Introduction
The perforin-dependent granule-mediated cytolysis of
cytotoxic lymphocytes (CLs), including natural killer cells and
cytotoxic T lymphocytes, is the key machinery in the clearance of
viral, and intracellular bacterial infections, as well as in the
prevention of tumor development (1,2). The
proteins encoded by perforin 1 (PRF1), unc-13 homolog D (UNC13D),
syntaxin 11 (STX11), and STXBP2 (syntaxin binding protein 2) serve
an essential role in this pathway. Mutations in these genes lead to
function defects of CLs and are causative of familial
hemophagocytic lymphohistiocytosis type 2 (FHL2), FHL3, FHL4, and
FHL5 (3–6). The clinical manifestation of X-linked
lymphoproliferative disease (XLP), which is caused by mutations in
SH2 domain containing 1A (SH2D1A) (7)
or X-linked inhibitor of apoptosis (XIAP) (8) genes, resembles hemophagocytic
lymphohistiocytosis. Furthermore, XLP2 due to XIAP deficiency has
been suggested to be classified as X-linked FHL (9).
A proportion of patients with lymphoma have been
reported to harbor mutations in PRF1, UNC13D, STX11, STXBP2 or
SH2D1A genes (10–14), indicating that genetic defective
function of CLs may increase susceptibility to lymphomagenesis. The
aim of the present study was to investigate the association between
mutations in genes involved in the cytotoxic function of CLs and
the development of lymphoma in Chinese patients.
Patients and methods
Cases and controls
In the present study, 68 and 34 patients with
lymphoma were admitted to Hebei Yanda Lu Daopei Hospital (Sanhe,
China) and Peking University First Hospital (Beijing, China),
respectively, between August 2013 and August 2015; 12/102 were
excluded due to poor DNA quality. A total of 90 (61 from Hebei
Yanda Lu Daopei Hospital and 29 from Peking University First
Hospital) unrelated patients with lymphoma (48 males and 42
females; age range, 3–60 years) were recruited in the present
study; 39 were diagnosed with Hodgkin lymphoma and 51 were
diagnosed with non-Hodgkin lymphoma according to the World Health
Organization classification (15).
Healthy donors of Han nationality (n=210) at the Hebei Yanda Lu
Daopei Hospital served as controls. The present study was approved
by the Ethics Committees of Hebei Yanda Lu Daopei Hospital and
Peking University First Hospital. Written informed consent was
obtained from all patients and healthy donors or their parents in
accordance with the 1964 Helsinki declaration, and its later
amendments or comparable ethical standards.
Amplification and sequence
analysis
Genomic DNA was isolated from peripheral blood and
bone marrow using the TIANamp Blood DNA kit (item no. DP318;
Tiangen Biotech Co., Ltd., Beijing, China) or from nails using the
TIANamp FFPE DNA kit (item no. DP331; Tiangen Biotech Co., Ltd.)
according to the manufacturer's protocol. Referenced coding
sequences of the PRF1 (NM_005041.4), UNC13D (NM_199242.2), STXBP2
(NM_003764.3), STX11 (NM_006949.2), SH2D1A (NM_002351.3), and XIAP
(NM_001167.2) were obtained from the National Center for
Biotechnology Information Consensus CDS database (https://www.ncbi.nlm.nih.gov/projects/CCDS/CcdsBrowse.cgi).
Primers were designed to amplify the coding exons and the flanking
intron sequences by polymerase chain reaction (PCR). The sequences
of primers are presented in Table I.
The PCR system comprised of 1 µl genomic DNA (10 ng/µl), 1 ml
forward primer (20 pmol/µl), 1 ml reverse primer (20 pmol/µl), 10
µl Phusion Flash High-Fidelity PCR Master mix (Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and 7 µl distilled water in a
total volume of 20 µl. Reaction conditions were 10 sec at 98°C
followed by 38 cycles of 10 sec at 98°C, 10 sec at 68°C, 15 sec at
72°C, and then 1 min at 72°C. The amplified PCR products were
purified with ExoSAP-IT (USB Co., Cleveland, OH, USA) and followed
by cycle sequencing PCR using a BigDye Terminator Sequencing Kit
version 3.1 (Thermo Fisher Scientific, Inc.). Fluorescent labeled
products were separated using an ABI 3500xL Genetic Analyzer
(Thermo Fisher Scientific, Inc.). Variations were analyzed using
Variant Reporter software (version 1.1; Thermo Fisher Scientific,
Inc.). Genetic polymorphism information from the Single Nucleotide
Polymorphism database (dbSNP; http://www.ncbi.nlm.nih.gov/snp/), 1000 Genomes
Project (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/)
and the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) were referenced to
obtain the frequencies of variants in large populations. Variants
with minor allele frequencies >1% in the 1000 Genomes Project
and/or ExAC were regarded as SNPs rather than mutations.
 | Table I.Primers used for amplification of the
coding exons and the flanking intron sequences of perforin 1,
unc-13 homolog D, syntaxin binding protein 2, syntaxin 11, SH2
domain containing 1A and X-linked inhibitor of apoptosis. |
Table I.
