Introduction
Primary liver cancer, predominantly hepatocellular
carcinoma (HCC) and hepatoblastoma (HB), one of the most common
solid tumors, is the most common cause of cancer-associated
mortality worldwide (1–4). HCC is the most common liver tumor in
adolescents and adults. HCC alone is the fifth most common newly
diagnosed cancer and the third leading cause of cancer mortality
worldwide (1,2), while HB is the most common liver
malignant tumor diagnosed by the age of 4 years, accounting for 80%
of liver cancers in children under the age of 15 years (5,6). Among
patients with localized HB or HCC, surgical resection is a common
treatment option. Liver transplantation is considered to be the
only curative therapy; however, the majority of patients with
advanced HB or HCC are not suitable for transplantation (3,4,6,7).
Currently, chemotherapy utilized to treat patients with
unresectable HCC results in a poor response and severe toxicity
(7). Thus, more effective agents to
combat primary liver cancer are in high demand.
Although the molecular pathogenesis of HCC and HB
remains unclear, liver cirrhosis is acknowledged as a premalignant
condition for developing HCC (8).
Deregulation of a number of oncogenes, including c-MYC, cyclin D1
and β-catenin, has been observed in HCC (2,9). The
expression of certain tumor-suppressor genes, including those
expressing retinoblastoma protein, deleted in liver cancer 1,
P16INK4A, P53 and E-cadherin, is changed in HCC
(2,10). HB is considered to arise from hepatic
progenitors or hepatoblasts (11,12).
Mutations in catenin beta 1 and other components of the β-catenin
degradation complex, including adenomatous polyposis coli and axis
inhibition protein 2, have been reported (13–17).
Recent studies indicated that genome-wide DNA hypomethylation and
promoter CpG island hypermethylation contribute to the initiation
and progression of HB and HCC, and are correlated with poor
survival (2,17–21).
Rapid cellular proliferation and abnormal
vasculature in solid tumors such as HCC result in a highly hypoxic
environment in which the expression of hypoxia inducible factor 1-α
(HIF-1α) is markedly increased (22).
HIF-1α serves key roles in cancer development by regulating the
expression of numerous genes involved in proliferation,
angiogenesis, metabolism, survival, cell migration and invasion
(22,23). Recently, numerous studies have shown
that DNA methylation is affected by hypoxia (24,25).
Methylation at the C-5 position of cytosine is specifically
mediated by DNA methyltransferases. The methylated cytosine (5-mC)
of CpG dinucleotides in the promoter of a gene represses the
transcription of this gene (26–28). The
ten-eleven-translocation 5-methylcytosine dioxygenase (TET) enzymes
catalyze the conversion of 5-mC to 5-hydroxymethylcytosine (5-hmC)
to demethylate mammalian DNA (29–31).
Elevated 5-hmC levels are associated with increased gene expression
(26).
In the present study, the effects of hypoxia on DNA
methylation and the expression of TET enzymes in HB HepG2 cells
were investigated. The expression of TET enzymes in HepG2 cells
exposed to various concentrations of oxygen was assessed using
Reverse transcription-quantitative polymerase chain reaction
(RT-qPCR), and 5-hmC was detected using immunochemistry and
quantified by dot blot. The results of the present study
demonstrated that hypoxia regulates DNA methylation through
HIF-1α-mediated TET enzymes in HepG2 cells.
Materials and methods
Materials
HepG2 cells were purchased from the American Type
Culture Collection (Manassas, VA, USA). Anti-β-actin antibody
(clone AC-15) and CoCl2 were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Rabbit anti-human
HIF-1α antibody was purchased from Abcam (Cambridge, MA, USA).
Pierce™ ECL Western Blotting substrate and goat
anti-rabbit immunoglobulin G (IgG) antibody conjugated to
horseradish peroxidase (HRP) were purchased from Pierce (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Polyvinylidene fluoride
(PVDF) membrane was purchased from EMD Millipore (Billerica, MA,
USA). Rabbit anti-5-hmC antibody was purchased from Active Motif
(Carlsbad, CA, USA). Dulbecco's modified Eagle's medium (DMEM)
(REF11965), fetal bovine serum (FBS) (REF16000), trypsin/EDTA, DAPI
and donkey anti-rabbit IgG antibody conjugated to Alexa Fluor 594
were purchased from Invitrogen (Thermo Fisher Scientific,
Inc.).
Cell culture and hypoxia
incubation
HepG2 cells were maintained at 37°C in an atmosphere
of 95% air and 5% CO2 in DMEM containing 10%
heat-inactivated FBS, 100 µg/ml penicillin and 100 µg/ml
streptomycin. Cells were subcultured every 5 days with trypsin/EDTA
and the medium was changed every other day. Hypoxia was achieved by
incubating the cells in an incubator in which the oxygen was
replaced by pure nitrogen. The gas proportions used were 21%
O2: 21% O2 and 5% CO2; 5%
O2: 5% O2, 5% CO2, and 90%
N2; and 1% O2: 1% O2, 5%
CO2, and 94% N2.
