Introduction
In Europe, urothelial bladder cancer (UBC) is the
second most common malignancy in the genitourinary tract and the
fifth most common type of cancer, with a high rate of morbidity and
mortality (1,2). The majority of initially diagnosed cases
of UBC is non-muscle invasive and can be effectively treated by
transurethral resection, combined with intravesical chemo- or
immunotherapy (3). However, at the
time of first diagnosis or at late visits, ~30% of UBC cases
exhibit an invasive growth pattern, being either muscle-invasive
(pT2) or more advanced, such as locally advanced (pT3-4 and/or
pN1-3 M0) or metastatic (M1) (3,4). The
overall prognosis of patients with UBC with this pattern remains
poor. Cisplatin (cis-diaminodichloroplatinum, CDDP)-based
chemotherapy is widely used in this group of patients and provides
a survival advantage (5). However,
the response rate to chemotherapy remains ~50% (6). In consideration of function of CDDP in
the currently employed chemotherapy regimens, the principal cause
of poor response involves resistance to CDDP. However, thus far,
the mechanisms underlying CDDP inhibition of UBC have not yet been
fully elucidated, which represents a great challenge for
physicians.
High-mobility group nucleosome-binding domain 5
(HMGN5), also termed NBP-45, GARP45 or NSBP1, is the latest member
of the HMGN family of proteins, which was identified by King et
al in 2001 (7,8). Since identification, the gene has been
reported to primarily function in embryonic development, regulation
of transcription and chromatin decompaction (8). In recent years, emerging studies have
confirmed that HMGN5 is overexpressed in various human tumors and
confers oncogenic effects in various cancer models (9). However, the effects of the gene on
chemosensitivity to commonly used chemotherapy regimens in cancer
cells remain largely unknown and controversial.
In a previous study by the present authors, it was
revealed that knockdown of HMGN5 suppressed the viability and
invasion of human UBC 5637 cells via regulating the expression of
E-cadherin, a marker of epithelial-mesenchymal transition (EMT),
and vascular endothelial growth factor (VEGF)-C, a marker of
lymphangiogenesis (10,11). It was reported that EMT and the
transcription factor slug directly contribute to CDDP resistance
(12,13). In addition, Zhu et al (14) reported that inhibition of VEGF-C
reversed resistance of UBC cells to CDDP. Therefore, the present
study aimed to investigate the involvement of HMGN5 in the
treatment of UBC using CDDP. However, more efforts are required to
elucidate the role of HMGN5 in cancer progression of UBC.
The present study examined the function of HMGN5 on
the sensitivity of UBC cells to CDDP in vitro and
investigated the underlying mechanisms. Results of the present
study demonstrated that the UBC cells expressing a low level of
HMGN5 are more sensitive to CDDP, and CDDP suppresses the growth of
UBC cells by inhibiting HMGN5. Furthermore, it was verified that
HMGN5 depletion increases the sensitivity of UBC 5637 cells to CDDP
via inhibiting PI3K/Akt signaling. These findings indicated that
HMGN5 is a potential therapy target in UBC treatment.
Materials and methods
Cell culture, transfection and drug
treatment
The human UBC 5637, UM-UC-3 and T24 cell lines were
obtained from Yingrun Biotechnologies, Inc. (Changsha, China). The
cells were maintained in RPMI-1640 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal calf serum (Thermo
Fisher Scientific, Inc.) in a humidified 5% CO2, 37°C
incubator.
HMGN5 short hairpin RNA (shRNA) sequences and
construction of lentivirus were the same as described in a previous
study by the present authors (10).
Briefly, the most effective shRNA sequences targeting HMGN5
(5′-GTTGTTGAAGAAGACTACAAT-3′) were synthesized and cloned into the
pYr-Lvsh vectors by Yingrun Biotechnologies, Inc. to generate the
lentiviral vectors against HMGN5. The other shRNA sequences with no
significant homology to any known human genes
(5′-GACTTCATAAGGCGCATGC-3′) were employed to generate the shRNA
control lentiviral vectors. UBC cells were placed on 6-well plates
(~5×104 cells/well) until sufficient cell fusion, then
the cells were infected with the recombinant lentivirus at a
multiplicity of infection of 50, recommended by the manufacturer,
for 24 h. Medium was subsequently replaced with RPMI-1640 medium
and 1 µg/ml puromycin (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) could be used for screening positively stable
transfectants. Blank controls (untransfected controls) were used in
experiments to demonstrate there was no significant difference
between untransfected controls and cells transfected with shControl
lentivirus.
CDDP was obtained from Sigma-Aldrich (Merck KGaA)
and diluted in sterile serum (Thermo Fisher Scientific, Inc.) as
indicated concentrations (0, 1, 2, 4, 8 and 16 µg/ml). The PI3K
signaling activator, insulin-like growth factor-1 (IGF-1), was
purchased from PeproTech, Inc. (Rocky Hill, NJ, USA) and dissolved
in dimethyl sulfoxide (DMSO) (15).
Different concentrations (0, 1, 2, 4, 8 and 16 µg/ml) of CDDP were
used to treat different types of UBC cells or cells infected with
lentivirus for dose-dependent cell proliferation assay for 72 h.
IGF-1 (50 ng/ml) was also added as indicated.