Primers used for amplification of the
coding exons and the flanking intron sequences of perforin 1,
unc-13 homolog D, syntaxin binding protein 2, syntaxin 11, SH2
domain containing 1A and X-linked inhibitor of apoptosis.
| Name of the
primer | Sequence 5′ to
3′ |
|---|
| UNC13D-1FS |
TGTAAAACGACGGCCAGTACTCGAGGAAGTGGGGTGAGA |
| UNC13D-1RS |
CAGGAAACAGCTATGACCGAGACCACAGTGCTCCCCAA |
| UNC13D-2FS |
TGTAAAACGACGGCCAGTCCTGTCCATCTGAGCCTGCTC |
| UNC13D-2RS |
CAGGAAACAGCTATGACCGGGACCCCACCCCATGCTCA |
| UNC13D-3FS |
TGTAAAACGACGGCCAGTGGTCAGGGAGCTTGAGGTAACC |
| UNC13D-3RS |
CAGGAAACAGCTATGACCAGACCCTGCTACCCAGGAAAG |
| UNC13D-4FS |
TGTAAAACGACGGCCAGTGCTCTGGGCTGTGGTCACTTAC |
| UNC13D-4RS |
CAGGAAACAGCTATGACCAGGCTCAGCTTTGTGAGGACAC |
| UNC13D-5FS |
TGTAAAACGACGGCCAGTCCTGGGGTCCACCTCCTGTC |
| UNC13D-5RS |
CAGGAAACAGCTATGACCGCTGGTGGCTCAGGGGTTC |
| UNC13D-6FS |
TGTAAAACGACGGCCAGTGGCAATTTCCTCCTCCCTGTC |
| UNC13D-6RS |
CAGGAAACAGCTATGACCCAGTGGTGCCAGTCTGTCGAC |
| UNC13D-7FS |
TGTAAAACGACGGCCAGTGCAGGGTCCTGGTACAGATGTG |
| UNC13D-7RS |
CAGGAAACAGCTATGACCGCCATGGAGAAGAGGTGGATC |
| UNC13D-8FS |
TGTAAAACGACGGCCAGTGGTGTATGCCACTGGGTGACA |
| UNC13D-8RS |
CAGGAAACAGCTATGACCAGGTCCAGGCAGAACCCAAG |
| UNC13D-9FS |
TGTAAAACGACGGCCAGTCTGGTGATGGTAGCTGCTCTATGA |
| UNC13D-9RS |
CAGGAAACAGCTATGACCCAGCTGGGACAGAGATGCAGA |
| UNC13D-10FS |
TGTAAAACGACGGCCAGTCCAGGCAGCCAACATGGTAA |
| UNC13D-10RS |
CAGGAAACAGCTATGACCAGAGAACATGCTTTGCCTGGTC |
| UNC13D-11FS |
TGTAAAACGACGGCCAGTCTACAAACTGCTCTCACAGAACGG |
| UNC13D-11RS |
CAGGAAACAGCTATGACCGGCTGCTACACCCCTCAGAAC |
| UNC13D-12FS |
TGTAAAACGACGGCCAGTGAGCGTCTTTGCTTCCTCCTC |
| UNC13D-12RS |
CAGGAAACAGCTATGACCGCTCACTGTCAAGGGTAACATGTC |
| UNC13D-13FS |
TGTAAAACGACGGCCAGTTCCCATGACCCAATACTTTCCA |
| UNC13D-13RS |
CAGGAAACAGCTATGACCGCACTGACCCCTCCTGGTAAC |
| UNC13D-14FS |
TGTAAAACGACGGCCAGTACTCATCCGGAAGTACTTCTGCA |
| UNC13D-14RS |
CAGGAAACAGCTATGACCCACATCCAGCTGCAAACTCTTG |
| UNC13D-15FS |
TGTAAAACGACGGCCAGTAGCTGGCTTTGCAGTCCAAA |
| UNC13D-15RS |
CAGGAAACAGCTATGACCTCAGACCGTTGCTGGTATCAAA |
| UNC13D-16FS |
TGTAAAACGACGGCCAGTGGAGAAGGGCCTGGATCTCA |
| UNC13D-16RS |
CAGGAAACAGCTATGACCCCTACAGGAAAGCCCTTGCA |
| STXBP2-1FS |
TGTAAAACGACGGCCAGTGACTCAACTTCCTGGGCCTG |
| STXBP2-1RS |
CAGGAAACAGCTATGACCGGAGCAGCTGAGGCCGGAACT |
| STXBP2-2FS |
TGTAAAACGACGGCCAGTTGGTGGGACCAGAGAACCAG |
| STXBP2-2RS |
CAGGAAACAGCTATGACCCACGCTCAGGTCCCATCTCA |
| STXBP2-3FS |
TGTAAAACGACGGCCAGTTGGTGGTCCCTAAGTGGGTTTC |
| STXBP2-3RS |
CAGGAAACAGCTATGACCGCATACACACACGCTCACTCATG |
| STXBP2-4FS |
TGTAAAACGACGGCCAGTCCATGTGGGTGCGACACTAGT |
| STXBP2-4RS |
CAGGAAACAGCTATGACCGCCCAGCCTCAGTGTCTGTTT |
| STXBP2-5FS |
TGTAAAACGACGGCCAGTCAACCCTGGTGCTTCTGTCC |
| STXBP2-5RS |
CAGGAAACAGCTATGACCGGAACCAGGTCAGTGGCAAG |
| STXBP2-6FS |
TGTAAAACGACGGCCAGTCTTGCCACTGACCTGGTTCC |
| STXBP2-6RS |
CAGGAAACAGCTATGACCGAACGCAGACAGAGCATGGG |
| STXBP2-7FS |
TGTAAAACGACGGCCAGTCCGCAGTACCAGAAGGAGCT |