RT-qPCR
Total RNA was extracted using the RNeasy Mini kit
according to the manufacturer's protocol (Qiagen China Co., Ltd.,
Shanghai, China). Further genomic DNA removal was performed using
the RNase-Free DNase kit, in accordance with manufacturer's
protocol (Qiagen China Co., Ltd.). First-strand complementary DNA
(cDNA) was synthesized with oligo-dT or random hexamers as primers,
using the SuperScript First-Strand Synthesis System (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. An equal volume mixture of the cDNA products (50 ng) was
used as templates for PCR amplification. Reactions were performed
in a 25-µl volume with iQ™ SYBR Green Supermix (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), and 200 nM each of forward
and reverse primers using an iCyler iQ instrument and iQ software
(Version 2.0; Bio-Rad Laboratories, Inc.). Each sample was analyzed
in triplicate. PCR conditions included an initial denaturation step
of 4 min at 95°C, followed by 40 cycles of PCR consisting of 30 sec
at 95°C, 30 sec at 60°C and 30 sec at 72°C. Mean quantification
cycle (Cq) values from the triplicate PCRs for a gene of interest
(GOI) were normalized against the average Cq values for GAPDH from
the same cDNA sample (32). The
following primers were used: TET1 forward,
5′-CCGAATCAAGCGGAAGAATA-3′ and reverse, 5′-ACTTCAGGTTGCACGGTCTC-3′;
TET2 forward, 5′-AGCCCCATCACGTACAAAAC-3′ and reverse,
5′-TGTGGTGGCTGCTTCTGTAG-3′; TET3 forward,
5′-CAGCAGCCGAGAAGAAGAAG-3′ and reverse, 5′-GGACAATCCACCCTTCAGAG-3′;
and GAPDH forward, 5′-GACAACAGCCTCAAGATCATCAG-3′ and reverse,
5′-ATGGCATGGACTGTGGTCATGAG-3′. GAPDH served as control.
Production of short hairpin RNA
(shRNA) lentiviruses and transduction
A shRNA against human HIF-1α was cloned into the
pLKO.1 vector according to the manufacturer's protocol (Addgene,
Inc., Cambridge, MA, USA). The target sequence is
5′-CTGATGACCAGCAACTTGA-3′. pLKO.1, scrambled shRNA (negative
control), pMD2.G (used for virus packaging) and psPAX2 (used for
virus packaging) were purchased from Addgene, Inc. All constructs
were verified by sequencing. Lentiviruses were produced by
co-transfecting 293FT cells (Invitrogen; Thermo Fisher Scientific,
Inc.) in 10-cm dishes with 10 µg pLKO.1-shRNA, 2.5 µg pMD2.G and
7.5 µg psPAX2 using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). Viruses were collected between 16 and 60 h after
transfection, and tittered for p24 levels using an ELISA kit
(ZeptoMetrix Corporation, Buffalo, NY, USA). HepG2 cells were
infected with HIF-1α shRNA at a multiplicity of infection of 20 in
the presence of 6 µg/ml polybrene. Virus-containing medium was
removed after 16 h and replaced with fresh DMEM. After 24 h, the
cells were used experimentally.
Immunocytochemistry
HepG2 cells (3×104 cells/cm2)
were plated onto poly-D-Lysine-coated 12-well plates. Following
treatment with 21 or 1% O2 for 24 h, the cells were
washed with PBS; fixed with 4% formaldehyde for 15 min at room
temperature; permeabilized in 0.1% Triton X-100 for 20 min; and
blocked with 5% goat serum (Invitrogen; Thermo Fisher Scientific,
Inc.) for 1 h at room temperature. Immunostaining with anti-5-hmC
antibody (dilution, 1:1,000; cat. no. 39791) was performed at 4°C
overnight. Next, donkey anti-rabbit IgG Alexa Fluor 594 (cat. no.
A-21207; dilution, 1:2,000) was incubated for 1 h at room
temperature and the nuclei were stained with DAPI for 10 min at
room temperature. Images were acquired on a Zeiss Axio Observer
inverted fluorescence microscope (Zeiss AG, Oberkochen, Germany)
with lenses corrected for plastic culture plates.
Western blot analysis
HepG2 cells (5×106 cells) were washed
three times with cold PBS and lysed in cold lysis buffer (1% Triton
X-100, 10 mM Tris pH 7.6, 50 mM NaCl, 30 mM sodium pyrophosphate,
50 mM NaF, 5 mM EDTA and 0.1 mM Na3VO4) with
protease inhibitor cocktail tablets for 20 min. Lysates were
centrifuged at 4°C at 16,000 × g for 30 min. Supernatant fractions
containing equal amounts of total protein (30 µg/lane) were
separated by SDS-PAGE (7.5% gel), transferred onto a PVDF membrane
and analyzed using western blot analysis. Protein concentration was
determined using the DC Protein Assay (Bio-Rad Laboratories, Inc.)
in accordance with manufacturer's protocol. The membranes were
blocked in 5% non-fat dry milk in Tris-Buffered Saline-Tween 20
(TBST) buffer for 1 h at room temperature, and immunostained with
anti-β actin antibody (dilution, 1:1,000; cat. no. A1978) or rabbit
anti-human HIF-1α antibody (1:1,000; cat. no. ab51608) in 5%
non-fat milk overnight at 4°C. Subsequently, the membranes were
washed in TBST buffer three times, 5 min each time, at room
temperature and the membranes were blotted in goat anti-mouse (cat.
no. A28177) or goat anti-rabbit (cat. no. A27036) IgG
antibody-conjugated to HRP (both: Dilution, 1:2,000; Thermo Fisher
Scientific, Inc.) in 5% non-fat milk, separately, for 2 h at room
temperature. Following washing with TBST, ECL detection
(Pierce™ ECL; Pierce; Thermo Fisher Scientific, Inc.)
was performed according to manufacturer's protocol.
5-hmC dot blot
Genomic DNA, from HepG2 cells exposed to either 21
or 1% oxygen, was extracted using a QIAamp DNA Mini kit in
according with manufacturer's protocol (Qiagen China Co., Ltd.). A
total of 200 or 500 ng genomic DNA was denatured in 0.1 M NaOH at
100°C for 10 min, followed by the addition of an equal volume of
cold 2 M ammonium acetate (pH 7.2). Denatured DNA samples were
spotted onto a nitrocellulose membrane and crosslinked using a
Stratalinker 2400 UV Crosslinker (Agilent Technologies, Inc., Santa
Clara, CA, USA) twice. The membrane was blocked with 5% non-fat
milk for 1 h and incubated with anti-5-hmC antibody for detection
by ECL.
