Introduction
Metformin has been employed as an anti-diabetic
agent for a number of years. There have been a number of studies on
the use of this agent for cancer chemoprevention or therapy;
epidemiological findings suggest a lower incidence of cancer in
diabetics receiving metformin compared with diabetics receiving
sulfonylurea or insulin (1,2). However a more recent epidemiologic
metanalysis which corrects for specifics of metformin diabetic
patients fails to confirm any efficacy of metformin (3).
Metformin has a number of potential mechanisms of
action through which it may impact the process of carcinogenesis.
Two major pathways are: i) Activation of adenosine triphosphate
(AMP)-activated protein kinase, which is downstream of metformin
effects on the mitochondria, and increased levels of liver kinase
B1 (LKB1) (4,5), which may secondarily affect the
expression of a wide variety of genes involved in gluconeogenesis
and the mammalian target of rapamycin (mTOR) pathway; and ii)
reduction in the levels of insulin-like growth factor-1 (IGF1),
which may be relevant for a variety of forms of cancer (6).
Although there have been hundreds of studies
discussing the use of metformin for cancer prevention, the majority
of these have involved cell culture studies (5,7). In fact,
in vivo animal data employing in situ cancer models
are limited. For this reason, a series of previous studies examined
the efficacy of metformin as a chemopreventive agent in animal
carcinogenesis models that have been used widely to screen for
chemopreventive agents. In our previous study (8), it was demonstrated that, despite
altering the pharmacodynamic biomarkers of drug action, metformin
was ineffective in preventing estrogen receptor (ER)+ or
ER− types of mammary cancer in two widely used animal
models. A similar lack of metformin activity was observed in a
third breast cancer model (9). In the
present study, the chemopreventive efficacy of metformin was
examined in: i) The 4-hydroxybutyl(butyl) nitrosamine
(OH-BBN)-induced model of urinary bladder cancer (10) in rats, a model that has significant
molecular congruity with invasive bladder cancer in humans
(11); ii) the
4-nitroquinoline-1-oxide (4-NQO)-induced model of oral squamous
cell carcinoma (OSCC) in rats, a model that displays numerous
molecular similarities with human oral cancer (12,13); and
iii) a modified Min mouse model [carrying a germline adenomatous
polyposis coli (APC) mutation] (14)
which develops multiple adenomas of the small intestine and is used
as a model for human familial adenomatous polyposis (FAP). The
results revealed that metformin was ineffective as a
chemopreventive agent in each of these widely used in situ
carcinogenesis models. By contrast, nonsteroidal anti-inflammatory
drugs (NSAIDs) and epidermal growth factor receptor (EGFR)
inhibitors have been demonstrated to be effective chemopreventive
agents in all three models (10,12,15).
Materials and methods
Ethical approval
All in vivo studies were performed in full
compliance with the Animal Welfare Act and United States Public
Health Service Policy on Humane Care and Use of Laboratory Animals.
Animal experiments were conducted in facilities at the University
of Alabama at Birmingham (Birmingham, AL, USA; urinary bladder
cancer studies), IIT Research Institute (Chicago, IL, USA; oral
cancer studies) or the Fox Chase Cancer Center (Philadelphia, PA,
USA; intestinal cancer studies). Prior to the initiation of in
vivo studies at any performing site, study protocols were
reviewed and approved by the appropriate Institutional Animal Care
and Use Committee. All animals were housed 5/cage in a room lighted
12 h/day and maintained at 22°C.
Rat urinary bladder cancer model
This model has been previously described (10,16). The
carcinogen OH-BBN was purchased from TCI America, Inc. (Portland,
OR, USA). OH-BBN (150 mg/gavage) was administered 2 times/week for
8 weeks, beginning when the female Fischer-344 rats (n=30/group)
were purchased from Envigo (Indianapolis, IN) at 56 days of age.
The carcinogen was administered in 0.5-ml ethanol:water (25:75,
v/v). Two weeks following the last dose of OH-BBN, the animals
received metformin (50 or 150 mg/kg body weight/day), obtained from
the National Cancer Institute Chemical Repository, Bethesda,
Maryland, USA) in saline or a saline control until the end of the
study. All animals received a Teklad (4%) mash diet (Envigo,
Indianapolis, IN, USA). Animals were observed daily, weighed weekly
and palpated for urinary bladder tumors 2 times/week. Rats were
sacrificed when they developed a large palpable bladder lesion or
were observed to have bloody urine. At necropsy, urinary bladders
were inflated with 10% buffered formalin. Following fixation, the
bladder was observed under a high-intensity light for gross
lesions. Each lesion was dissected and processed for pathological
classification. For immunofluorescence, bladder cancers were fixed
in formalin for 24 h and the switched to 70% ethanol. After
histological processing, the blocks were sectioned at 5 microns and
forwarded to the laboratory of Dr. A. Bode for further analyses.
The multiplicity and weight of the urinary bladder tumors were
determined at the end of the study. Statistical analysis of bladder
tumors between groups was performed by the Wilcoxon rank-sum test
and the final cancer weights by the Fischer's exact test. The
method for determination of metformin concentration in urine was
described in our previous study (8).
Urinary bladder
immunofluorescence
Paraffin embedded bladder cancer tissue samples on
slides were baked in a 60° oven for 2 h. Paraffin was removed using
4 changes of xylene at 5 min Rehydration of the samples was
performed using decreasing percentages of ethanol: three changes of
100% at 5 min each, and 5–10 min using the following percentages
respectively: 95, 90, 70%, and DI Water. Final rinse was performed
using 1xPBS 2×3 min.