| STXBP2-7RS |
CAGGAAACAGCTATGACCCCCTCCACCTCTCCACAAGC |
| STXBP2-8FS |
TGTAAAACGACGGCCAGTCCTTGAGAGACCTGGTGCTGAG |
| STXBP2-8RS |
CAGGAAACAGCTATGACCGTGGGAGACGCTGGCAAATG |
| STXBP2-9FS |
TGTAAAACGACGGCCAGTCCAGGTTTCCCACTCTTGCTC |
| STXBP2-9RS |
CAGGAAACAGCTATGACCGACCAGACCCGAAACACTGC |
| STXBP2-10FS |
TGTAAAACGACGGCCAGTTCTGTGACCAGCCTCCTTCC |
| STXBP2-10RS |
CAGGAAACAGCTATGACCCCTCAGCAGAGCAGATCGGT |
| STXBP2-11FS |
TGTAAAACGACGGCCAGTCAGAGGCAGGAGGTGGAGATG |
| STXBP2-11RS |
CAGGAAACAGCTATGACCTGTCCCTGTCCCTCAGCAAA |
| STXBP2-12FS |
TGTAAAACGACGGCCAGTAAGTGGGAGGTGCTCATTGG |
| STXBP2-12RS |
CAGGAAACAGCTATGACCAAGTCCAAGTTCTTAACCTCCATGA |
| STX11-1FS |
TGTAAAACGACGGCCAGTTTGCCCACACCGAGGAATAC |
| STX11-1RS |
CAGGAAACAGCTATGACCCTCGCTCAGCTCCTTCATGG |
| STX11-2FS |
TGTAAAACGACGGCCAGTGCGAGGTCATCCACTGCAAG |
| STX11-2RS |
CAGGAAACAGCTATGACCCTTTGGTGCGTCCTTCCCAG |
| PRF1-1FS |
TGTAAAACGACGGCCAGTCCTTCCATGTGCCCTGATAA |
| PRF1-1RS |
CAGGAAACAGCTATGACCGCCAGGATTGCAGTTTCTTC |
| PRF1-2FS |
TGTAAAACGACGGCCAGTCCCTGGGTTCCAGTCCTAGT |
| PRF1-2RS |
CAGGAAACAGCTATGACCGCCCTGTCCGTCAGGTACT |
| PRF1-3FS |
TGTAAAACGACGGCCAGTCTGCACGTGCTGCTGGACA |
| PRF1-3RS |
CAGGAAACAGCTATGACCCTGGTCCTTTCCAAGCTCAC |
| SH2D1A-1FS |
TGTAAAACGACGGCCAGTGCTCGATCGAACCAAGCTAC |
| SH2D1A-1RS |
CAGGAAACAGCTATGACCGGATTGAGGCGAAAGTGTGT |
| SH2D1A-2FS |
TGTAAAACGACGGCCAGTTCTCACTGGAAACTGTGGTTGG |
| SH2D1A-2RS |
CAGGAAACAGCTATGACCGCTAAACAGGACTGGGACCAAA |
| SH2D1A-3FS |
TGTAAAACGACGGCCAGTACTTCTCTTAGCATCCCTAGCAC |
| SH2D1A-3RS |
CAGGAAACAGCTATGACCCTGGCTACATCTACTTTCTCACTGC |
| SH2D1A-4FS |
TGTAAAACGACGGCCAGTAGGCTCAGGCATAAACTGAC |
| SH2D1A-4RS |
CAGGAAACAGCTATGACCGCATTTGTAGCTCACCGAACTGT |
| XIAP-1FS |
TGTAAAACGACGGCCAGTAGAATGTTTCTTAGCGGTCGTGTAG |
| XIAP-1RS |
CAGGAAACAGCTATGACCGTTCCTCGGGTATATGGTGTCTGATAT |
| XIAP-2FS |
TGTAAAACGACGGCCAGTTCTGGGAAGCAGAGATCATTTTG |
| XIAP-2RS |
CAGGAAACAGCTATGACCCCTGGCATACTTGGGAAGCT |
| XIAP-3FS |
TGTAAAACGACGGCCAGTAGTGTGTATTTCTTCCTCAAAGGATAA |
| XIAP-3RS |
CAGGAAACAGCTATGACCCTCCCACTGCATGCTATCCAA |
| XIAP-4FS |
TGTAAAACGACGGCCAGTCAGTGGGATAGGGAATTGGGTA |
| XIAP-4RS |
CAGGAAACAGCTATGACCCACTGCCCAGCTAGCTCTCAT |
| XIAP-5FS |
TGTAAAACGACGGCCAGTGGTGGCCAAGGCATCAGTAA |
| XIAP-5RS |
CAGGAAACAGCTATGACCGCGCATCACAAGATCAGGAGT |
| XIAP-6FS |
TGTAAAACGACGGCCAGTACCCGCTCTGCTACAGAAAC |
| XIAP-6RS |
CAGGAAACAGCTATGACCCACATCTGGCCCTTTCTTGCTTT |
| XIAP-7FSa |
TGTAAAACGACGGCCAGTCAGATGCCACGGGTGAGTCA |
| XIAP-7RSa |
CAGGAAACAGCTATGACCATTGCCAACTAAAACACTGCCAT |
Confirmation of germline derivation of
mutations
For patients determined to harbor mutations, the
same mutation was detected in the DNA isolated from peripheral
blood of their parents. In the absence of one or both parents, the
detection of the same mutation in DNA extracted from nails of the
patients could be of value. This was performed in order to confirm
that the mutations were germline-derived.