Western blot analysis
UBC cells were treated with CDDP (6 µg/ml), with or
without IGF-1 (50 ng/ml), and then lysed with lysis buffer
supplemented with protease inhibitor (catalog no. P0013B; Beyotime
Institute of Biotechnology, Haimen, China). Western blotting was
performed as previously described (10). In the present study, antibody against
HMGN5 (dilution, 1:1,000; catalog no. ab18601) was obtained from
Abcam (Cambridge, MA, USA), while antibodies against Akt (catalog
no. 4685S), phosphorylated (p)-Akt (Ser473; catalog no.
4060P), slug (catalog no. 9585P), E-cadherin (catalog no. 3195S),
VEGF-C (catalog no. 2445S), cytochrome c (catalog no. 19940S),
cleaved-caspase-3 (catalog no. 9664P), cleaved-PARP (catalog no.
5625P) and β-actin (catalog no. 4967; all 1:1,000) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). The
proteins were visualized using the ECL Plus Kit (Thermo Fisher
Scientific, Inc.). β-actin served as a loading control. Experiments
were performed in triplicate.
Cell proliferation assay
Cell proliferation under CDDP treatment, with or
without IGF-1, was analyzed by the MTT assay as previously
described (10). Briefly, the cells
were seeded on 96-well plates (4×103 cells/well) and
treated with CDDP as aforementioned. After 24 h, MTT (5 mg/ml;
Sigma-Aldrich; Merck KGaA) was added to cells for 4 h at 37°C.
Subsequently, DMSO was used to dissolve the purple-blue formazan
crystals produced by living cells. Absorbance was measured at 490
nm.
Colony formation assay
After 72 h infection, 5637 cells were seeded at a
low density of 400 cells/well onto a 6-well plate. RPMI-1640 medium
was added with CDDP (6 µg/ml), with or without IGF-1 (50 ng/ml) as
indicated, and changed every 3 days for 2 weeks. Colonies (≥50
cells in a colony) were counted under a light microscope
(magnification, ×200; Leica TCS-SP5; Leica Microsystems GmbH,
Wetzlar, Germany) subsequent to being fixed with pure methanol for
15 min at room temperature and stained with 0.005% gentian violet
for 20 min at room temperature. Experiments were tested in
triplicate.
Cell invasion assay
Cell invasion was evaluated using the Transwell
assay as described previously (10).
Briefly, the upper chamber of each Transwell insert (pore size, 8
µm; Corning Incorporated, Corning, NY, USA) was paved with the
Matrigel gelatum (BD Biosciences, San Jose, CA, USA), in which
approximately 1×105 bladder cancer 5637 cells with
different HMGN5 expression were seeded. The cells were incubated at
37°C in serum-free RPMI-1640 medium medium and treated with CDDP (6
µg/ml) as well as IGF-1 (50 ng/ml). Subsequent to culturing for 24
h to allow cell migration, adherent cells in the upper chamber were
removed with a cotton swab and the invasive cells attached on the
lower surface were fixed with 4% paraformaldehyde for 30 min at
room temperature. The cells were stained with 0.1% crystal violet
for 20 min at room temperature and counted using a confocal
microscope (magnification, ×200, Leica TCS-SP5).
Cell apoptosis assay
Annexin V-fluorescein isothiocyanate apoptosis kit
(Nanjing KeyGen Biotech Co. Ltd., Nanjing, China) was used to
measure apoptosis as reported previously (10). Briefly, following infection and drug
treatment with CDDP (6 µg/ml) and IGF-1 (50 ng/ml) for 24 h, the
cells were collected and labeled with annexin V as well as
propidium iodide (PI) in the dark. Cell apoptosis was then detected
using a FACSCalibur flow cytometry (BD Biosciences, San Jose, CA,
USA). The results were analyzed using BD FACSDiva 6.1.3 software
(BD Biosciences).
Hoechst 33342 dye
Following transfection, 5637 cells were cultured on
a 24-well plate and treated with CDDP and IGF-1 as aforementioned.
Hoechst 33342 (1 µg/ml; Thermo Fisher Scientific, Inc.) was then
added, and the cells were stained for 20–30 min at 37°C.
Subsequently, the cells were washed twice with ice-cold PBS and
observed under a fluorescence microscope (magnification, ×200,
Leica TCS-SP5). Thick and dense fluorescence was observed in the
apoptotic cells. The percentage of apoptotic cells was calculated
according to the following formula: (Apoptotic cells in five random
visual fields)/(total cells in five random visual fields).
Statistical analysis
All data were collected from three independent
experiments and analyzed using GraphPad Prism 5.01 software
(GraphPad Software, Inc., La Jolla, CA, USA). The quantitative data
are presented as the mean ± standard deviation. Differences among
the means of multiple groups were compared by one-way analysis of
variance, followed by a Newman-Keuls multiple comparison test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
HMGN5 protein determines the response
of UBC cells to CDDP
The expression of HMGN5 protein was determined using
western blotting in UBC cell lines 5637, T24 and UM-UC-3 cells. The
results confirmed that the level of HMGN5 protein in 5637 cells was
the highest among the three cell lines, while the level in UM-UC-3
cells was the lowest (Fig. 1A). Cell
proliferation MTT assays were then employed to investigate the
chemosensitivity of the three cell lines to CDDP. Following
incubation with the indicated concentrations of CDDP for 72 h, it
was revealed that the 5637 cells were relatively more resistant to
CDPP compared with the other two types of cells (Fig. 1B). Notably, the UM-UC-3 cells
exhibited the highest sensitivity in the three cell types (Fig. 1B).