Antigen retrieval was performed using 10 mM sodium
citrate buffer (pH 6.0). Slides were heated in a 1,200 W microwave
for 10 min, allowing to cool at room temperature for 20 min
afterward. Rinse slides with DI water for 2×3 min and then 1xPBS
2×3 min.
Specimens were blocked using 10% normal donkey serum
(cat. no., 017-000.121; lot #121615 Jackson Immuno Research
Laboratories, Inc). 1x TPBS for 1 h at room temperature. Primary
antibodies were diluted and samples were labeled as follows:
Anti-p53 (total; monoclonal; cat. No. 2524; lot #4; Cell Signaling
Technology Inc., Danvers, MA, USA) used at 1:100 dilution, and
anti-phosphorylated (p)-p53 (serine 392; goat; cat. no., sc-7997;
lot #L1809; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) used
at 1.25 dilution, and anti-p-p53 (serine 20; goat; cat. no.,
sc-18078; lot #C0510; Santa Cruz Biotechnology, Inc.) used at 1.50
dilution. Antibodies were left on overnight at 4°C.
Samples were rinsed with 1xPBS. Secondary antibodies
were all diluted are 1:200 using 5% normal donkey serum-1xTPBS and
added as follows: anti-cyanine (Cy) 3 (Goat cat. no., 705-165-147;
lot #110658; Jackson Immuno Research Laboratories, Inc.) for p-p53
(ser20), anti-cyanine (Cy) 3 (mouse; cat. No., 715-166-151; lot
#102107; Jackson Immuno Research Laboratories Inc.) for p53 Total
and anti-cyanine (Cy) 2 (goat; cat. no., 705-225-147; lot #103215;
Jackson Immuno Research Laboratories, Inc.) for p-P53 (s392).
Secondary antibodies were left on for 1 h and 45 min and then
rinsed with 1xPBS followed by 1×5 min 1x PBS High Salt (23.38 g
NaCl in 1 liter 1x PBS). Coverslip were put on slides using
Fluoro-Gel II Dapi (cat. no., 17985-50; lot #130218; Electron
Microscopy Sciences) and seal using clear nail polish. The
integrated optical density was calculated as pixel area × mean
density. The units listed on the Image-Pro Premier program (Media
Cybernetics, Offline Version 9.0, S/N 050900000-1104) are
lum/pix2.
Rat oral cancer model
We have previously described the rat oral cancer
model (12,13,15). Male
Fischer 344 rats (n=30/group) were obtained from Envigo and were
placed into quarantine for a minimum of 1 week. Beginning at 8
weeks of age, rats were exposed to drinking water containing 20 ppm
4-NQO (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) for a
period of 10 weeks. Rats were administered a diet of Purina 5001
Laboratory diet. Dietary administration of metformin (250 or 500
ppm) was initiated 1 day following the final administration of
4-NQO. Alternatively, control rats were administered standard
Purina diet without Metformin. In addition, the protocol included a
delayed administration group that received 500 ppm metformin
beginning 6 weeks upon completion of 4-NQO administration. Animals
were monitored twice daily for general health status and were
weighed weekly. Animals were euthanized by CO2 overdose
if they demonstrated a large exophytic oral lesion or lost weight
for two successive weeks; the remaining rats were euthanized 14
weeks following the last administration of 4-NQO. All rats
underwent a gross necropsy that focused on the tongue and oral
cavity. The tongues from each animal were excised and grossly
visible oral lesions were charted. Tongues were then bisected
longitudinally. Gross lesions and phenotypically normal oral
tissues were dissected from one half of each tongue, and were
frozen in liquid nitrogen for molecular analyses. The remaining
half of each tongue was fixed in 10% neutral buffered formalin,
processed by routine histological methods, and stained with
hematoxylin and eosin (H&E) for pathological classification
(12). Cancer invasion was classified
using a semiquantitative grading system: Lesions scored as +1
extended through the mucosal epithelial basement membrane and into
the lamina propria only; lesions scored as +2 extended through the
lamina propria and into the upper muscle layers; and lesions with
the highest invasion score of +3 demonstrated extensive
infiltration into underlying muscle. Evidence of metformin activity
was defined as a statistically significant (P<0.05) reduction in
oral cancer incidence, reduction in oral cancer invasion score or
increase in survival in a group treated with metformin vs. that in
the carcinogen-treated dietary control group. Inter-group
comparisons of oral cancer incidence and survival at the
termination of the study were performed using χ2
analysis. As oral cancer invasiveness was evaluated using a
semiquantitative scoring system, comparisons of invasion scores
were performed using the nonparametric Wilcoxon rank-sum analysis.
Body weights and other continuous data were compared using analysis
of variance, with post hoc comparisons conducted using the
Dunnett's test.
Mouse intestinal tumor model
The methods used for this modified Min mouse assay
have been previously described (14,16). Male
Apc+/Min-FCCC mice (n=10/group, 8 weeks old) were
obtained from the Laboratory Animal Facility at Fox Chase Cancer
Center. Apc+/Min-FCCC mice demonstrate increased numbers
of colorectal adenomas (16) compared
with Apc+/Min mice. Animals were acclimated to a
modified American Institute of Nutrition-76A semi-purified diet for
1 week prior to starting treatment with metformin. At 9 weeks of
age, mice were randomized to receive the control diet or a diet
supplemented with 400 or 1,200 ppm metformin. Body weights were
recorded weekly to monitor toxicity. Animals were maintained on the
experimental diets for 45 days.
At study termination, mice were euthanized
(CO2) and the entire small intestine was excised,
divided into three equal sections (proximal, middle and distal),
opened lengthwise and rinsed with PBS. Gross lesions were counted
and recorded. Each segment of the small intestine was jelly-rolled,
fixed in neutral buffered formalin and embedded entirely in
paraffin. One slide of each intestinal segment (5 micron sections)
was stained with H&E (and evaluated pathologically in a blinded
manner. Adenomas were defined as circumscribed neoplasms composed
of tubular and villous structures and lined with dysplastic
epithelium. Cancer tissues were required to meet the following
three criteria: i) Invasion into at least the submucosa; ii) able
to elicit a desmoplastic reaction; and iii) exhibition of
cytological features of neoplasia.