Methylene blue staining
The nitrocellulose membranes contained genomic DNA
were immersed in 0.1% methylene blue (Sigma-Aldrich; Merck KGaA)
solution in 0.5 M sodium acetate (pH 5.2) and agitated for 10 min
at room temperature. The staining solution was removed and the
membranes were washed with successive changes of water until the
background was reduced sufficiently to observe the DNA dot.
Statistical analysis
All statistical analyses were performed using SPSS
10.0 (SPSS, Inc., Chicago, IL, USA). Data are expressed as mean ±
standard error of the mean. Unpaired, two-tailed Student's t-tests
were performed to evaluate whether two groups were significantly
different from each other. For comparison of multiple groups,
analysis of variance by Tukey's test was used. P<0.05 was
considered to indicate a statistically significant difference.
Results
Hypoxia increases the expression of
TET enzymes in HepG2 cells
The TET enzymes are comprised of three members:
TET1, TET2 and TET3 (26). The
expression of TET1, TET2 and TET3 was assessed using RT-qPCR of
HepG2 cells cultured under 21, 5 or 1% oxygen for 24 h. The HepG2
cell line is one of the most utilized cell lines in in vitro
studies on HB, since it is well characterized and retains numerous
hepatocyte-associated features (33,34). As
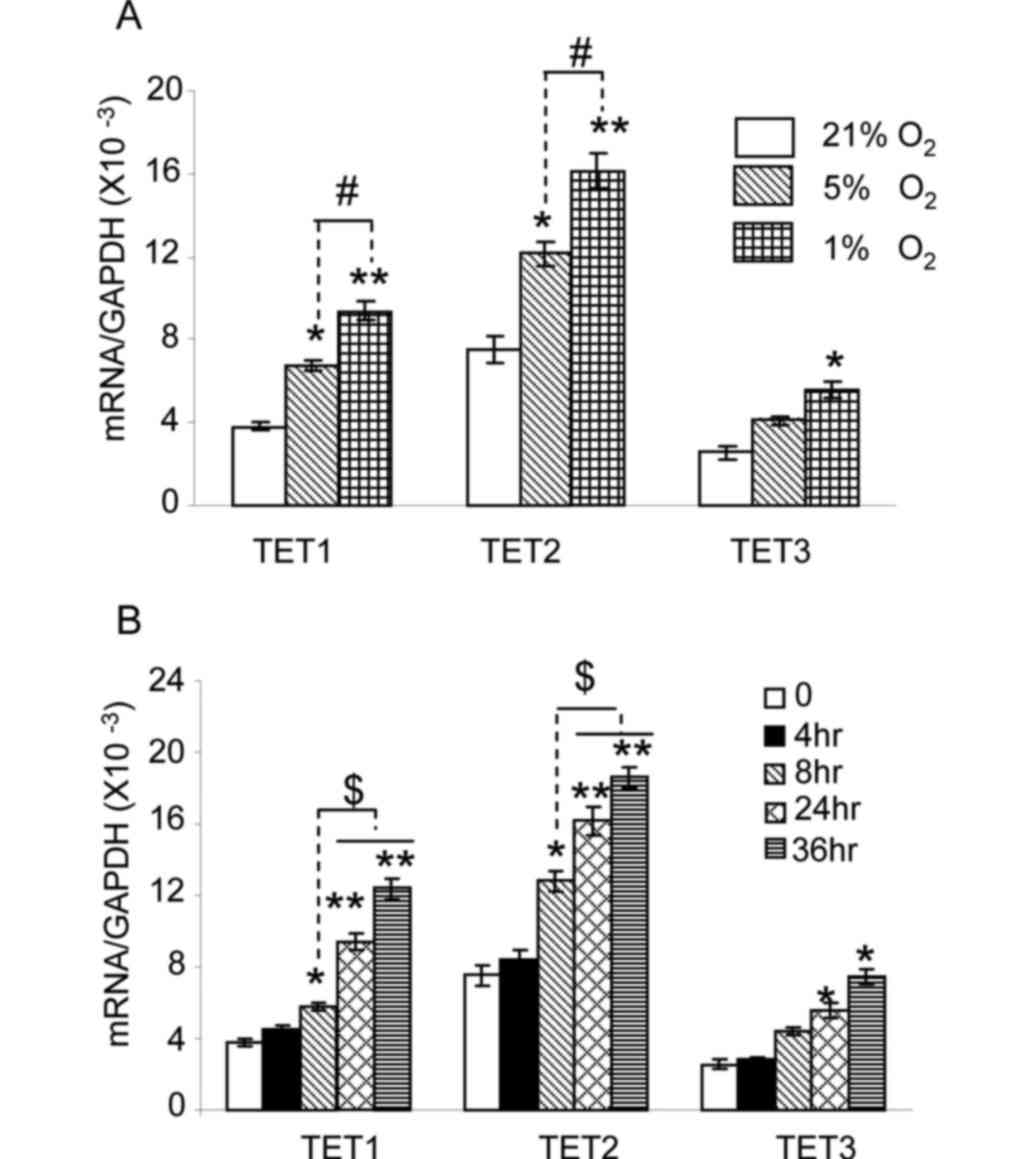
shown in Fig. 1A, the level of TET1
and TET2 messenger RNA (mRNA) in HepG2 cells cultured under 5%
oxygen was significantly increased compared with that in cells
cultured under 21% oxygen (P<0.05). No significant changes were
observed for TET3 (P>0.05). When cells were cultured under 1%
oxygen, a significant increase in TET1 and TET2 expression was
observed compared with the expression under 21 and 5% oxygen
(P<0.01 and P<0.05, respectively). TET3 expression under 1%
oxygen was also markedly increased compared with that under 21%
oxygen (P<0.05). Next, the expression of TET enzymes was
measured at various time intervals following exposure to 1% oxygen.