In silico analysis
Two bioinformatics tools were used to predict
whether an amino acid substitution was benign or deleterious:
Sorting Intolerant From Tolerant (SIFT; http://sift.jcvi.org/) predicts whether an amino acid
substitution affects protein function based on the degree of
conservation of amino acid residues in multiple sequence alignments
derived from closely associated sequences (16); and Polymorphism Phenotyping version
2.0 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph/) predicts the
possible impact of an amino acid substitution on the structure and
function of a human protein using straightforward physical and
comparative analyses (17). Iterative
Threading ASSEmbly Refinement (I-TASSER; http://zhanglab.ccmb.med.umich.edu/I-TASSER/) was
also used to predict and simulate the influence of the variants in
protein tertiary structures.
Statistical analysis
Comparisons of mutant frequencies as well as
genotype distributions between patients with lymphoma and controls
were performed using the Chi-square test with SPSS software
(version 20.0; IBM Corp., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Analysis of the gene mutations
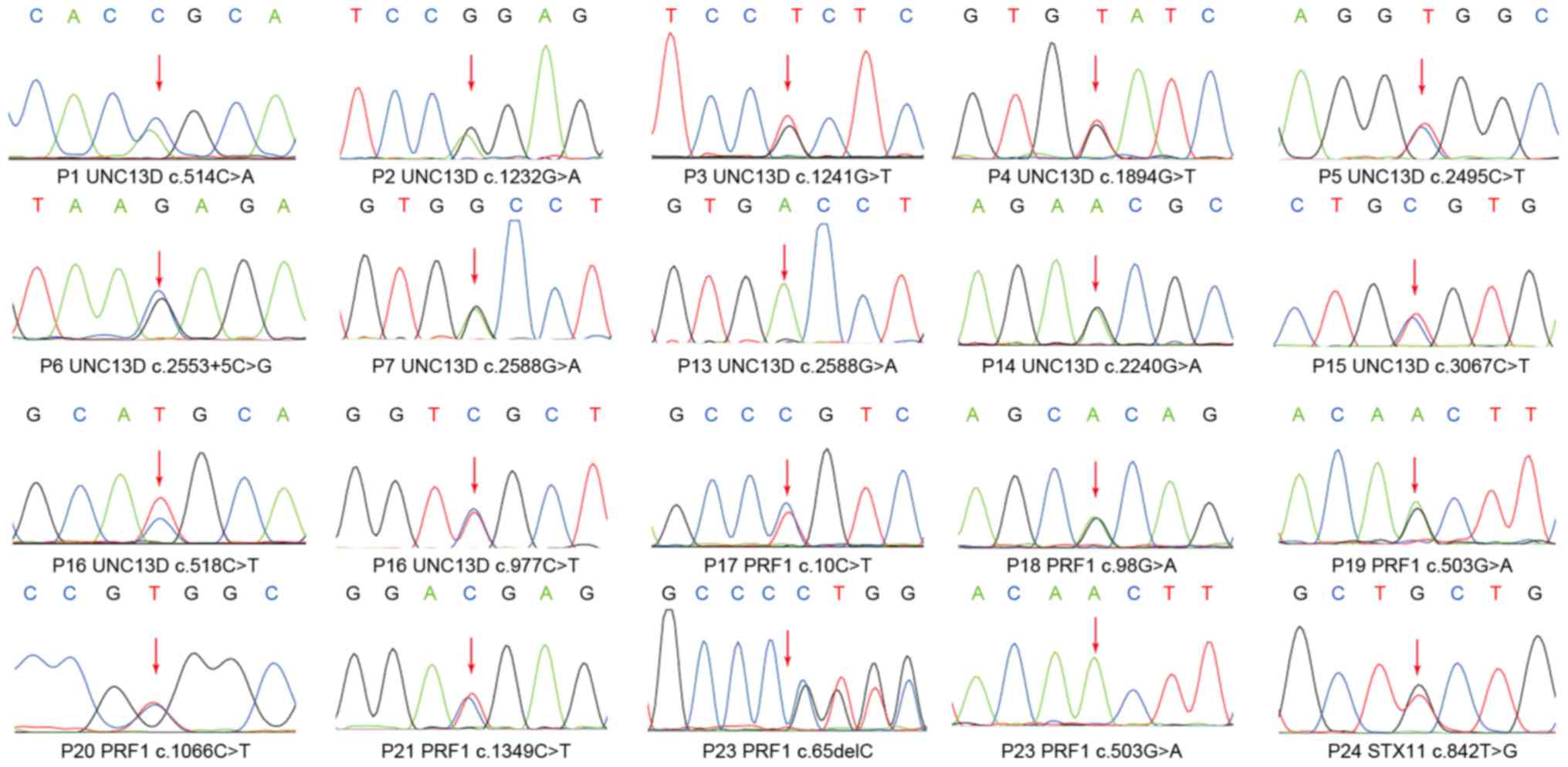
A total of 18 different mutations were identified in
24 unrelated patients (26.67%) (Fig.
1). A total of 16 patients (17.78%) carried mutations in
UNC13D, including 12 with monoallelic mutations, 1 with homozygous
mutation and 3 with compound heterozygous mutations. Seven patients
(7.78%) had PRF1 mutations, including 4 with monoallelic mutations,
1 with homozygous mutation and 2 with compound heterozygous
mutations. One patient (1.11%) was detected to carry STX11
monoallelic mutation (Table II). All
mutations were confirmed to be germline-derived.
| Table II.Gene mutations observed in 24
patients with lymphoma. |
Table II.