Body weights and tumor multiplicities were compared
among treatment groups using the Wilcoxon rank-sum test [NCSS
Statistical Software (Salstat, 3rd addition): Wilcoxon
Rank Sum Test]. The P-values of the pair-wise comparisons were
adjusted using the Bonferroni multi-comparison correction.
Results
Efficacy evaluation of metformin in
the OH-BBN rat urinary bladder cancer model
Metformin was administered to rats daily by gavage
at 50 or 150 mg/kg body weight, beginning 2 weeks after the last
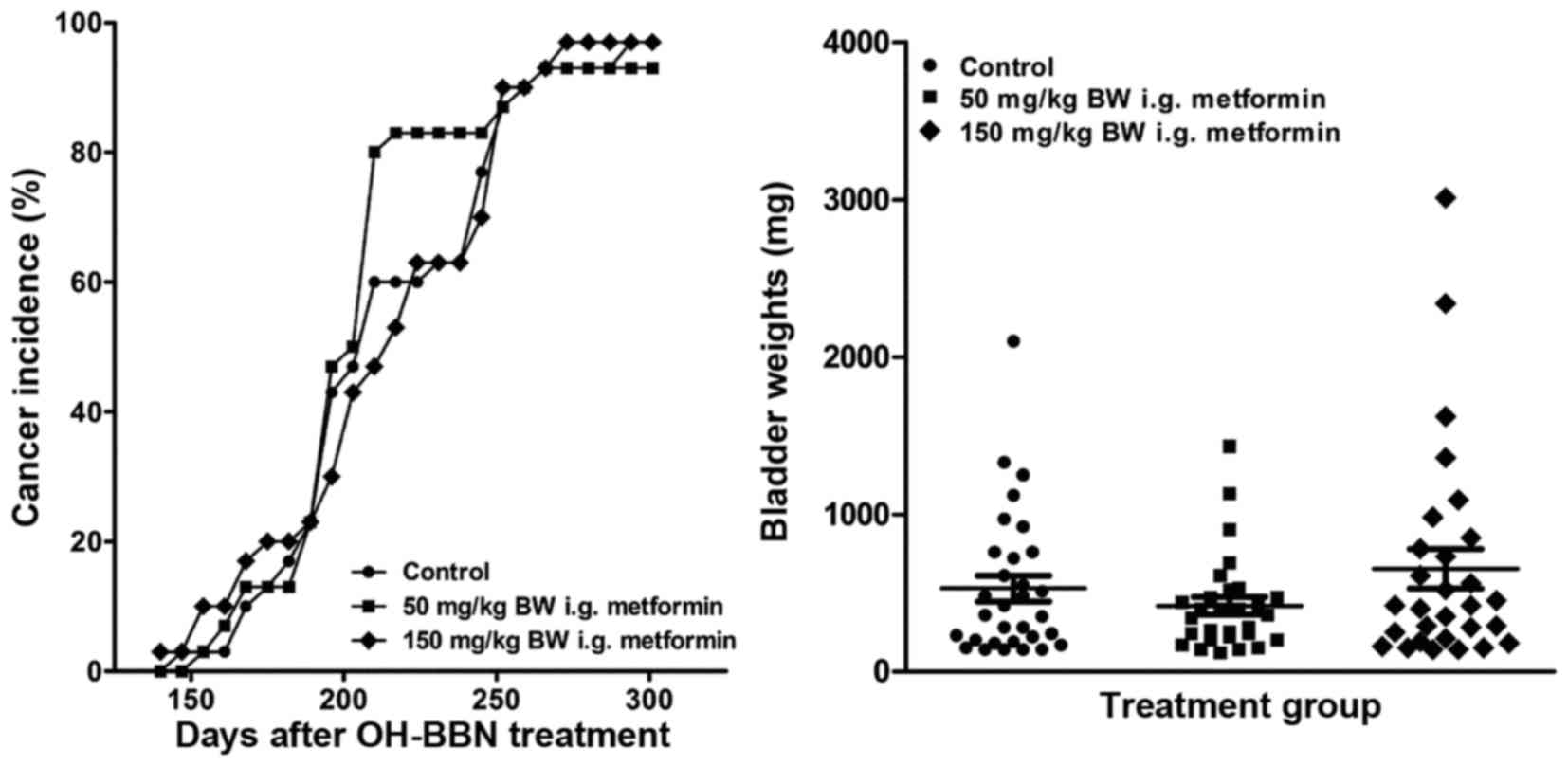
dose of OH-BBN. When compared with OH-BBN-treated rats receiving
vehicle only, neither dose of metformin reduced bladder cancer
incidence or increased bladder cancer latency (P>0.05 for the
two comparisons; Fig. 1A). Similarly,
neither dose of metformin decreased the final incidence of large
palpable cancer. Furthermore, the mean bladder weight, a surrogate
measure of tumor weight, did not differ between groups (P>0.05;
Fig. 1B). This lack of
chemopreventive activity against urinary bladder carcinogenesis was
observed despite the fact that concentrations of metformin in the
urine were markedly elevated compared with the concentrations in
plasma (data not shown). The doses of metformin employed did not
alter final body weights (vehicle, 303 g; high dose metformin, 290
g; low dose metformin, 293 g).