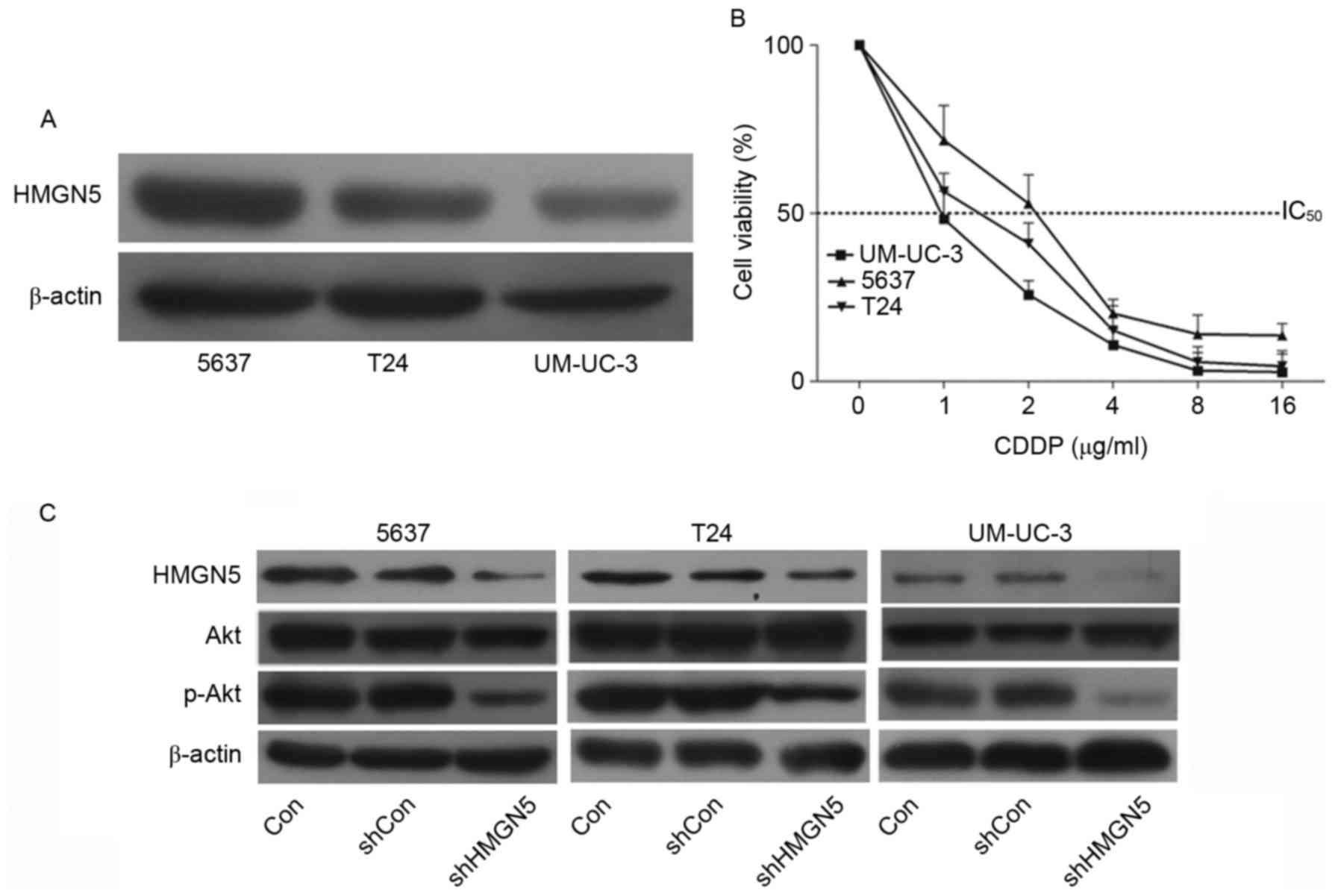 | Figure 1.HMGN5 determines the responses of UBC
cell lines to CDDP via the phosphoinositide 3-kinase/Akt signaling
pathway. (A) Western blotting of HMGN5 expression in 5637, T24 and
UM-UC-3 cell lines. β-actin served as a loading control. (B)
Different concentrations of CDDP as indicated were used to treat
5637, T24 and UM-UC-3 cells for dose-dependent cell proliferation
MTT assay for 72 h. (C) 5637, T24 and UM-UC-3 cells were infected
with lentivirus for HMGN5 depletion or negative control, the
expression of HMGN5, Akt and p-Akt was subsequently analyzed by
western blotting after 72 h. β-actin served as a loading control.
Results are expressed as the mean ± standard deviation (n=3).
HMGN5, high mobility group nucleosome-binding domain 5; CDDP,
cisplatin; p-, phosphorylated; IC50, Half-maximal
inhibitory concentration; sh, short hairpin; Con, control. |
The PI3K/Akt signaling pathway is associated with
the development of resistance to CDDP in human malignancies
(16). Zhou et al (17) first reported that HMGN5 is involved in
the progression of osteosarcoma through PI3K/Akt signaling. The
present study also confirmed that, following successful knockdown
of HMGN5 in UBC cell lines, the expression of p-Akt was
subsequently inhibited (Fig. 1C).
These results indicated that HMGN5 might be involved in the
negative regulation of chemosensitivity in UBC cell lines to CDDP
via the PI3K/Akt signaling pathway.
HMGN5 is a molecular target of CDDP
treatment in 5637 cells
To further investigate the molecular mechanisms
underlying the involvement of HMGN5 in CDDP treatment, the
expression of HMGN5, Akt, p-Akt, VEGF-C, slug and E-cadherin was
analyzed in 5637 cells during CDDP treatment for 24 h.