Gene mutations observed in 24
patients with lymphoma.
| Author, name | Patient | Sex | Age at diagnosis,
years | Diagnosis | Gene | Mutation | Genotype | (Refs.) |
|---|
|
| P1 | M | 7 | HL | UNC13D |
c.514C>A/p.R172S | Het. | Novel
observation |
| Tong et al,
2011; | P2 | M | 26 | HL | UNC13D |
c.1232G>A/p.R411Q | Het. | (12,20) |
| Zhang et al,
2014 |
|
|
|
|
|
|
|
|
| Sieni et al,
2011 | P3 | M | 32 | HL | UNC13D |
c.1241G>T/p.R414L | Het. | (21) |
|
| P4 | M | 17 | B-NHL | UNC13D |
c.1894G>T/p.D632Y | Het. | Novel
observation |
|
| P5 | M | 3 | HL | UNC13D |
c.2495C>T/p.A832V | Het. | Novel
observation |
| Tong et al,
2011; | P6 | F | 35 | B-NHL | UNC13D | c.2553+5C>G | Het. | (12,22) |
| Zhang et al,
2011 |
|
|
|
|
|
|
|
|
| Tong et al,
2011 | P7 | F | 54 | NK/T-NHL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
| Tong et al,
2011 | P8 | M | 46 | NHL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
| Tong et al,
2011 | P9 | F | 12 | NHL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
| Tong et al,
2011 | P10 | M | 40 | B-NHL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
| Tong et al,
2011 | P11 | F | 30 | NK/T-NHL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
| Tong et al,
2011 | P12 | M | 28 | NHL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
| Tong et al,
2011 | P13 | M | 9 | HL | UNC13D |
c.2588G>A/p.G863D | Hom. | (12) |
| Tong et al,
2011; | P14 | M | 38 | HL | UNC13D |
c.2240G>A/p.S747N | Het. | (12,22) |
| Zhang et al,
2011 |
|
|
|
|
|
|
|
|
| Tong et al,
2011; | P14 | M | 38 | HL | UNC13D | c.2553+5C>G | Het. | (12,22) |
| Zhang et al,
2011 |
|
|
|
|
|
|
|
|
| Tong et al,
2011 | P15 | M | 29 | HL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
|
|
|
|
|
| UNC13D |
c.3067C>T/p.R1023C | Het. | Novel
observation |
| Tong et al,
2011 | P16 | M | 12 | HL | UNC13D |
c.2588G>A/p.G863D | Het. | (12) |
|
|
|
|
|
| UNC13D |
c.518C>T/p.T173M | Het. | Novel
observation |
|
|
|
|
|
| UNC13D |
c.977C>T/p.S326L | Het. | Novel
observation |
| Zhang et al,
2011 | P17 | F | 36 | HL | PRF1 |
c.10C>T/p.R4C | Het. | (22) |
| Zhang et al,
2011 | P18 | M | 10 | HL | PRF1 |
c.98G>A/p.R33H | Het. | (22) |
| Lu et al,
2009 | P19 | F | 34 | NK/T-NHL | PRF1 |
c.503G>A/p.S168N | Hom. | (23) |
| Trizzino et
al, 2008 | P20 | M | 29 | B-NHL | PRF1 |
c.1066C>T/p.R356W | Het. | (24) |
| Trizzino et
al, 2008 | P21 | F | 19 | HL | PRF1 |
c.1349C>T/p.T450M | Het. | (24) |
| Zhang et al,
2011 | P22 | M | 24 | NK/T-NHL | PRF1 |
c.10C>T/p.R4C | Het. | (22) |
| Zhang et al,
2011 | P22 | M | 24 | NK/T-NHL | PRF1 |
c.98G>A/p.R33H | Het. | (22) |
| Tong et al,
2011 | P23 | M | 56 | NK/T-NHL | PRF1 |
c.65delC/p.P22Rfs*29 | Het. | (12) |
| Lu et al,
2009 | P23 | M | 56 | NK/T-NHL | PRF1 |
c.503G>A/p.S168N | Het. | (23) |
| Tong et al,
2011 | P24 | M | 15 | HL | STX11 |
c.842T>G/p.F281C | Het. | (12) |
Sixty unrelated healthy donors were sequenced for
these 6 genes with the same methods and 5 of them (8.33%) were
detected to harbor mutations. All 5 individuals were heterozygous
for UNC13D mutations (c.680G>A/p.R227H; c.3134C>T/p.T1045M;
c.3229_3235del/p.Arg1077SerfsTer48; c.2553+5C>G;
c.602A>G/p.H201R).
The Chi-square test revealed that the difference
between mutant frequencies of patients with lymphoma and healthy
donors was of statistical significance (P=0.005). Individuals
carrying mutations of these genes were more likely to develop
lymphoma compared with those without mutations [odds ratio (OR),
4.000; 95% confidence interval (CI), 1.431–11.180].
Statistical analysis of UNC13D
c.2588G>A mutation
UNC13D c.2588G>A/p.G863D was the most frequent
mutation identified in the current study, which was identified in 9
patients (10.00%), including 1 homozygous and 8 heterozygous. This
genetic variation was annotated as rs140184929 in dbSNP without
frequency data. Data in the 1000 Genomes Project demonstrated that
the c.2588A allele existed predominantly in the Chinese (0.83%),
and rarely in the Japanese (0.48%) and Bengali (0.58%) populations.
Other populations did not carry this variant (Table III). Data in ExAC also demonstrated
that the allelic frequency of c.2588A was increased in East Asian
populations (37/8,638; 0.43%) compared with that in South Asian
populations (5/16,504; 0.03%). Only one individual out of 32,962
Europeans was heterozygous for c.2588G>A variant. This variation
was not observed among 14,554 individuals analyzed from other
populations. Considering the high allele frequency of this mutation
in the present patient cohort and the distinctly different allele
frequencies among diverse populations, genotyping of the c.2588
allele was performed in 210 unrelated healthy donors of Chinese Han
nationality (Table III).