Effects of metformin on p53
phosphorylation in bladder tumor
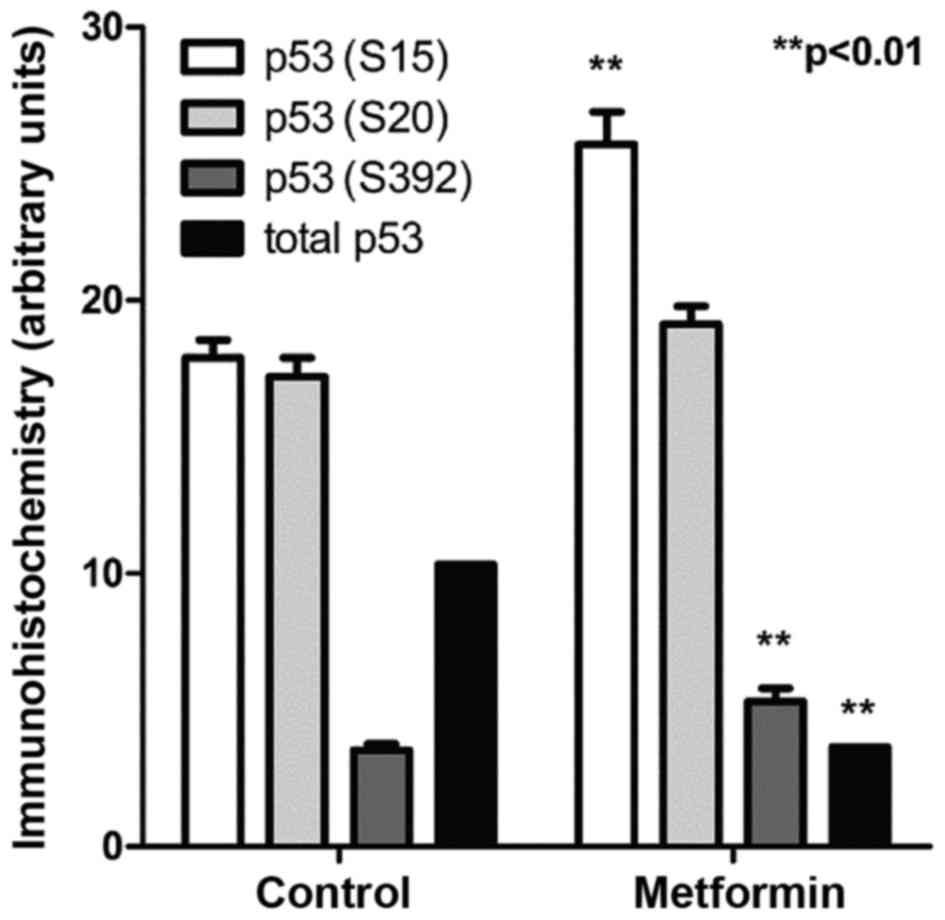
Control OH-BBN-treated rats bearing palpable bladder
tumors were treated with metformin for 5 days. Immunohistochemistry
for specific p53 phosphorylation sites revealed that metformin
increased the phosphorylation at two separate sites on p53, serine
15 and serine 392 (P<0.01 for the two comparisons; Fig. 2). These phosphorylations are
considered to be secondary changes downstream of the activation of
AMP kinase (17).
Efficacy evaluation of metformin in
the 4-NQO rat oral cancer model
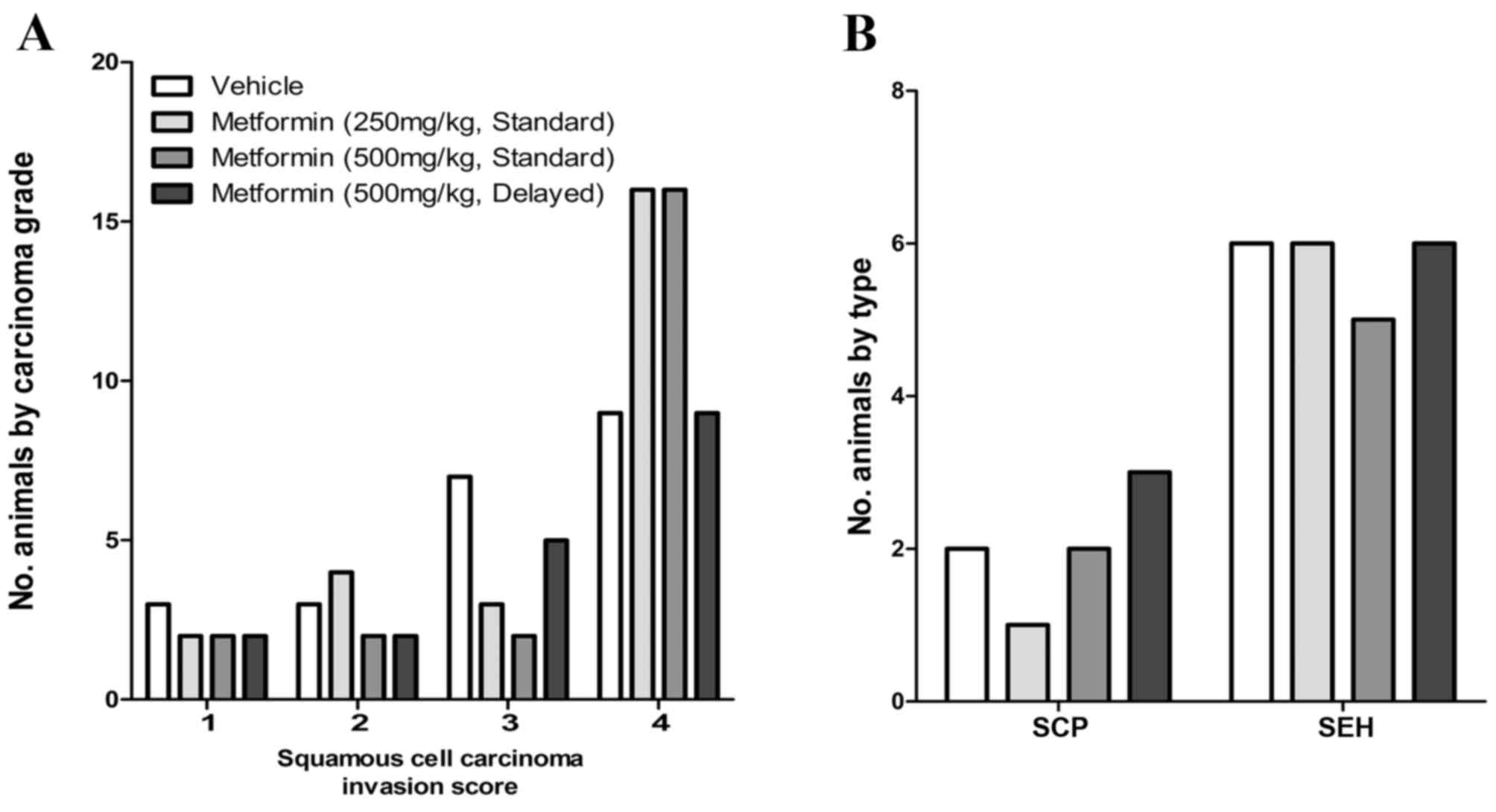
Metformin was administered in the diet (vehicle, 250
or 500 ppm) beginning 1 day following the final dose of 4-NQO. An
additional group of rats was exposed to metformin (500 ppm)
beginning 6 weeks after 4-NQO administration. The final incidence
of OSCC in OH-BBN-treated rats receiving vehicle only was 73%