As shown in Fig. 1B, the expression
of TET1 and TET2 was significantly increased in cells exposed to 1%
oxygen for 8 h (P<0.05), 24 h (P<0.01) and 36 h (P<0.01).
The expression of TET1 and TET2 in cells exposed to 1% oxygen for
24 and 36 h was higher than that at 8 h (P<0.05). TET3
expression in cells exposed to 1% oxygen for 24 and 36 h was
significantly increased (P<0.05). These results indicate that
hypoxia upregulates TET1, TET2 and TET3 expression in HepG2 cells.
TET1 and TET2 expression is more sensitive to hypoxia than TET3
expression.
Hypoxia elevates the cellular 5-hmC
level in HepG2 cells
The TET enzymes catalyze the oxidation of 5-mC to
5-hmC. The present study evaluated whether the 5-hmC level was
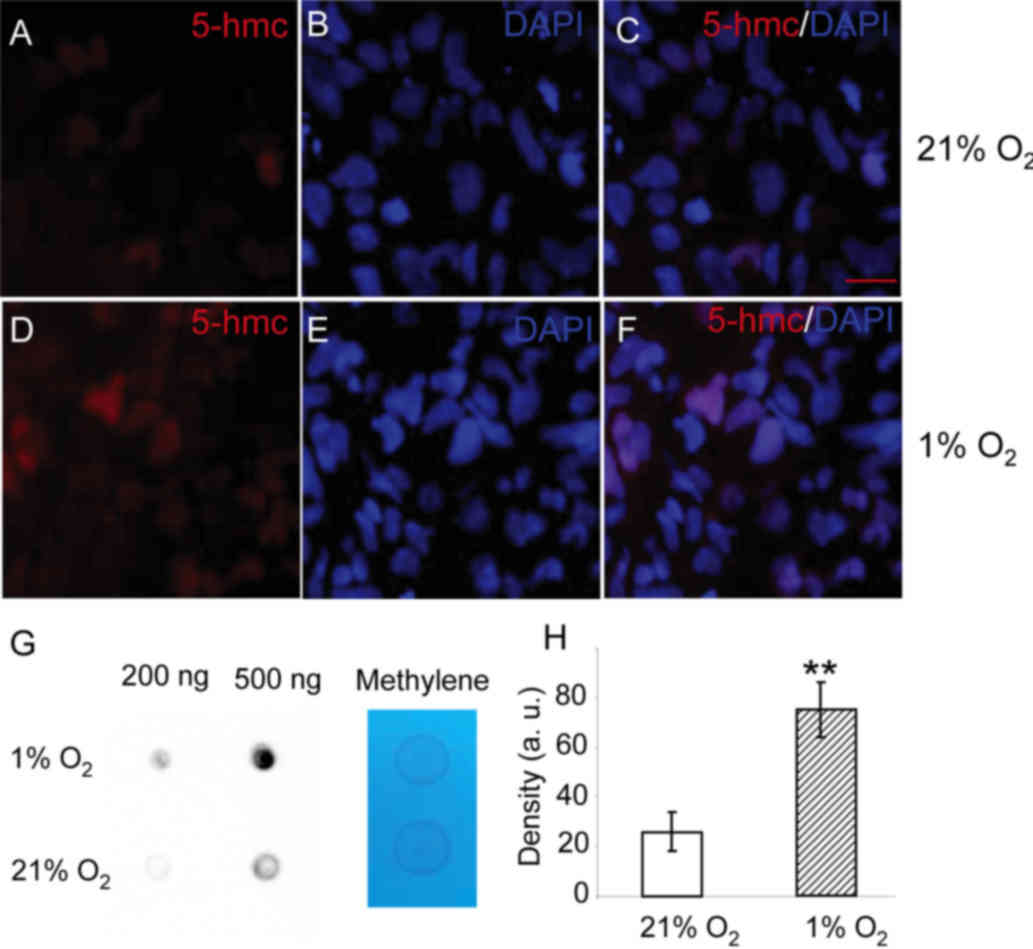
changed upon hypoxia. As shown in Fig.
2, 5-hmC can be detected in the nucleus of HepG2 cells cultured
under 21% oxygen. When HepG2 cells were cultured in 1% oxygen for
24 h, greater levels of 5-hmC were observed in the nucleus
(Fig. 2D). To quantify the levels of
5-hmC, a dot-blot assay was performed. The 5-hmC levels in the
total DNA from cells exposed to 1% oxygen for 24 h were markedly
higher than those in cells cultured in 21% oxygen (Fig. 2G and H). The results show that levels
of 5-hmC in HepG2 cells are increased upon hypoxia.
CoCl2 increases the
expression of TET enzymes in HepG2 cells
Hypoxia induces biological changes in the cell
mainly through stabilizing HIF-1α (35). CoCl2 is a known chemical
inducer of HIF-1α expression (35,36). The
mRNA levels of TET1, TET2 and TET3 were measured in HepG2 cells
treated with 0, 100, 150 or 200 µM of CoCl2 for 24 h in
21% oxygen (Fig. 3A). TET1 expression
was markedly sensitive to CoCl2, and a significant
increase in TET1 mRNA expression was observed at 100 µM
CoCl2 treatment (P<0.05). TET1 and TET2 expression
were significantly increased in cells treated with 150 and 200 µM
CoCl2, compared with that observed in untreated cells
(P<0.01) or in cells treated with 100 µM CoCl2
(P<0.05). TET3 expression was significantly increased in cells
treated with 150 and 200 µM CoCl2, compared with that
observed in untreated cells (P<0.05).