Heterozygous c.2588G>A was observed in 4 of them. Combined with
data in the 1000 Genomes Project (a total of 301 Chinese), a
control cohort of 511 individuals, 9 of whom harbored c.2588A
allele in a heterozygous state was obtained. The Chi-square test
revealed that the allele frequency of c.2588A in patients was
significantly increased compared with that in the control group
(P<0.001; OR, 6.621; 95% CI, 2.652–16.532), suggesting an
association between the c.2588G>A mutation, and the risk of
developing lymphoma.
| Table III.Allele frequencies of PRF1 c.272T and
UNC13D c.2588A among different populations. |
Table III.
Allele frequencies of PRF1 c.272T and
UNC13D c.2588A among different populations.
|
| Allele
frequencies |
|---|
|
|
|
|---|
|
Populations/samples | PRF1 c.272T | UNC13D c.2588A |
|---|
| 1000G-all
populations | 0.0132
(66/5008) | 0.0014
(7/5008) |
| 1000G-CHB | 0 (0/206) | 0.0097 (2/206) |
| 1000G-CHS | 0.0048 (1/210) | 0.0048 (1/210) |
| 1000G-CDX | 0 (0/186) | 0.0108 (2/186) |
| 1000G-JPT | 0 (0/208) | 0.0048 (1/208) |
| 1000G-BEB | 0 (0/172) | 0.0058 (1/172) |
| 1000G-FIN | 0.0253 (5/198) | 0 (0/198) |
| 1000G-GBR | 0.0385 (7/182) | 0 (0/182) |
| 1000G-TSI | 0.0561
(12/214) | 0 (0/214) |
| Patients in the
present study | 0 (0/180) | 0.0556
(10/180) |
| Controls in the
present study | 0 (0/120) | 0.0095 (4/420) |
In silico analysis of UNC13D
c.2588G>A mutation
The UNC13D c.2588G>A/p.G863D mutation resulted in
a substitution of the nonpolar and hydrophobic glycine (often
involved in the formation of the turn structure) in the Munc13
homology domain 2 of protein UNC13D by the polar, and neutral
aspartic acid (often involved in the formation of the coil
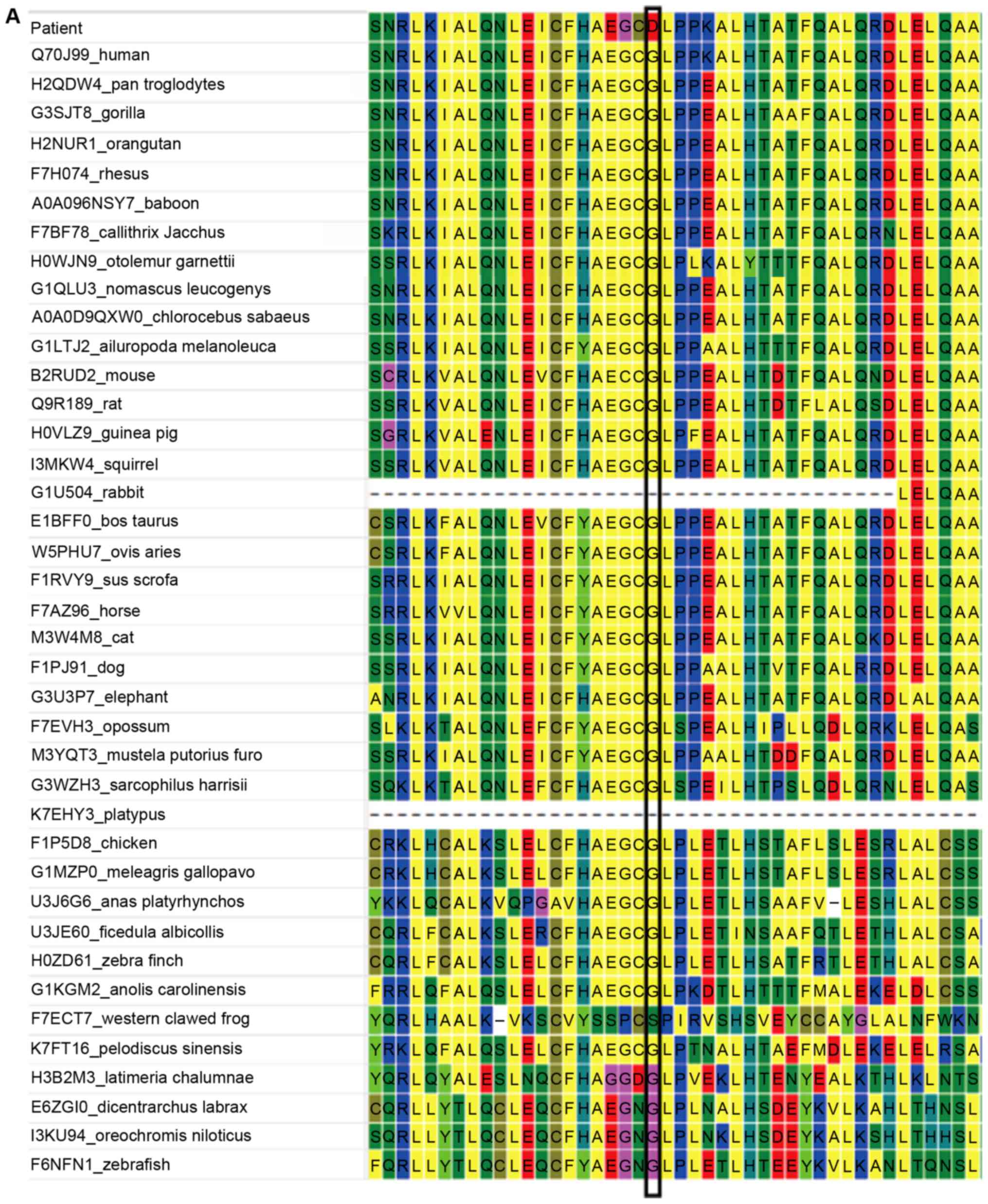
structure). Multiple sequence alignment demonstrated that the amino
acid at this position was highly conserved in available vertebrate
species (Fig. 2A) and the alteration
is predicted to be possibly damaging using PolyPhen-2 (Fig. 2B), and deleterious with SIFT in
silico analysis. I-TASSER also demonstrated significant
differences in the 3D structures of the wild-type and mutant-type
proteins (Fig. 2).