(22/30 rats). In comparison, OSCC incidences in rats receiving
4-NQO and metformin were 77% (23/30; low-dose metformin), 73%
(22/30; high-dose metformin) and 67% (18/27; high-dose metformin,
delayed administration); none of these incidences was significantly
different from vehicle controls. When compared with vehicle
controls, metformin also failed to alter the invasion score of
induced OSCC (P>0.05 for all comparisons) and did not alter the
incidence of preneoplastic lesions (squamous cell papillomas or
squamous cell hyperplasias; P>0.05 for all comparisons; Fig. 3). The doses of metformin employed did
not alter the final body weights (vehicle, 24 g; high does
metformin, 22.8 g; low dose metformin, 24.1 g).
Efficacy evaluation of metformin in
the Min mouse intestinal tumor model
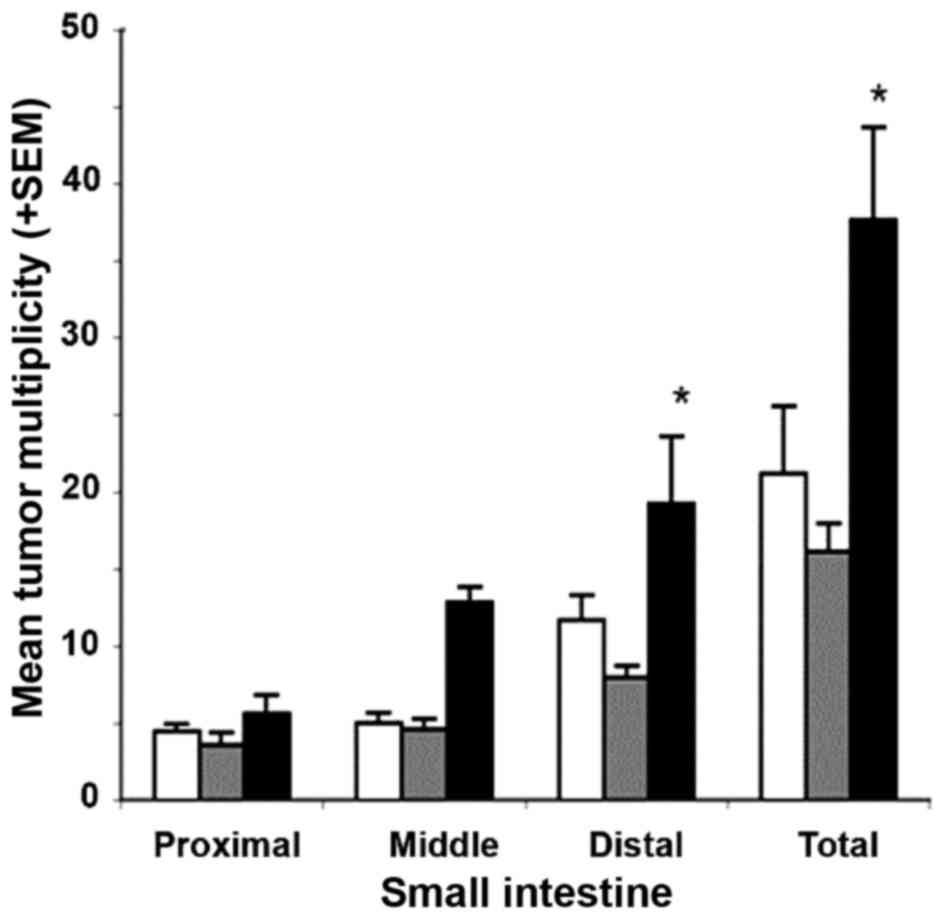
The survival rate for mice receiving diets
supplemented with 400 or 1,200 ppm metformin was ≥95%; this
survival was comparable to that of the untreated control group.