Additionally, considering the participation of mitochondrial
apoptosis pathway in cellular response in tumor cells to numerous
anticancer drugs, including CDDP (18), the expression of cytochrome c,
cleaved-caspase-3 and cleaved-PARP was also detected.
The results from western blot analysis indicated
that CDDP impaired the expression of HMGN5 protein in a
dose-dependent manner (Fig. 2).
Furthermore, consistent with the change in HMGN5 protein
expression, the active form of Akt, p-Akt, was also downregulated
by CDDP treatment (Fig. 2). According
to previous studies, the expression of VEGF-C, slug and the process
of EMT can be positively regulated by PI3K/Akt signaling (19–21).
Consistent with these findings, in the present study, the
expression of VEGF-C and slug was observed to be inhibited by CDDP
treatment, while the expression of the epithelial marker,
E-cadherin, was increased (Fig. 2).
Furthermore, the expression of cytochrome c, cleaved-caspase-3 and
cleaved-PARP was increased in a dose-dependent manner during CDDP
treatment, indicating the activation of the mitochondrial apoptosis
pathway by CDDP treatment (Fig.
2).
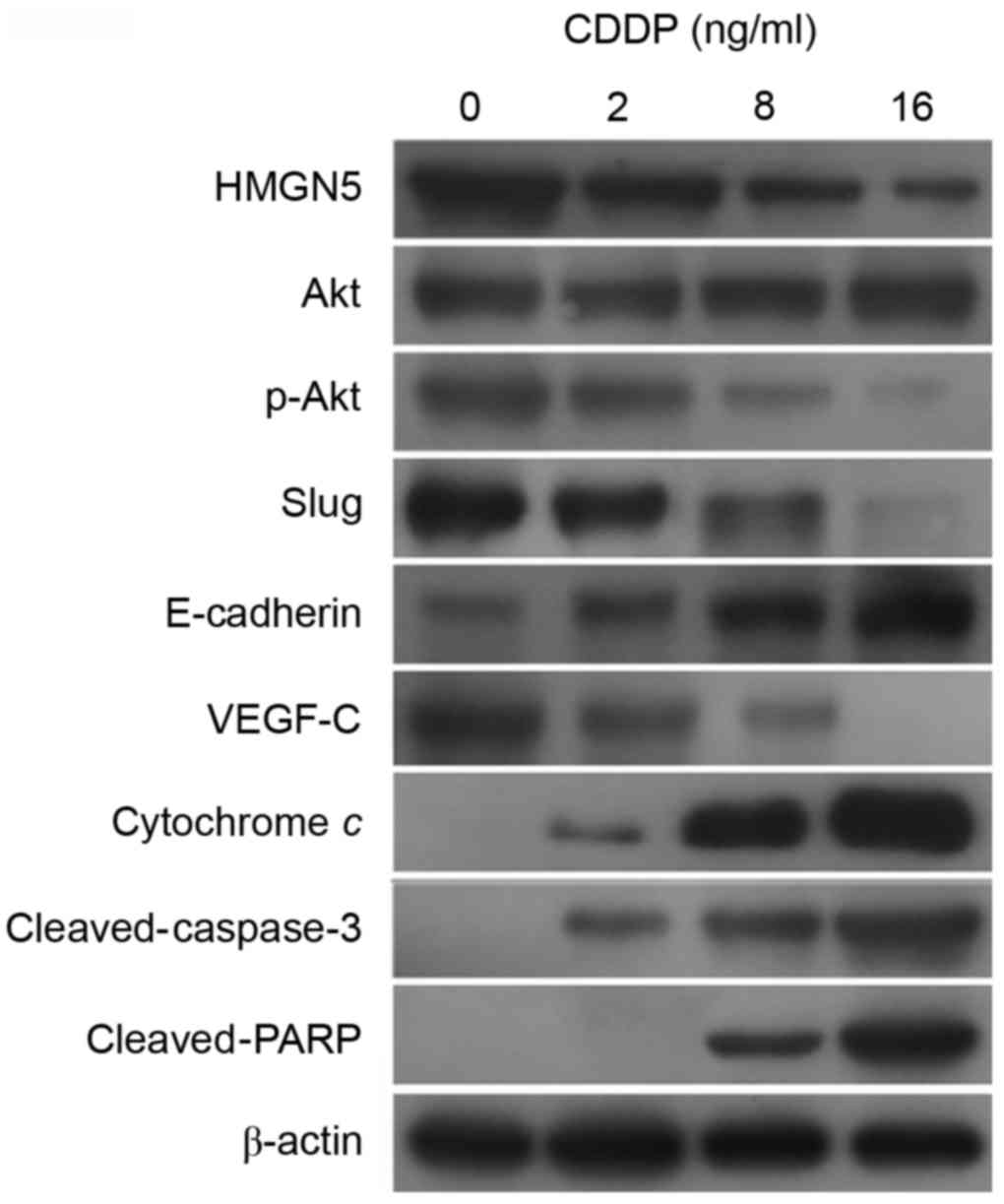 | Figure 2.HMGN5 serves as a target for CDDP
treatment in UBC cells. Different concentrations (0, 1, 2, 8 and 16
µg/ml) of CDDP were used to treat UBC 5637 cells for 24 h. The
expression of HMGN5 and its downstream signal molecules, including
Akt, p-Akt, slug, E-cadherin and VEGF-C, was then analyzed by
western blot analysis. The expression of a number of markers in the
mitochondrial apoptosis pathway, including cytochrome c,
cleaved-caspase-3 and cleaved-PARP, was also analyzed. β-actin
served as a loading control. HMGN5, high mobility group