Discussion
In 2005, Clementi et al (10) first reported that 8/29 (27.6%)
unrelated Italian patients with lymphoma carried PRF1 mutations and
5 of them carried PRF1 c.272C>T/p.A91V heterozygous mutation. In
2014, Ciambotti et al (11)
observed mutations in 23/84 (27.4%) Italian patients with
anaplastic large cell lymphoma following genotype analysis of PRF1,
UNC13D and SH2D1A. Twenty-one patients (25%) carried PRF1 mutations
and the other 2 patients had mutations of UNC13D. PRF1
c.272C>T/p.A91V mutation was also the most common mutant
genotype (11/84).
In the present study 6 genes, which are all involved
in cytotoxic function of natural killer cells and cytotoxic T
lymphocytes, were identified in 90 Chinese patients with lymphoma.
The results demonstrated the association of germline defective
mutations and development of lymphoma. The majority of mutations
detected in the current study were heterozygous missense mutations,
which were consistent with previous reports (10,11). This
may explain why these patients developed lymphoma later in life
rather than outbreak fatal FHL during infancy. Such monoallelic
mutations may contribute to the pathogenesis of the disease, but
are not sufficient to initiate the disease phenotype alone.
Additional unidentified genetic defects, or possibly even
environmental factors, may contribute to the development of
lymphoma (10). What was different
from reports in Europe was that the most common mutant gene in the
present study was UNC13D while PRF1 was less frequently involved,
indicating a distinct mutation spectrum in Chinese patients with
lymphoma.
Notably, no hot spot region or predominant
pathogenic mutation in UNC13D had been previously identified
(18). In the current study; however,
9/16 UNC13D mutation carriers exhibited c.2588G>A/p.G863D
mutation, including 1 homozygous and 8 heterozygous. This single
amino acid substitution occurred in an evolutionary conserved
position and was predicted to be pathogenic using PolyPhen-2, SIFT,
and I-TASSER. Furthermore, statistical analysis revealed that this
mutation was significantly associated with the risk of developing
lymphoma. In addition, none of our patient harbored the PRF1
c.272C>T/p.A91V mutation, which was most frequently reported in
European populations (10,11). In the present consecutive cohort of
>500 patients with diagnosed or suspected FHL, the PRF1
c.272C>T mutation was not identified (data not shown).
Data in the 1000 Genomes Project demonstrated that
the allele frequency of PRF1 c272T was significantly higher in
European population compared with that in Chinese and Japanese,
supporting the concept of a Mediterranean origin of the mutation
(11). However, the UNC13D c.2588A
allele existed predominantly in Chinese, less in Japanese and
Bengali, and was not identified in any other populations listed in
this database (Table III). In
regards to Korea, where UNC13D is the predominant causative gene in
Korean patients with FHL, c.2588G>A was not reported (19). Collectively, the data obtained from
the present study and the databases suggest that UNC13D
c.2588G>A/p.G863D is a founder mutation of Chinese patients.
In conclusion, the current study provides a
relatively comprehensive mutation spectrum of defective
cytotoxicity associated genes in Chinese patients with lymphoma.
Monoallelic germline mutations were identified to be most frequent
in the present cohort, suggesting that partially impaired cytotoxic
machinery may represent a predisposing factor for the development
of lymphoma. In addition, UNC13D was identified as the predominant
causative gene, while PRF1 was less frequently involved.
Furthermore, UNC13D c.2588G>A/p.G863D, which is not reported in
other populations, is a founder mutation in Chinese patients.
References
|
1
|
Vesely MD, Kershaw MH, Schreiber RD and
Smyth MJ: Natural innate and adaptive immunity to cancer. Annu Rev
Immunol. 29:235–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tang YM and Xu XJ: Advances in
hemophagocytic lymphohistiocytosis: Pathogenesis, early
diagnosis/differential diagnosis and treatment.
ScientificWorldJournal. 11:697–708. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stepp SE, Dufourcq-Lagelouse R, Le Deist
F, Bhawan S, Certain S, Mathew PA, Henter JI, Bennett M, Fischer A,
de Saint Basile G and Kumar V: Perforin gene defects in familial
hemophagocytic lymphohistiocytosis. Science. 286:1957–1959. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feldmann J, Callebaut I, Raposo G, Certain
S, Bacq D, Dumont C, Lambert N, Ouachée-Chardin M, Chedeville G,
Tamary H, et al: Munc13-4 is essential for cytolytic granules
fusion and is mutated in a form of familial hemophagocytic
lymphohistiocytosis (FHL3). Cell. 115:461–473. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
zur Stadt U, Schmidt S, Kasper B, Beutel
K, Diler AS, Henter JI, Kabisch H, Schneppenheim R, Nürnberg P,
Janka G and Hennies HC: Linkage of familial hemophagocytic
lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and
identification of mutations in syntaxin 11. Hum Mol Genet.