Gross tumor counts in the small intestine (either per segment or
total) in mice that were administered a diet containing 400 ppm
metformin were comparable to those of untreated control mice
(P=0.49; Fig. 4). Although not
statistically significant, animals administered a diet supplemented
with 1,200 ppm metformin exhibited a ~2-fold increase in the mean
multiplicity of gross small intestinal tumors, particularly in the
mid and distal segments (P=0.069 and P=0.097, respectively;
Fig. 4). These results demonstrate
non-significant increases in tumor multiplicity in mice receiving
the high dose of metformin, and non-significant decreases in tumor
incidence in mice receiving the low dose of metformin. In the
colon, a dose-dependent increase in tumor multiplicity was
observed; however, this trend was not statistically significant
(P>0.05). The doses of metformin employed did not alter final
body weights (vehicle, 24 g; high does metformin, 22.8 g; low dose
metformin, 24.1 g).
Discussion
As a result of its relatively low toxicity and its
ability to alter energy metabolism pathways that are important for
neoplastic growth, metformin has been the subject of numerous
studies as a potential cancer chemopreventive and therapeutic agent
(1,2,6). The
results of several early epidemiology studies suggested that
individuals taking metformin had a reduced risk of cancer; however,
several more recent epidemiologic studies have failed to confirm
these findings (3,17,18). It
has been proposed that metformin acts directly on mitochondria,
which secondarily affects LKB1 and cyclic AMP kinase levels
(4), finally altering the mTOR
pathway. Additionally, the systemic effects of metformin on IGF-1
signaling may be mediated through the liver. Thus there are
multiple direct and indirect mechanisms by which metformin may
target cancer cells (19,20). In vitro studies have supported
these expected mechanistic results and shown that metformin is
preferentially effective in tumor cells (6).
Our previous study reported that metformin was
ineffective in preventing neoplastic development in animal models
of ER+ and ER− breast cancer (8), in which metformin provided no
significant protection against tumor development in the
N-methyl-N-nitrosourea (MNU)-induced ER+ mammary cancer
model in rats or in the MMTV-Neu/P53KO transgenic mouse model for
ER− mammary tumors. The results of the present study
provide additional chemoprevention efficacy data for metformin in
three well-characterized animal models of cancer in organ sites
that are not associated with the mammary gland. Metformin failed to
provide protection against carcinogenesis in any of the three
models tested.
Using standard interspecies scaling factors based on
mg/m2 equivalent doses, the metformin dose of 150 mg/kg
body weight/day was selected for use in our previous ER+
breast cancer chemoprevention study (8) as the rat equivalent of a daily human
dose of ~1.5 g. Notably, the Cmax (maximal concentration) and area
under the curve (AUC) values of this dose in rats were 2-3-fold
greater than the Cmax (3.1±0.9 mg/ml; 20 µM) and AUC (18.4±6.5
mg/ml/h) values reported for humans. Despite the fact that the
metformin Cmax and AUC in rodent models were higher compared with
those achieved in humans (21,22),
metformin demonstrated no chemopreventive activity in the
ER+ rat mammary cancer model (8). In this regard, the peak concentration of
metformin achieved in serum of rats (7.5 mg/l) is equivalent to a
concentration of 60 mM (8); these
concentrations (20 µM in humans or 60 mM in rats) are notably lower
than the metformin concentrations of 2.0 mM that are routinely used
in cell culture experiments (4,5).
In the urinary bladder study, metformin
concentrations were measured in overnight urine samples collected
from vehicle or metformin-treated rats. Using the analytic methods
described previously (8) serum levels
of 55 µM, 900 µM and 2800 µM (overnight urines at days 1 and 14)
were observed. This reflects the fact that metformin is routinely
excreted unmetabolized in urine in rodents and humans. Despite
these high urinary concentrations, metformin was ineffective as a
chemopreventive agent in the OH-BBN-induced bladder cancer
model.
Parenthetically, our previous studies have reported
that tumors in this model have strong molecular congruity with
invasive human bladder cancer (11,23). In
addition, cyclooxygenase (COX) inhibitors and EGFR inhibitors
(10) have been demonstrated to be
highly effective in bladder cancer prevention in this rat model,
even when their administration is initiated in the presence of
microscopic lesions. However, metformin was completely ineffective
in the present study. This negative result is somewhat unexpected
considering that i) very high levels of metformin were measured in
the urine of treated rats and ii) phosphorylation of p53 (serine 15
and serine 392) was demonstrated by immunohistochemistry in urinary
bladder cancer tissues in metformin-treated rats. These data
suggest that neither metformin pharmacokinetics nor
pharmacodynamics were limiting in this model. There have been a
number of papers dealing with the use of metformin in bladder
models in rodents (24–26). The first two studies required
intraperitoneal or intravesicular administration of metformin to
achieve efficacy in bladder graft models and therefore are quite
unlike the present studies. The results from Liu et al
(26) appear to be in greater
conflict with the present study. This study was performed in an
in situ model of bladder cancer induced by MNU, and
metformin was administered to rats in water. The authors argued
that the moderate inhibition of bladder cancer observed was due to
inhibition of prostaglandin E levels secondary to the inhibition of
COX-2. However, the model we employed in the current study is
profoundly sensitive to COX-2 inhibition (10) but was not inhibited by metformin.
The present study also examined the chemopreventive
activity of metformin in the 4-NQO-induced model of OSCC. Oral
cancers induced in this model are phenotypically similar to
invasive oral cancers, develop at a site that is a common location
for human oral cancer (the tongue) and demonstrate significant
molecular congruity with human oral cancers (12,13). These
highly invasive lesions, although sensitive to the preventive
effects of several NSAIDs (12),
peroxisome proliferator-activated receptor γ agonists (15) and EGFR tyrosine kinase inhibitors
(15), were not affected by treatment
with metformin. Metformin had no effect on the incidence of OSCC,
did not impact the OSCC invasion score and had no effect on the
incidence of preneoplastic lesions in this animal model.