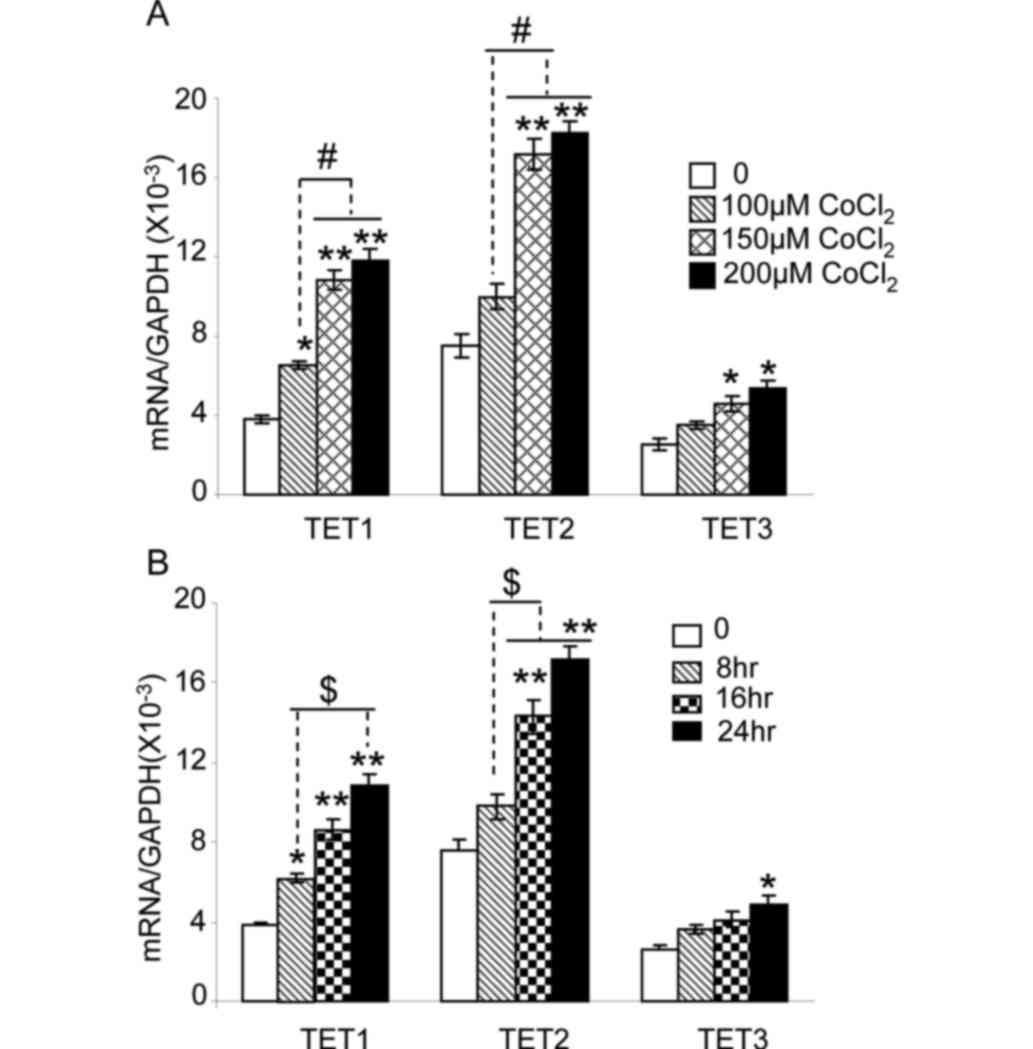 | Figure 3.CoCl2 increases the
expression of TET1, TET2 and TET3 in HepG2 cells. (A) HepG2 cells
were cultured in 21% oxygen, and then treated with 100, 150 or 200
µM CoCl2 for 24 h. The mRNA expression level of TET1,
TET2 or TET3 was quantified by reverse transcription-quantitative
polymerase chain reaction. *P<0.05 and **P<0.01 vs. untreated
cells. #P<0.05 vs. 100 µM CoCl2 treatment.
(B) HepG2 cells were cultured under 21% oxygen, and then treated
with 150 µM CoCl2 for 0, 8, 16 or 24 h. The expression
levels of TET1, TET2 and TET3 mRNA are shown. *P<0.05 and
**P<0.01 vs. cells without treatment (0 h).
$P<0.05 vs. CoCl2 treatment for 8 h. n=9
from three independent experiments. TET, ten-eleven-translocation
5-methylcytosine dioxygenase; mRNA, messenger RNA. |
The expression of TET enzymes in HepG2 cells exposed
to 150 µM CoCl2 was assessed at various time intervals
(Fig. 3B). TET1 expression was
increased following 8 h of CoCl2 treatment, compared
with the expression in untreated cells (P<0.05). Expression of
TET1 and TET2 following 16 and 24 h of exposure to CoCl2
was significantly higher than that observed in untreated cells
(P<0.01) and in cells treated for 8 h (P<0.05). TET3
expression after 24 h of 150 µM CoCl2 treatment was
significantly increased compared with that observed in untreated
cells (P<0.05). These results indicate that, as hypoxia,
CoCl2 treatment increases the expression of TET1, TET2
and TET3 in HepG2 cell in a dose-dependent manner.
HIF-1α knockdown attenuates the
hypoxia-induced expression of TET enzymes
The results of the present study demonstrated that
the induced expression of HIF-1α, either by hypoxia or
CoCl2, increases the expression of TET1, TET2 and TET3.
