (Fig. 6C).
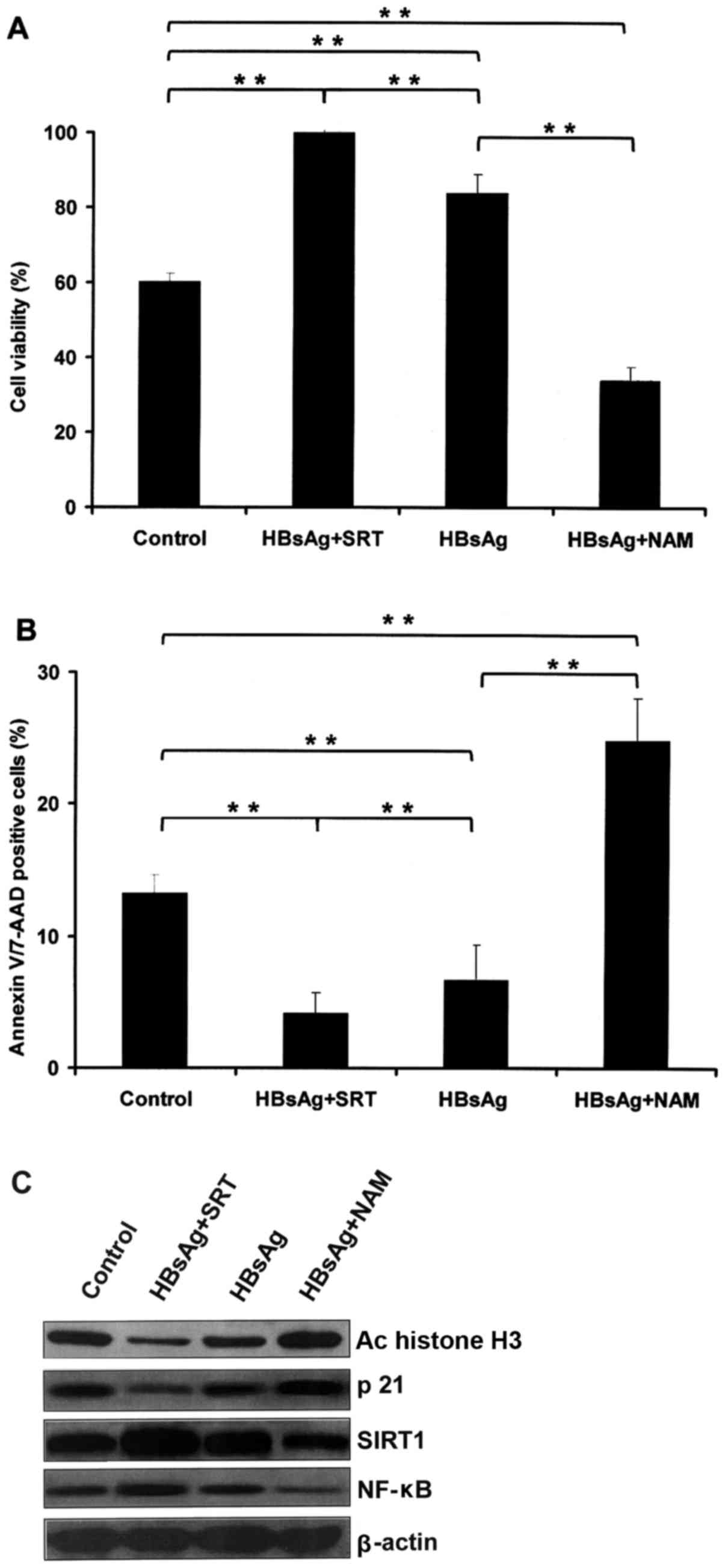 | Figure 6.Inhibition of SIRT1 suppresses the
effects induced by HBsAg. (A) Cell viability was determined by MTS
assay, which revealed that the increase in cell viability induced
by HBsAg was promoted by SRT and inhibited by NAM. (B) The
anti-apoptosis activity induced by HBsAg was promoted by SRT and
inhibited by NAM, as determined by flow cytometry. (C)
Downregulation of histone H3 acetylation and p21 expression induced
by HBsAg was increased by SRT and inhibited by NAM, as determined
by western blotting. **P<0.01, with comparisons indicated by
lines. SIRT1, sirtuin 1; HBsAg, hepatitis B surface antigen; SRT,
SRT1720; NAM, nicotinamide; Ac, acetylated; NF-κB, nuclear
factor-κB. |
Discussion
HBV is well-known to be associated with NHL.