nucleosome-binding domain 5; CDDP, cisplatin; p-, phosphorylated;
PARP, poly[ADP-ribose] synthase 1; VEGF, vascular endothelial
growth factor. |
Knockdown of HMGN5 increases the
chemosensitivity of 5637 cells to CDDP
To verify the function of HMGN5 in controlling
sensitivity of UBC cells to CDDP and elucidate the mechanisms, the
5637 cell line was used as the cell model and the loss-of-function
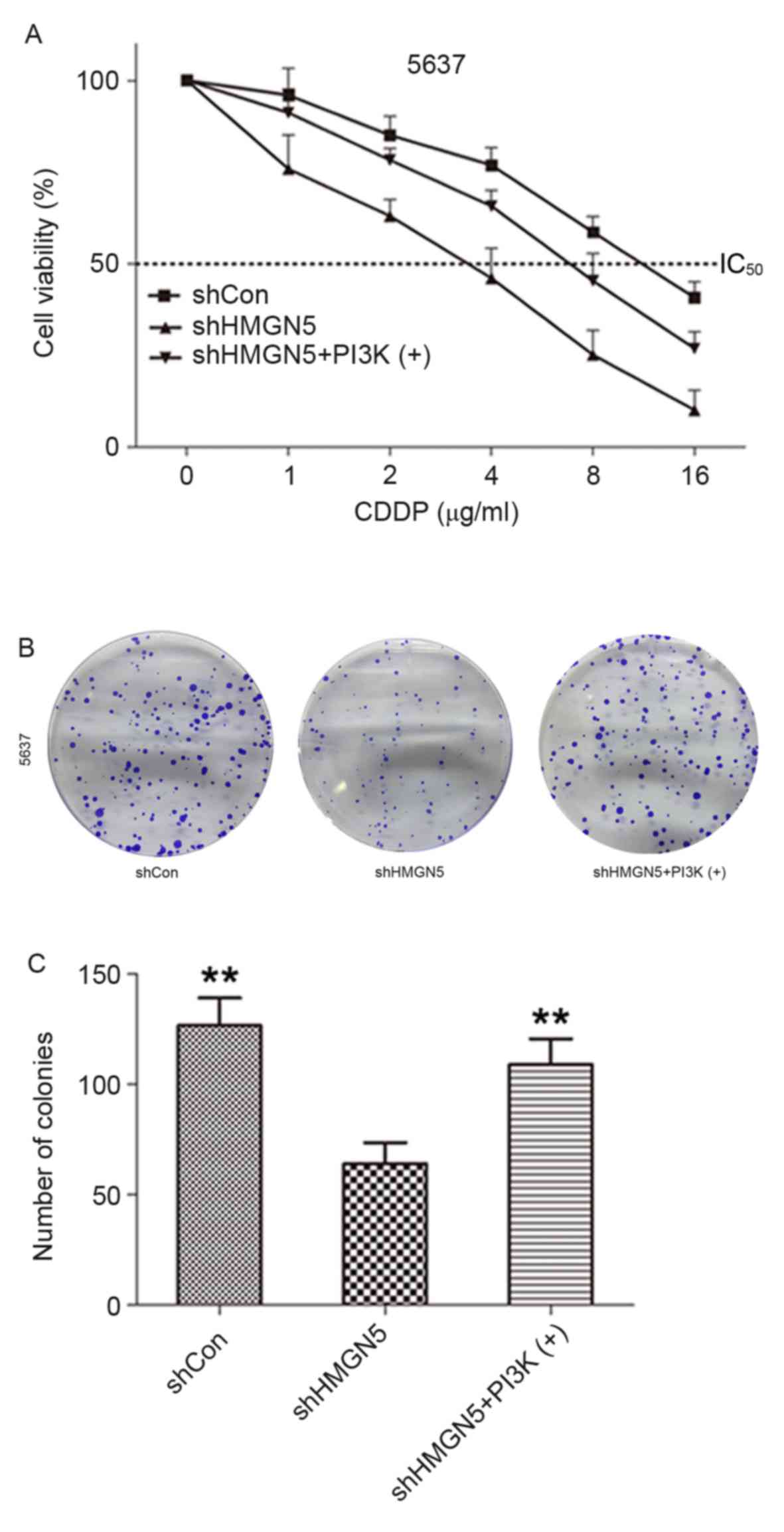
method was employed. As demonstrated in Fig. 3A, following incubation with various
concentrations (1–16 µg/ml) of CDDP for 24 h, knockdown of HMGN5
markedly decreased viability of 5637 cells compared with negative
control cells. As expected, the effect could be reversed to a
certain extent by treatment with the PI3K signaling activator,
IGF-1. Similarly, the results from colony formation assay indicated
that 5637 cells where HMGN5 was knocked down formed fewer and
smaller colonies compared with the negative control cells in
monolayer culture for 14 days, which could also be rescued to a
certain extent by IGF-1 treatment (Fig.
3B and C). These results indicated that HMGN5-knockdown might
increase the therapeutic effects of CDDP in inhibiting the
proliferation and tumorigenesis of 5637 cells.
Transwell invasion assays were subsequently employed
to investigate the effects of HMGN5-knockdown on invasion of UBC
cells following CDDP treatment for 24 h. HMGN5-knockdown was found
to significantly decrease the number of 5637 cells crossing the
Matrigel, indicating that HMGN5-knockdown inhibits the invasiveness
of UBC cells during CDDP treatment (Fig.
4A). In addition, this effect on invasion can be blocked by
IGF-1 to a certain extent (Fig. 4A).