14:827–834. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
zur Stadt U, Rohr J, Seifert W, Koch F,
Grieve S, Pagel J, Strauss J, Kasper B, Nürnberg G, Becker C, et
al: Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is
caused by mutations in Munc18-2 and impaired binding to syntaxin
11. Am J Hum Genet. 85:482–492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nichols KE, Harkin DP, Levitz S, Krainer
M, Kolquist KA, Genovese C, Bernard A, Ferguson M, Zuo L, Snyder E,
et al: Inactivating mutations in an SH2 domain-encoding gene in
X-linked lymphoproliferative syndrome. Proc Natl Acad Sci USA.
95:13765–13770. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rigaud S, Fondanèche MC, Lambert N,
Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux-Laucat
F, Revy P, et al: XIAP deficiency in humans causes an X-linked
lymphoproliferative syndrome. Nature. 444:110–114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marsh RA, Madden L, Kitchen BJ, Mody R,
McClimon B, Jordan MB, Bleesing JJ, Zhang K and Filipovich AH: XIAP
deficiency: A unique primary immunodeficiency best classified as
X-linked familial hemophagocytic lymphohistiocytosis and not as
X-linked lymphoproliferative disease. Blood. 116:1079–1082. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clementi R, Locatelli F, Dupré L,
Garaventa A, Emmi L, Bregni M, Cefalo G, Moretta A, Danesino C,
Comis M, et al: A proportion of patients with lymphoma may harbor
mutations of the perforin gene. Blood. 105:4424–4428. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ciambotti B, Mussolin L, d'Amore ES,
Pillon M, Sieni E, Coniglio ML, Ros MD, Cetica V, Aricò M and
Rosolen A: Monoallelic mutations of the perforin gene may represent
a predisposing factor to childhood anaplastic large cell lymphoma.
J Pediatr Hematol Oncol. 36:e359–e365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tong CR, Liu HX, Xie JJ, Wang F, Cai P,
Wang H, Zhu J, Teng W, Zhang X, Yang JF, et al: The study of gene
mutations in unknown refractory viral infection and primary
hemophagocytic lymphohistiocytosis. Zhonghua Nei Ke Za Zhi.
50:280–283. 2011.(In Chinese). PubMed/NCBI
|
|
13
|
Machaczka M, Klimkowska M, Chiang SC,
Meeths M, Müller ML, Gustafsson B, Henter JI and Bryceson YT:
Development of classical Hodgkin's lymphoma in an adult with
biallelic STXBP2 mutations. Haematologica. 98:760–764. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sandlund JT, Shurtleff SA, Onciu M,
Horwitz E, Leung W, Howard V, Rencher R and Conley ME: Frequent
mutations in SH2D1A (XLP) in males presenting with high-grade
mature B-cell neoplasms. Pediatr Blood Cancer. 60:E85–E87. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
World Health Organization (WHO): WHO
classification of tumours of haematopoietic and lymphoid tissues.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H,
Thiele J and Vardiman JW: 2. 4th. International Agency for Research
on cancer (IARC) Press; Lyon: 2008
|
|
16
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ramensky V, Bork P and Sunyaev S: Human
non-synonymous SNPs: Server and survey. Nucleic Acids Res.
30:3894–3900. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zur SU, Beutel K, Kolberg S, Schneppenheim
R, Kabisch H, Janka G and Hennies HC: Mutation spectrum in children
with primary hemophagocytic lymphohistiocytosis: Molecular and
functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat.
27:62–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoon HS, Kim HJ, Yoo KH, Sung KW, Koo HH,
Kang HJ, Shin HY, Ahn HS, Kim JY, Lim YT, et al: UNC13D is the
predominant causative gene with recurrent splicing mutations in
Korean patients with familial hemophagocytic lymphohistiocytosis.
Haematologica. 95:622–626. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang K, Chandrakasan S, Chapman H,
Valencia CA, Husami A, Kissell D, Johnson JA and Filipovich AH:
Synergistic defects of different molecules in the cytotoxic pathway
lead to clinical familial hemophagocytic lymphohistiocytosis.
Blood. 124:1331–1334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sieni E, Cetica V, Santoro A, Beutel K,
Mastrodicasa E, Meeths M, Ciambotti B, Brugnolo F, zur Stadt U,
Pende D, et al: Genotype-phenotype study of familial
haemophagocytic lymphohistiocytosis type 3. J Med Genet.
48:343–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang K, Jordan MB, Marsh RA, Johnson JA,
Kissell D, Meller J, Villanueva J, Risma KA, Wei Q, Klein PS and
Filipovich AH: Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2
are associated with adult-onset familial HLH. Blood. 118:5794–5798.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu G, Xie ZD, Shen KL, Ye LJ, Wu RH, Liu
CY, Jin YK and Yang S: Mutations in the perforin gene in children
with hemophagocytic lymphohistiocytosis. Chin Med J (Engl).
122:2851–2855. 2009.PubMed/NCBI
|
|
24
|
Trizzino A, zur Stadt U, Ueda I, Risma K,
Janka G, Ishii E, Beutel K, Sumegi J, Cannella S, Pende D, et al:
Genotype-phenotype study of familial haemophagocytic
lymphohistiocytosis due to perforin mutations. J Med Genet.
45:15–21. 2008. View Article : Google Scholar : PubMed/NCBI
|