However, whether these associations reveal causal relationships
remains open to discussion. There are three possibilities that may
account for these relationships. First, as a result of that the
carcinoma itself directly suppressing the immune system, the risk
of viral infection or reactivation increases. Unfortunately, this
explanation lacks evidence. According to a number of studies
concerning patients with newly diagnosed disease, significant
immune deficiency was not discovered prior to any treatment
intervention (26,27). Secondly, another unknown virus sharing
a similar transmission mechanism with the hepatitis virus may
represent the actual oncogenic stimulus. Due to the lack of
evidence, this hypothesis remains unclarified. The third
possibility is that hepatitis viruses serve an oncogenic function
in the development of NHL. This possibility and applicable
mechanisms are explored in the next section. In fact, the
association between NHL and HBV is not strong, because the RR is
within the 2–3 range. This is less than the relationship between
HBV infection and HCC (12). For
example, one of the highest RR of HBV carriers for HCC is ~200
(28).
As discussed in a previous publication, the causal
association between HBV and NHL has not been conclusively
demonstrated, but the evidence suggests the existence of a positive
relationship between them (6). This
causal relationship may be HBV-driven. The first potential
mechanism is that HBV infection directly leads to the development
of carcinoma. HBV is known to infect and replicate within
lymphocytes, but although infection may bring about neoplastic
transformation, this is not the inevitable result of viral
replication. HBV that integrate in the host genome may result in
high expression of cellular oncogenes or low expression of tumor
suppressor genes (29,30). The second potential mechanism is
similar to a commonly explained model of HCV-driven lymphomagenesis
(6). This is known as chronic
antigenic stimulation. This mechanism does not need to involve the
infection of lymphocytes. With the infection of hepatocytes there
is sufficient infective virus and viral antigens to support
chronic, antigen-driven proliferation and stimulation of
lymphocytes. This is likely to autonomously induce neoplastic
transformation and proliferation, because chronic antigenic
stimulation may lead to a predisposition to genetic aberrations,
induction of double stranded DNA breaks and translocation or
overexpression of proto-oncogenes (3). However, whether HBV is merely involved
in the initial stages of neoplastic or contributes to continuous
neoplastic viability remains to be demonstrated.
In the present study, the human peripheral
lymphoblastoid cell line, IM-9, was cultured with HBsAg to imitate
chronic antigenic stimulation. HBsAg was demonstrated to promote
cell viability and to reduce apoptosis in a time- and
dose-dependent manner in IM-9 cells. The underlying mechanism was
the dose-dependent decrease in histone H3 acetylation and p21
expression. Furthermore, HBsAg upregulated the expression of
anti-apoptotic Bcl-2 family proteins, and inactivated the intrinsic
apoptosis pathway. Caspase-3 and −8 increased following treatment
with HBsAg. HBsAg also induced the upregulation of SIRT1 and NF-κB
in dose-dependent manner, and induced the increased expression of
the anti-apoptotic Bcl-xL and Bcl-2 proteins. In addition, HBsAg
inactivated the intrinsic apoptosis pathway through upregulating
the expression of NF-κB and SIRT1 and decreasing the expression of
Bax.
Dysregulated cytokines in NHL may cause the immune
system to fail to respond against carcinoma cells (24). A Human Thirty-Plex Bead kit was used
to analyze the impact of HBsAg on cytokine and chemokine
production. The results demonstrated that HBsAg regulated the level
of multiple chemokines and cytokines, including IL-4, IL-10, IL-12,
IP-10, and RANTES. HBsAg increased levels of IL-4 and IL-10 while
it decreased levels of RANTES, IP-10 and IL-12.