Flow cytometry and cell nuclei Hoechst 33342 staining were also
utilized to detect cell apoptosis following CDDP treatment for 24
h. The results from flow cytometry demonstrated that the early
apoptotic rate was increased by HMGN5-knockdown in 5637 cells, and
this may be reversed by treatment IGF-1 to a certain extent
(Fig. 4B). Furthermore, as shown in
Fig. 4C, morphological changes in the
nuclei of apoptotic cells were analyzed using Hoechst 33342
staining, which are characterized by pyknosis, shrinkage and
karyorrhexis, with marked fluorescence (Fig. 4C). It was revealed that the percentage
of apoptotic nuclei in the HMGN5 knocked down group was
significantly higher compared with the percentage in the negative
control group and the IGF-1 treatment group (Fig. 4C).
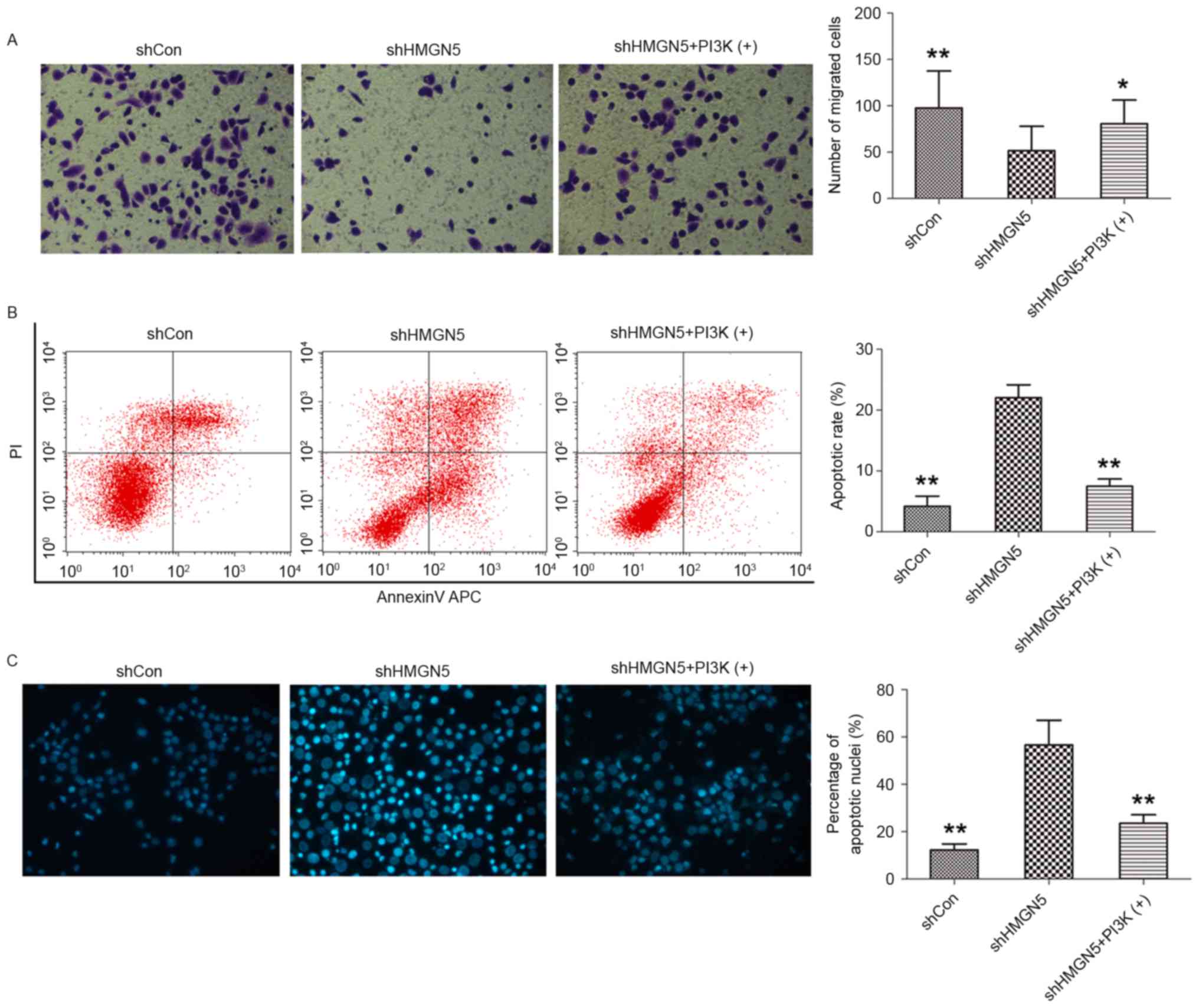 | Figure 4.HMGN5-knockdown inhibits invasion but
increases apoptosis in UBC cells. (A) Following infection with
lentivirus, 5637 cells were treated with CDDP, with or without
IGF-1 (left panels). The Transwell assay was then used to detect
cell invasion. The cells penetrating the Matrigel were stained with
crystal violet and shown as blue under a light microscope
(magnification, ×200). Quantitative analysis of the number of
migrated cells in different groups (right panel). (B) After
infection, the cells were treated with indicated agents for 24 h,
followed by staining with Annexin V-fluorescein isothiocyanate and
PI and analyzed by flow cytometry (left panels). Quantitative
analysis of the rate of early apoptosis in different groups (right
panel). (C) Hoechst 33342 staining was employed to observe the
morphological change of apoptotic cells (left panels;
magnification, ×200). Cell nuclei with apoptosis were characterized
by pyknosis, shrink and karyorrhexis and exhibited the marked
fluorescence under fluorescence microscope. Quantitative analysis
of the percentage of apoptotic nuclei in different groups (right
panel). Results are expressed as the mean ± standard deviation
(n=3). *P<0.05, **P<0.01 compared with HMGN5 knocked down
5637 cells. HMGN5, high mobility group nucleosome-binding domain 5;
PI3K, phosphoinositide 3-kinase; sh, short hairpin; Con, control;
PI, propidium iodide. |
Additionally, western blotting indicated that
HMGN5-knockdown decreased the expression of p-Akt and VEGF-C
compared with control, and increased the expression of E-cadherin,
cytochrome c, cleaved-caspase-3 and cleaved-PARP during CDDP
treatment. Notably, it was possible to reverse these changes in
protein expression by IGF-1 treatment (Fig. 5).