Finally, the chemopreventive efficacy of metformin
was evaluated in the Min mouse model of intestinal cancer in the
current study. Min mice have a germline mutation in the APC gene
similar to humans with FAP. Furthermore, mutations in the APC or
β-catenin genes are observed in the preponderance of sporadic colon
cancers in mice (14). The Min model,
by contrast to human FAP, yields multiple intestinal lesions rather
than colonic lesions. As was the case with the urinary bladder
cancer and oral cancer studies, metformin failed to confer any
protection against intestinal carcinogenesis in the Min mouse model
in the present study. Reductions in tumor multiplicity of 60–80%
have been reported in other Min mouse chemoprevention studies with
NSAIDs, COX-2 inhibitors and difluoromethylornithine (27). It is of note that one study has
reported a positive result for metformin in the Min mouse model
(28). However, these investigators
identified virtually no decrease in tumor multiplicity, as the
reported positive result was based on a small, yet significant,
effect on tumor size (28).
Pharmacokinetic data revealing high urinary
concentrations of metformin and demonstration of the modulation of
a pharmacodynamics endpoint (increased p53 phosphorylation) in the
urinary bladder cancer model may suggest potential chemopreventive
activity. However, in none of the three models used in the present
study did metformin demonstrate a beneficial effect on tumor
incidence, multiplicity or growth. The observed lack of correlation
between p53 activation and chemopreventive efficacy may be
interpreted as suggesting that clinical trials at the phase-IIA
level may use biomarkers that are more directly related to
preventive efficacy, including cell proliferation or apoptosis
(29,30) and not biochemical parameters,
including activated AMP kinase, and pharmacokinetic data alone.
In the present study, metformin demonstrated no
statistically significant protective activity against neoplastic
development in animal models for cancer of the urinary bladder,
oral cavity or intestinal tract. The lack of metformin
chemopreventive activity in these studies, when considered with our
previous data showing lack of chemopreventive efficacy in two
mammary cancer models, argues that metformin is not a high-priority
candidate for clinical study as a chemopreventive agent in persons
without diabetes or exhibiting insulin resistance.
One major caveat regarding these three studies (as
well as our previous mammary cancer studies) is that these studies
were conducted in non-obese, non-diabetic rodent models. Results
may have been altered in diabetic or insulin-resistant animals, in
which physiological effects on the underlying disease may have
effects on the tumor outcome. A previous study has indicated
preferable efficacy for metformin in rodents with diabetes
(31). The results of the current
study are, nevertheless, relevant to the preponderance of ongoing
clinical trials that specifically exclude diabetics and administer
metformin orally. A previous metformin biomarker-based study using
proliferation as a potential biomarker identified that a large
placebo-controlled trial exhibited differences in efficacy based on
insulin resistance (32). However,
even in this example, the decrease in proliferation in lesions
observed in individuals with insulin resistance treated with
metformin are not nearly as notable as those routinely induced by
selective estrogen receptor modulators or aromatase inhibitors in
similar lesions (29). Furthermore,
if the majority of the effect occurs in persons with diabetes, then
one cannot randomize such individuals to a placebo-controlled trial
or even easily develop a trial comparing two alternative
anti-diabetic drugs. Future work may be performed in normal and
pre-diabetic animals to further interrogate the potential use of
metformin for cancer prevention.
Acknowledgements
The funding for the current study was provided in
part by the National Cancer Institute (Bethesda, MD, USA; grant
nos. HHSN261200433001C, HHSN261201200021I and
HHSN261200433003C).
References
|
1
|
Noto H, Goto A, Tsujimoto T and Noda M:
Cancer risk in diabetic patients treated with metformin: A
systematic review and meta analysis. PLoS One. 7:e334112012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: A systematic review and meta analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gandini S, Puntoni M, Heckman-Stoddard BM,
Dunn BK, Ford L, DeCensi A and Szabo E: Metformin and cancer risk
and mortality: A systematic review and meta-analysis taking into
account biases and confounders. Cancer Prev Res (Phila). 7:867–885.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homoestasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pollak MN: Investigating metformin for
cancer prevention and treatment: The end of the beginning. Cancer
Discov. 2:778–790. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pollak M: The insulin and insulin-like
growth factor receptor family in neoplasia; An update. Nat Rev
Cancer. 12:159–169. 2012.PubMed/NCBI
|
|
7
|
Gallagher EJ and Le Roith D: Diabetes,
cancer, and metformin: Connections of metabolism and cell
proliferation. Ann N Y Acad Sci. 1243:54–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thompson MD, Grubbs CJ, Bode AM, Reid JM,
McGovern R, Bernard PS, Stijleman IJ, Green JE, Bennett C, Juliana
MM, et al: Lack of effect of metformin on mammary carcinogenesis in
nondiabetic rat and mouse models. Cancer Prev Res (Phila).
8:231–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu Z, Jiang W, Thompson MD, Echeverria D,
McGinley JN and Thompson HJ: Effects of metformin, buformin, and
phenformin on the post-initiation stage of chemically induced
mammary carcinogenesis in the rat. Cancer Prev Res (Phila).
8:518–527. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lubet RA, Steele VE, Juliana MM and Grubbs
CJ: Screening agents for preventive efficacy in a bladder cancer
model: Study design, end points, and gefitinib and naproxen
efficacy. J Urol. 183:1598–1603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu Y, Liu P, Wen W, Grubbs CJ, Townsend
RR, Malone JP, Lubet RA and You M: Cross-species comparison of
orthologous gene expression in human bladder cancer and
carcinogen-induced rodent models. Am J Transl Res. 3:8–27.