Mammalian SIRT1, the ortholog of yeast Sir2, is a
class III histone deacetylase whose activation is dependent on
nicotinamide adenine dinucleotide in the nucleus (31,32). It
deacetylates histones and a large number of non-histone substrates,
including the forkhead box class O family (33–35). High
expression of SIRT1 is often detected in prostate cancer,
glioblastoma and primary colon cancer, and it inactivates proteins
that participate in DNA damage repair and tumor suppression
(36). Consequently, SIRT1 is
considered to be a tumor promoter.
In the present study, IM-9 cells were cultured with
PBS or HBsAg (200 µg/ml) for 48 h, and then treated with activator
of SRT or NAM for a further 48 h (25). Cell viability was then determined, and
the data revealed that the increase in cell viability induced by
HBsAg was promoted by SRT and inhibited by NAM. Cell apoptosis
levels were then determined, and the anti-apoptotic effect of HBsAg
was promoted by SRT and inhibited by NAM. The downregulation of
histone H3 acetylation and p21 expression induced by HBsAg was
increased by SRT and inhibited by NAM. Furthermore, the
upregulation of SIRT1 and NF-κB induced by HBsAg was increased by
SRT and inhibited by NAM.
In conclusion, HBsAg was demonstrated to induce an
anti-apoptotic activity effect in IM-9 cells via a number of
mechanisms, including promotion of cell viability, inhibition of
apoptosis, regulation of chemokines and cytokines, alteration of
SIRT1 and NF-κB expression and a decrease of histone H3
acetylation. Consecutive stimulation with HBsAg promoted the
viability of a human B lymphoblastoid cell line, IM-9, through
regulating the SIRT1-NF-κB pathway. This may be a potential
mechanism underlying HBV-associated NHL.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant no. 30600524),
the National Natural Science Foundation of Hainan Province, China
(grant no. 817351) and the Clinical Support and Scientific Research
Projects of Chinese People's Liberation Army General Hospital
(grant no. 2013 FC-ZHCG-1001).
References
|
1
|
Kew MC: Epidemiology of chronic hepatitis
B virus infection, hepatocellular carcinoma and hepatitis B
virus-induced hepatocellular carcinoma. Pathol Biol (Paris).
58:273–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mizuguchi Y, Takizawa T and Uchida E: Host
cellular microRNA involvement in the control of hepatitis B virus
gene expression and replication. World J Hepatol. 7:696–702. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marcucci F, Spada E, Mele A, Caserta CA
and Pulsoni A: The association of hepatitis B virus infection with
B-cell non-Hodgkin lymphoma-a review. Am J Blood Res. 2:18–28.
2012.PubMed/NCBI
|
|
4
|
Ciesek S, Helfritz FA, Lehmann U, Becker
T, Strassburg CP, Neipp M, Ciner A, Fytili P, Tillmann HL, Manns MP
and Wedemeyer H: Persistence of occult hepatitis B after removal of
the hepatitis B virus-infected liver. J Infect Dis. 197:355–360.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yoffe B, Burns DK, Bhatt HS and Combes B:
Extrahepatic hepatitis B virus DNA sequences in patients with acute
hepatitis B infection. Hepatology. 12:187–192. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marcucci F and Mele A: Hepatitis viruses
and non-Hodgkin lymphoma: Epidemiology, mechanisms of
tumorigenesis, and therapeutic opportunities. Blood. 117:1782–1789.