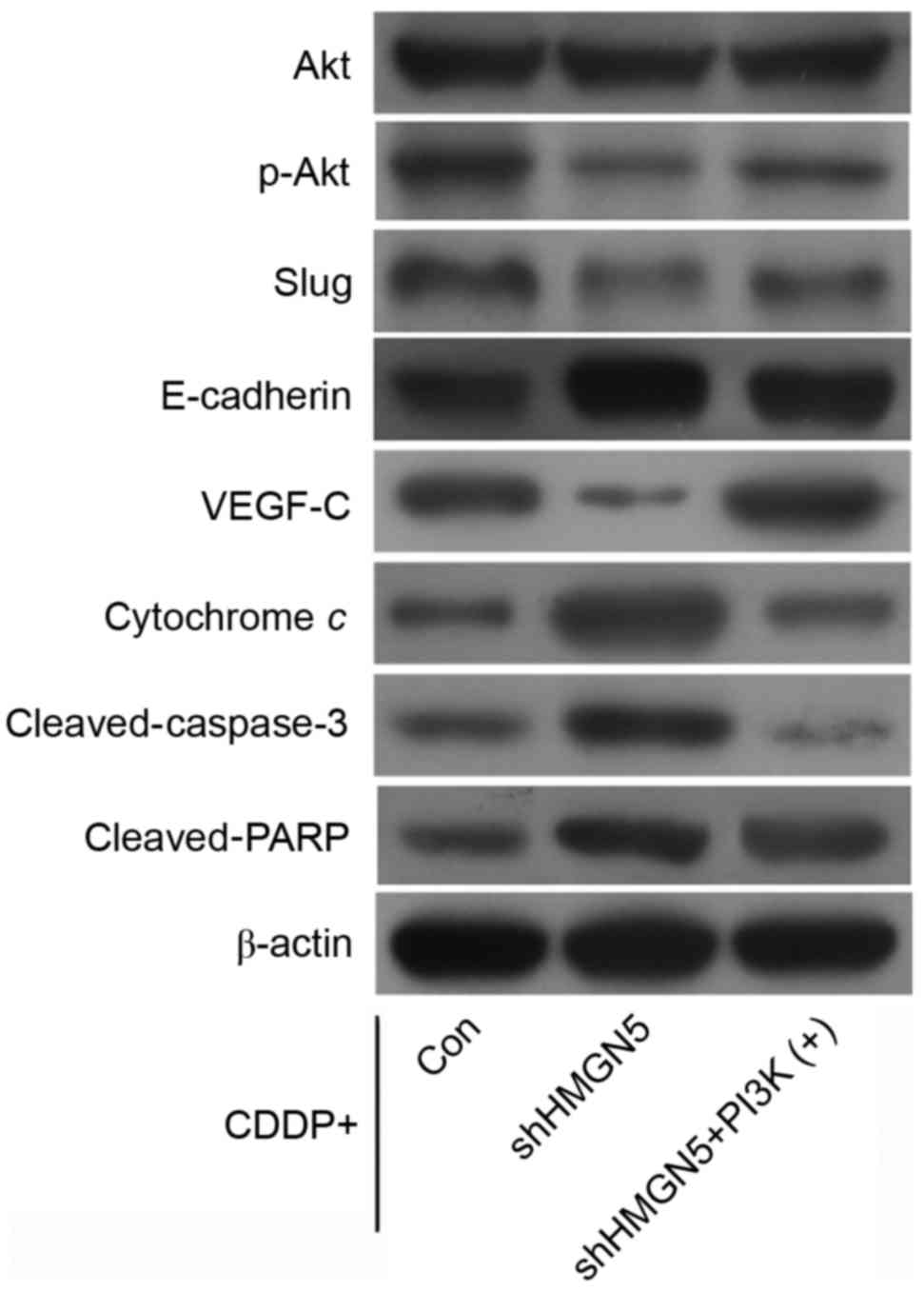 | Figure 5.Effect of HMGN5 depletion on the
expression of Akt, p-Akt, slug, E-cadherin, VEGF-C, cytochrome c,
cleaved-caspase-3 and cleaved-PARP. After infection with lentivirus
as indicated, 5637 cells were treated with CDDP, with or without
IGF-1. The expression of Akt, p-Akt, slug, E-cadherin, VEGF-C,
cytochrome c, cleaved-caspase-3 and cleaved-PARP was then analyzed
by western blotting. β-actin served as a loading control. CDDP,
cisplatin; IGF-1, insulin-like growth factor-1; PARP, poly ADP
ribose polymerase; VEGF, vascular endothelilal growth factor;
HMGN5, high mobility group nucleosome-binding domain 5. |
Discussion
HMGN5 is ubiquitously expressed in human tissues and
is classified as a member of the HMGN family of proteins, based on
the presence of the three important functional domains: The
nucleosome-binding domain, the nuclear localization signal and the
negatively charged C-terminus (8). On
account of these domains, HMGN5 is able to enter the nucleus, bind
to nucleosomes, unfold chromatin and increase the accessibility of
DNA (8). As a result, factors
associated with DNA lesion and repair, replication, transcription
and recombination may combine with DNA more easily and take action
and thereby modulate the cellular epigenetic profile (for example,
ultraviolet light can cause more DNA lesions when chromatin is
unfolded) (8,22,23). In
the last few years, ectopic high expression level of HMGN5 has been
confirmed in several malignant tumors, including prostate cancer,
renal cancer, lung cancer, breast cancer, osteosarcoma and glioma,
indicating the potential role of this protein as a target in cancer
therapy (9,11). The present authors previously
identified the carcinogenic role of HMGN5 in UBC and reported the
regulation of the gene in the expression of E-cadherin and VEGF-C
(10,11,24). In
the present study, the mechanisms underlying the modulation were
further investigated and the possible application of HMGN5 in UBC
therapy was examined.