To investigate whether the hypoxia-induced changes in TET
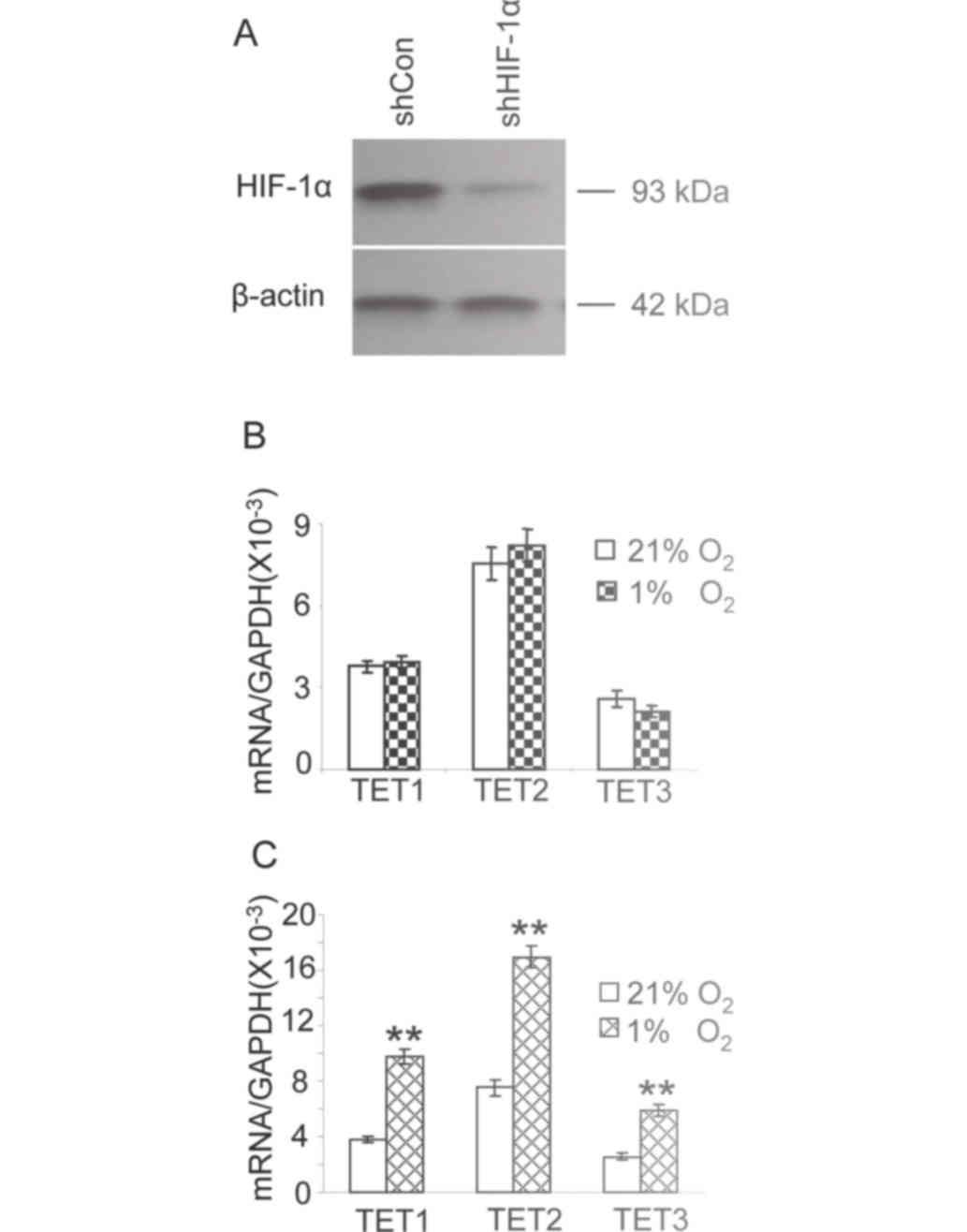
expression are dependent on HIF-1α, the expression of HIF-1α in
HepG2 cells was inhibited with a specific shRNA. HepG2 cells were
treated with a shRNA targeted against HIF-1α for 24 h; the cells
were then cultured in 1% oxygen for a further 24 h and the
expression of HIF-1α was assessed by western blot analysis. HIF-1α
expression was markedly reduced compared with that observed in
cells transfected with a scrambled shRNA (Fig. 4A). The expression of TET1, TET2 and
TET3 was at a similar level in cells transfected with the shRNA
against HIF-1α cultured in either 21 or 1% oxygen (Fig. 4B). The expression of TET1, TET2 and
TET3 in HepG2 cells transfected with scrambled shRNA (control) in
1% oxygen was significantly increased compared with that observed
in cells under 21% oxygen (Fig. 4C,
P<0.01). These results suggest that HIF-1α knockdown attenuates
the increased expression of TET enzymes in hypoxic HepG2 cells.
Discussion
Aberrant DNA methylation associated with epigenetic
modification is a hallmark of cancer pathogenesis (37,38). When
a tumor is growing, certain tumor regions, such as those in the
center of the tumor, are starved of oxygen due to abnormal
vascularization, resulting in a hypoxic microenvironment (22,23).
Hypoxia has been demonstrated to induce DNA hypomethylation in
normal and hepatoma cells (24,25). The
current study demonstrated that hypoxia increases the expression of
TET enzymes and elevates cellular 5-hmC levels in HB HepG2 cells.
CoCl2, a chemical inducer of HIF-1α (35,36), also
increases the expression of TET enzymes. HIF-1α knockdown with a
specific shRNA attenuates the hypoxia-induced expression of TET
enzymes. The current results indicate that hypoxia controls DNA
methylation through HIF-1α-mediated TET enzymes regulation in HepG2
cells.