2011. View Article : Google Scholar
|
|
7
|
Engels EA, Cho ER and Jee SH: Hepatitis B
virus infection and risk of non-Hodgkin lymphoma in South Korea: A
cohort study. Lancet Oncol. 11:827–834. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yi HZ, Chen JJ, Cen H, Yan W and Tan XH:
Association between infection of hepatitis B virus and onset risk
of B-cell non-Hodgkin's lymphoma: A systematic review and a
meta-analysis. Med Oncol. 31:842014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Galun E, Ilan Y, Livni N, Ketzinel M,
Nahor O, Pizov G, Nagler A, Eid A, Rivkind A, Laster M, et al:
Hepatitis B virus infection associated with hematopoietic tumors.
Am J Pathol. 145:1001–1007. 1994.PubMed/NCBI
|
|
10
|
Kim JH, Bang YJ, Park BJ, Yoo T, Kim CW,
Kim TY, Heo DS, Lee HS and Kim NK: Hepatitis B virus infection and
B-cell non-Hodgkin's lymphoma in a hepatitis B endemic area: A
case-control study. Jpn J Cancer Res. 93:471–477. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marcucci F, Mele A, Spada E, Candido A,
Bianco E, Pulsoni A, Chionne P, Madonna E, Cotichini R, Barbui A,
et al: High prevalence of hepatitis B virus infection in B cell
non-Hodgkin lymphoma. Haematologica. 91:554–557. 2006.PubMed/NCBI
|
|
12
|
Nath A, Agarwal R, Malhotra P and Varma S:
Prevalence of hepatitis B virus infection in non-Hodgkin's
lymphoma. A systematic review and meta-analysis. Intern Med J.
40:633–641. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bréchot C, Thiers V, Kremsdorf D, Nalpas
B, Pol S and Paterlini-Bréchot P: Persistent hepatitis B virus
infection in subjects without hepatitis B surface antigen:
Clinically significant or purely ‘occult’? Hepatology. 34:194–203.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuki N, Nagaoka T, Yamashiro M, Mochizuki
K, Kaneko A, Yamamoto K, Omura M, Hikiji K and Kato M: Long-term
histologic and virologic outcomes of acute self-limited hepatitis
B. Hepatology. 37:1172–1179. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rehermann B, Ferrari C, Pasquinelli C and
Chisari FV: The hepatitis B virus persists for decades after
patients' recovery from acute viral hepatitis despite active
maintenance of a cytotoxic T-lymphocyte response. Nat Med.
2:1104–1108. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen MH, Hsiao LT, Chiou TJ, Liu JH, Gau
JP, Teng HW, Wang WS, Chao TC, Yen CC and Chen PM: High prevalence
of occult hepatitis B virus infection in patients with B-cell
non-Hodgkin's lymphoma. Ann Hematol. 87:475–480. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rossi D, Sala L, Minisini R, Fabris C,
Falleti E, Cerri M, Burlone ME, Toniutto P, Gaidano G and Pirisi M:
Occult hepatitis B virus infection of peripheral blood mononuclear
cells among treatment-naive patients with chronic lymphocytic
leukemia. Leuk Lymphoma. 50:604–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang F, Xu RH, Han B, Shi YX, Luo HY,
Jiang WQ, Lin TY, Huang HQ, Xia ZJ and Guan ZZ: High incidence of
hepatitis B virus infection in B-cell subtype non-Hodgkin lymphoma
compared with other cancers. Cancer. 109:1360–1364. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sie L, Loong S and Tan EK: Utility of
lymphoblastoid cell lines. J Neurosci Res. 87:1953–1959. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sugimoto M, Tahara H, Ide T and Furuichi
Y: Steps involved in immortalization and tumorigenesis in human
B-lymphoblastoid cell lines transformed by Epstein-Barr virus.
Cancer Res. 64:3361–3364. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hussain T and Mulherkar R: Lymphoblastoid
cell lines: A continuous in vitro source of cells to study
carcinogen sensitivity and DNA repair. Int J Mol Cell Med. 1:75–87.