2010.PubMed/NCBI
|
|
12
|
McCormick DL, Phillips JM, Horn TL,
Johnson WD, Steele VE and Lubet RA: Overexpression of
cyclooxygenase-2 in rat oral cancers and prevention of oral
carcinogenesis in rats by selective and nonselective COX
inhibitors. Cancer Prev Res (Phila). 3:73–81. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng X, Li W, Johnson WD, Torres KE and
McCormick DL: Overexpression of lipocalins and pro-inflammatory
chemokines and altered methylation of PTGS2 and APC2 in oral
squamous cell carcinomas induced in rats by
4-nitroquinoline-1-oxide. PLoS One. 10:e01162852015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cooper HS, Chang WC, Coudry R, Gary MA,
Everly L, Spittle CS, Wang H, Litwin S and Clapper ML: Generation
of a unique strain of multiple intestinal neoplasia
(Apc(+/Min-FCCC)) mice with significantly increased numbers of
colorectal adenomas. Mol Carcinog. 44:31–41. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCormick DL, Horn TL, Johnson WD, Peng X,
Lubet RA and Steele VE: Suppression of rat oral carcinogenesis by
agonists of peroxisome proliferator activated receptor γ. PLoS One.
10:e01418492015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lubet RA, Clapper ML, McCormick DL,
Pereira MA, Chang WC, Steele VE, Fischer SM, Juliana MM and Grubbs
CJ: Chemopreventive efficacy of Targretin in rodent models of
urinary bladder, colon/intestine, head and neck and mammary
cancers. Oncol Rep. 27:1400–1406. 2012.PubMed/NCBI
|
|
17
|
Giovannucci E, Harlan DM, Archer MC,
Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG and
Yee D: Diabetes and cancer: A consensus report. CA Cancer J Clin.
60:207–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Evans JM, Donnelly LA, Emslie-Smith AM,
Alessi DR and Morris AD: Metformin and reduced risk of cancer in
diabetic patients. BMJ. 330:1304–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones RG, Plas DR, Kubek S, Buzzai M, Mu
J, Xu Y, Birnbaum MJ and Thompson CB: AMP-activated protein kinase
induces a p53-dependent metabolic checkpoint. Mol Cell. 18:283–293.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pollak M: Metformin and other biguanides
in oncology: Advancing the research agenda. Cancer Prev Res
(Phila). 3:1060–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tucker GT, Casey C, Phillips PJ, Connor H,
Ward JD and Woods HF: Metformin kinetics in healthy subjects and in
patients with diabetes mellitus. Br J Clin Pharmacol. 12:235–246.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Martin-Castillo B, Vazquez-Martin A,
Oliveras-Ferraros C and Menendez JA: Metformin and cancer: Doses,
mechanisms and the dandelion and hormetic phenomena. Cell Cycle.
9:1057–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Williams PD, Lee JK and Theodorescu D:
Molecular credentialing of rodent bladder carcinogenesis models.
Neoplasia. 10:838–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang T, Guo P, Zhang Y, Xiong H, Yu X, Xu
S, Wang X, He D and Jin X: The antidiabetic drug metformin inhibits
the proliferation of bladder cancer cells in vitro and in vivo.
Intl J Mol Sci. 14:24603–24618. 2013. View Article : Google Scholar
|
|
25
|
Peng M, Su Q, Zeng Q, Li L, Liu Z, Xue L,
Cheng Y, Huang Y, Tao T, Lv H, et al: High efficacy of
intravescicular treatment of metformin on bladder cancer in
preclinical model. Oncotarget. 7:9102–9117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu Q, Yuan W, Tong D, Liu G, Lan W, Zhang
D, Xiao H, Zhang Y, Huang Z, Yang J, et al: Metformin represses
bladder cancer progression by inhibiting stem cell repopulation via
COX2/PGE2/STAT3 axis. Oncotarget. 7:28235–28246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fischer SM, Hawk ET and Lubet RA: Coxibs
and other nonsteroidal anti-inflammatory drugs in animal models of
cancer chemoprevention. Cancer Prev Res (Phila). 4:1728–1735. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tomimoto A, Endo H, Sugiyama M, Fujisawa
T, Hosono K, Takahashi H, Nakajima N, Nagashima Y, Wada K, Nakagama
H and Nakajima A: Metformin suppresses intestinal polyp growth in
ApcMin/+ mice. Cancer Sci. 99:2136–2141. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dowsett M, Nielsen TO, A'Hern R, Bartlett
J, Coombes RC, Cuzick J, Ellis M, Henry NL, Hugh JC, Lively T, et
al: Assessment of Ki67 in breast cancer: Recommendations from the
International Ki67 in breast cancer working group. J Natl Cancer
Inst. 103:1656–1664. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hadad S, Iwamoto T, Jordan L, Purdie C,
Bray S, Baker L, Jellema G, Deharo S, Hardie DG, Pusztai L, et al:
Evidence for biological effects of metformin in operable breast
cancer: A pre-operative, window-of-opportunity, randomized trial.
Breast Cancer Res Treat. 128:783–794. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Algire C, Amrein L, Zakikhani M, Panasci L
and Pollak M: Metformin blocks the stimulative effect of a
high-energy diet on colon carcinoma growth in vivo and is
associated with reduced expression of fatty acid synthase. Endocr
Relat Cancer. 17:351–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bonanni B, Putoni M, Cazzaniga M, Pruneri
G, Serrano D, Guerrieri-Gonzaga A, Gennari A, Trabacca MS,
Galimberti V, Veronesi P, et al: Dual effect of metformin on breast
cancer proliferation in a randomized presurgical trial. J Clin
Oncol. 30:2593–2600. 2012. View Article : Google Scholar : PubMed/NCBI
|