HepG2 cells were isolated from a human liver biopsy
of a 15-year old male with HB, and were originally considered to be
a HCC cell line, but were later shown to be a HB cell line
(39–41). This cell line is widely utilized in
vitro to study HB due to preserving numerous
hepatocyte-associated features (33,34). The
present study demonstrates that three TET enzyme family members,
namely TET1, TET2 and TET3, are expressed in HepG2 cells and are
involved in maintaining the balance of DNA methylation. TET enzymes
sequentially oxidize the methyl group of 5-mC to form 5-hmC, and
then catalyze the oxidation of 5-hmC to generate 5-formylcytosine
and 5-carboxylcytosine (26,30). It has been demonstrated that 5-hmC, as
a major epigenetic modification marker, serves a notable role in
regulating gene expression in hepatocytes (42). Recent studies have revealed that the
expression levels of 5-hmC and TET enzymes are significantly
reduced in HCC tissue samples compared with those in non-cancerous
liver tissue in the same patient (18,43).
Furthermore, the loss of 5-hmC is associated with the progression
of HCC (18,43). Cui et al (21) has demonstrated that the methylation of
genomic DNA in HB tissues is significantly lower than that in the
adjacent non-tumor tissues, and the hypomethylation in the CpG
sites of the alpha-fetoprotein (AFP) promoter in HB tissues
negatively correlates with the expression of AFP. However, the
expression of TET enzymes in HB tissues was not reported.
Hypoxia has been regarded as an important factor of
the microenvironment, which can induce epigenetic changes in solid
tumor cells (24,25). Hypoxia induces the expression of TET1,
TET2 and TET3, and elevates the level of cellular 5-hmC in HepG2
cells, which is consistent with a previous report stating that
hypoxia induces genomic DNA hypomethylation in HCC Hep3B cells
(24). Hypoxia modulates the
malignant phenotype of tumor cells through HIF-1α, which regulates
the expression of numerous target genes (23,25). The
HIF-1α chemical inducer CoCl2 increases the expression
of TET1, TET2 and TET3 in a dose-dependent manner in HepG2 cells.
To substantiate the hypothesis that HIF-1α upregulates the
expression of TET enzymes, HIF-1α expression was attenuated using a
specific shRNA in HepG2 cells. HIF-1α knockdown significantly
inhibited the transcriptional upregulation of TET enzymes upon
hypoxia. These results provide direct evidence that hypoxia
regulates TET enzymes expression via HIF-1α.
An increase in TET1 expression and in the level of
5-hmC upon hypoxia has been reported in the neuroblastoma SK-N-BE,
NBL-WN and LA1-55n (44) cell lines.
Wu et al (45) demonstrated
that hypoxia increases cellular 5-hmC levels, and upregulates the
expression of TET1 and TET3 in breast cancer MCF7 and MDA-MB-231
cell lines, and in primary breast cancer cells. In line with these
studies, the results of the present study demonstrate that hypoxia
increases cellular 5-hmC levels and the expression of all three TET
enzymes in HepG2 cells. TET1 and TET2 expression was more sensitive
to hypoxia than TET3 was. The differences on the expression of TET
enzymes upon hypoxia among the above studies may be due to the
different patterns of gene expression and/or the different
signaling pathway involved in cell lines used.
In conclusion, the results of the present study
demonstrate that hypoxia induces expression of TET enzymes, a
process mediated by HIF-1α, thus increasing cellular 5-hmC levels
in HepG2 cells, which could inform on novel strategies for the
future development of therapeutic plans.
References
|
1
|
Thomas MB and Zhu AX: Hepatocellular
carcinoma: The need for progress. J Clin Oncol. 23:2892–2899. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Lee JS, Conner EA, Schroeder I, Factor VM and Thorgeirsson SS:
Mechanistic and prognostic significance of aberrant methylation in
the molecular pathogenesis of human hepatocellular carcinoma. J
Clin Invest. 117:2713–2722. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Altekruse SF, McGlynn KA and Reichman ME:
Hepatocellular carcinoma incidence, mortality, and survival trends
in the United States from 1975 to 2005. J Clin Oncol. 27:1485–1491.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sia D, Villanueva A, Friedman SL and
Llovet JM: Liver cancer cell of origin, molecular class, and
effects on patient prognosis. Gastroenterology. 152:745–761. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herzog CE, Andrassy RJ and Eftekhari F:
Childhood cancers: Hepatoblastoma. Oncologist. 5:445–453. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Darbari A, Sabin KM, Shapiro CN and
Schwarz KB: Epidemiology of primary hepatic malignancies in U.S.
Children. Hepatology. 38:560–566. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seeff LB: Introduction: The burden of
hepatocellular carcinoma. Gastroenterology. 127(5): S1–S4. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bisteau X, Caldez MJ and Kaldis P: The
complex relationship between liver cancer and the cell cycle: A
story of multiple regulations. Cancers (Basel). 6:79–111. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dan YY, Riehle KJ, Lazaro C, Teoh N, Haque
J, Campbell JS and Fausto N: Isolation of multipotent progenitor
cells from human fetal liver capable of differentiating into liver
and mesenchymal lineages. Proc Natl Acad Sci USA. 103:pp.
9912–9917. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bell D, Ranganathan S, Tao J and Monga SP:
Novel Advances in Understanding of Molecular Pathogenesis of
Hepatoblastoma: A Wnt/β-Catenin Perspective. Gene Expr. 17:141–154.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen
L, Li Q, Huang Y, Zhang Y, Zhang Z, et al: Exome sequencing of
hepatoblastoma reveals novel mutations and cancer genes in the Wnt
pathway and ubiquitin ligase complex. Hepatology. 60:1686–1696.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eichenmuller M, Trippel F, Kreuder M, Beck
A, Schwarzmayr T, Haberle B, Cairo S, Leuschner I, von Schweinitz
D, Strom TM and Kappler R: The genomic landscape of hepatoblastoma
and their progenies with HCC-like features. J Hepatol.