2012.PubMed/NCBI
|
|
22
|
Georgakis GV, Li Y, Rassidakis GZ,
Martinez-Valdez H, Medeiros LJ and Younes A: Inhibition of heat
shock protein 90 function by
17-allylamino-17-demethoxy-geldanamycin in Hodgkin's lymphoma cells
down-regulates Akt kinase, dephosphorylates extracellular
signal-regulated kinase, and induces cell cycle arrest and cell
death. Clin Cancer Res. 12:584–590. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gui CY, Ngo L, Xu WS, Richon VM and Marks
PA: Histone deacetylase (HDAC) inhibitor activation of p21WAF1
involves changes in promoter-associated proteins, including HDAC1.
Proc Natl Acad Sci USA. 101:pp. 1241–1246. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maggio E, van den Berg A, Diepstra A,
Kluiver J, Visser L and Poppema S: Chemokines, cytokines and their
receptors in Hodgkin's lymphoma cell lines and tissues. Ann Onc. 13
Suppl 1:52–56. 2002. View Article : Google Scholar
|
|
25
|
Côté CD, Rasmussen BA, Duca FA,
Zadeh-Tahmasebi M, Baur JA, Daljeet M, Breen DM, Filippi BM and Lam
TK: Resveratrol activates duodenal Sirt1 to reverse insulin
resistance in rats through a neuronal network. Nat Med. 21:498–505.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Simms-Waldrip TR, Sunkersett G, Coughlin
LA, Savani MR, Arana C, Kim J, Kim M, Zhan X, Greenberg DE, Xie Y,
et al: Antibiotic-induced depletion of anti-inflammatory clostridia
is associated with the development of graft-versus-host disease in
pediatric stem cell transplantation patients. Biol Blood Marrow
Transplant. 23:820–829. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jain N, Oswal N, Chawla AS, Agrawal T,
Biswas M, Vrati S, Rath S, George A, Bal V and Medigeshi GR: CD8 T
cells protect adult naive mice from JEV-induced morbidity via lytic
function. PLoS Negl Trop Dis. 11:e00053292017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Feitelson MA and Duan LX: Hepatitis B
virus × antigen in the pathogenesis of chronic infections and the
development of hepatocellular carcinoma. Am J Pathol.
150:1141–1157. 1997.PubMed/NCBI
|
|
29
|
Natoli G, Avantaggiati ML, Chirillo P,
Puri PL, Ianni A, Balsano C and Levrero M: Ras- and Raf-dependent
activation of c-jun transcriptional activity by the hepatitis B
virus transactivator pX. Oncogene. 9:2837–2843. 1994.PubMed/NCBI
|
|
30
|
Wang XW, Gibson MK, Vermeulen W, Yeh H,
Forrester K, Stürzbecher HW, Hoeijmakers JH and Harris CC:
Abrogation of p53-induced apoptosis by the hepatitis B virus X
gene. Cancer Res. 55:6012–6016. 1995.PubMed/NCBI
|
|
31
|
Imai S, Armstrong CM, Kaeberlein M and
Guarente L: Transcriptional silencing and longevity protein Sir2 is
an NAD-dependent histone deacetylase. Nature. 403:795–800. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith JS, Brachmann CB, Celic I, Kenna MA,
Muhammad S, Starai VJ, Avalos JL, Escalante-Semerena JC, Grubmeyer
C, Wolberger C and Boeke JD: A phylogenetically conserved
NAD+-dependent protein deacetylase activity in the Sir2 protein
family. Proc Natl Acad Sci USA. 97:pp. 6658–6663. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang H and Tindall DJ: Dynamic FoxO
transcription factors. J Cell Sci. 120:2479–2487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Salminen A, Kauppinen A, Suuronen T and
Kaarniranta K: SIRT1 longevity factor suppresses NF-kappaB-driven
immune responses: Regulation of aging via NF-kappaB acetylation?
Bioessays. 30:939–942. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Leeuwen I and Lain S: Sirtuins and
p53. Adv Cancer Res. 102:171–195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deng CX: SIRT1, is it a tumor promoter or
tumor suppressor? Int J Biol Sci. 5:147–152. 2009. View Article : Google Scholar : PubMed/NCBI
|