61:1312–1320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koch A, Denkhaus D, Albrecht S, Leuschner
I, von Schweinitz D and Pietsch T: Childhood hepatoblastomas
frequently carry a mutated degradation targeting box of the
beta-catenin gene. Cancer Res. 59:269–273. 1999.PubMed/NCBI
|
|
16
|
Koch A, Weber N, Waha A, Hartmann W,
Denkhaus D, Behrens J, Birchmeier W, von Schweinitz D and Pietsch
T: Mutations and elevated transcriptional activity of conductin
(AXIN2) in hepatoblastomas. J Pathol. 204:546–554. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tomlinson GE and Kappler R: Genetics and
epigenetics of hepatoblastoma. Pediatr Blood Cancer. 59:785–792.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu C, Liu L, Chen X, Shen J, Shan J, Xu
Y, Yang Z, Wu L, Xia F, Bie P, et al: Decrease of
5-hydroxymethylcytosine is associated with progression of
hepatocellular carcinoma through downregulation of TET1. PLoS One.
8:e628282013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishida N and Goel A: Genetic and
epigenetic signatures in human hepatocellular carcinoma: A
systematic review. Curr Genomics. 12:130–137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rumbajan JM, Maeda T, Souzaki R, Mitsui K,
Higashimoto K, Nakabayashi K, Yatsuki H, Nishioka K, Harada R, Aoki
S, et al: Comprehensive analyses of imprinted differentially
methylated regions reveal epigenetic and genetic characteristics in
hepatoblastoma. BMC Cancer. 13:6082013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui X, Liu B, Zheng S, Dong K and Dong R:
Genome-wide analysis of DNA methylation in hepatoblastoma tissues.
Oncol Lett. 12:1529–1534. 2016.PubMed/NCBI
|
|
22
|
Majmundar AJ, Wong WJ and Simon MC:
Hypoxia-inducible factors and the response to hypoxic stress. Mol
Cell. 40:294–309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Semenza GL: Molecular mechanisms mediating
metastasis of hypoxic breast cancer cells. Trends Mol Med.
18:534–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Q, Liu L, Zhao Y, Zhang J, Wang D,
Chen J, He Y, Wu J, Zhang Z and Liu Z: Hypoxia induces genomic DNA
demethylation through the activation of HIF-1α and transcriptional
upregulation of MAT2A in hepatoma cells. Mol Cancer Ther.
10:1113–1123. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shahrzad S, Bertrand K, Minhas K and
Coomber BL: Induction of DNA hypomethylation by tumor hypoxia.
Epigenetics. 2:119–125. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu H and Zhang Y: Mechanisms and functions
of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev.
25:2436–2452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Das PM and Singal R: DNA methylation and
cancer. J Clin Oncol. 22:4632–4642. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kohli RM and Zhang Y: TET enzymes, TDG and
the dynamics of DNA demethylation. Nature. 502:472–479. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ito S, Shen L, Dai Q, Wu SC, Collins LB,
Swenberg JA, He C and Zhang Y: Tet proteins can convert
5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine.
Science. 333:1300–1303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tahiliani M, Koh KP, Shen Y, Pastor WA,
Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L and
Rao A: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qiu GH, Xie X, Xu F, Shi X, Wang Y and
Deng L: Distinctive pharmacological differences between liver
cancer cell lines HepG2 and Hep3B. Cytotechnology. 67:1–12. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rikhi RR, Spady KK, Hoffman RI, Bateman
MS, Bateman M and Howard LE: Hepatoblastoma: A Need for Cell Lines
and Tissue Banks to Develop Targeted Drug Therapies. Front Pediatr.
4:222016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang GL and Semenza GL: General
involvement of hypoxia-inducible factor 1 in transcriptional
response to hypoxia. Proc Natl Acad Sci USA. 90:pp. 4304–4308.
1993; View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pugh CW, O'Rourke JF, Nagao M, Gleadle JM
and Ratcliffe PJ: Activation of hypoxia-inducible factor-1;
definition of regulatory domains within the alpha subunit. J Biol
Chem. 272:11205–11214. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gopalakrishnan S, Van Emburgh BO and
Robertson KD: DNA methylation in development and human disease.
Mutat Res. 647:30–38. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
40
|
Capes-Davis A, Theodosopoulos G, Atkin I,
Drexler HG, Kohara A, MacLeod RA, Masters JR, Nakamura Y, Reid YA,
Reddel RR and Freshney RI: Check your cultures! A list of
cross-contaminated or misidentified cell lines. Int J Cancer.
127:1–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aden DP, Fogel A, Plotkin S, Damjanov I
and Knowles BB: Controlled synthesis of HBsAg in a differentiated
human liver carcinoma-derived cell line. Nature. 282:615–616. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ivanov M, Kals M, Kacevska M, Barragan I,
Kasuga K, Rane A, Metspalu A, Milani L and Ingelman-Sundberg M:
Ontogeny, distribution and potential roles of
5-hydroxymethylcytosine in human liver function. Genome Biol.
14:R832013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu
J, Xu ZD, Zhu HG, Ling ZQ, Ye D, et al: Tumor development is
associated with decrease of TET gene expression and
5-methylcytosine hydroxylation. Oncogene. 32:663–669. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mariani CJ, Vasanthakumar A, Madzo J,
Yesilkanal A, Bhagat T, Yu Y, Bhattacharyya S, Wenger RH, Cohn SL,
Nanduri J, et al: TET1-mediated hydroxymethylation facilitates
hypoxic gene induction in neuroblastoma. Cell Rep. 7:1343–1352.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu MZ, Chen SF, Nieh S, Benner C, Ger LP,
Jan CI, Ma L, Chen CH, Hishida T, Chang HT, et al: Hypoxia Drives
Breast Tumor Malignancy through a TET-TNFα-p38-MAPK Signaling Axis.
Cancer Res. 75:3912–3924. 2015. View Article : Google Scholar : PubMed/NCBI
|