Zhou et al (17) highlighted that, in osteosarcoma U2-OS
and SaO2 cell lines, HMGN5 positively regulates the expression of
PI3Kp85α and p-Akt and contributes to the malignant potential of
tumor cells. Similar to the results from the study by Zhou et
al (17), in the present study it
was observed that the expression of p-Akt was subsequently
decreased with no change in Akt expression following the silencing
of HMGN5 in UBC 5637, T24 and UM-UC-3. These findings indicate that
HMGN5 may positively regulate PI3K/Akt signaling. The possible
mechanism may be that HMGN5 can act as a protein kinase and lead to
phosphorylation of Akt (25).
However, the mechanism can also be indirect as HMGN5 may amplify
the transcriptional level of the catalytic subunit in PI3K,
therefore, additional studies are required to completely explain
this effect (23). Furthermore, as
reported, the progression of EMT and the expression of VEGF-C can
be regulated by PI3K/Akt signaling, therefore, it was hypothesized
that HMGN5 may modulate E-cadherin and VEGF-C expression through
PI3K/Akt signaling (19–21).
Chemotherapy represents an important approach in the
comprehensive treatment of solid tumors, while the efficacy usually
depends on the sensitivity of tumor cells to chemotherapeutics. In
fact, numerous studies support the pivotal role of certain
oncogenes in the regulation of chemosensitivity, such as
transcription factor AP-2α in UBC cells and REV3-like DNA-directed
polymerase ζ catalytic subunit in cervical cancer cells (26,27). The
role of HMGN5 in tumorigenesis in a number of types of cancer has
been highlighted (9). However,
information regarding HMGN5 in the regulation of chemosensitivity
is insufficient and disputable. He et al (28) reported that the inhibition of HMGN5
sensitized the malignant meningioma IOMM-Lee and CH157 cells to
temozolomide-induced cytotoxicity through modulation of the
expression of multidrug resistance-associated protein-1, B cell
lymphoma-2 and cleaved-caspase-3. Similarly, in a study on
osteosarcoma cells, Zhou et al (17) observed that the knockdown of HMGN5
enabled tumor cells to be significantly more sensitive to
doxorubicin-induced cell injury via activating the apoptotic
signaling pathways. These findings indicated that HMGN5 knockdown
may contribute to the increase in chemosensitivity to antitumor
drugs. Guo et al (29)
revealed that HMGN5 expression was positively associated with
chemosensitivity of prostate cancer cell lines to gemcitabine. In
addition, they also demonstrated that HMGN5 depletion reduced the
sensitivity of PC-3 cells to gemcitabine, while HMGN5
overexpression in DU145 cells increased the sensitivity to the
agent (29). Therefore, a high
expression level of HMGN5 may be beneficial to patients with
chemotherapy.
As one of the most effective anticancer drugs, CDDP
has been extensively used for the treatment of different types of
cancer, including UBC (18). The
present data supported the view from Zhou et al (17) and He et al (28), as it was revealed from MTT assays that
UBC cells with lower levels of HMGN5 expression were relatively
more sensitive to CDDP compared with cells with higher levels of
expression. Furthermore, the inhibition of HMGN5 was able to
markedly decrease proliferation, colony formation and invasion of
5637 cells during CDDP treatment, indicating that depletion of the
HMGN gene in UBC cells with relatively high HMGN5 expression may
increase the sensitivity of tumor cells to CDDP.
Abnormal activation of the PI3K/Akt signaling
pathway has been reported to have critical roles in the development
of resistance to CDDP, while inhibition of PI3K/Akt signaling
increases the effect of CDPP (30,31). In
addition, the progression of EMT and overexpression of its
activators, such as slug, are positively associated with CDDP
resistance (12,13). VEGF-C, a marker of lymphangiogenesis,
is also involved in the regulation of CDDP sensitivity (14). In the present study, western blotting
results indicated that the level of p-Akt, slug, E-cadherin and
VEGF-C proteins was inhibited in 5637 cells where HMGN5 was knocked
down and treated with CDDP, indicating the involvement of PI3K/Akt
signaling, EMT and lymphangiogenesis. As reported, activation of
cell apoptosis also serves as a major mechanism underlying the
anticancer effects of CDDP (18,32). In
the present study, the results from flow cytometric analysis and
Hoechst 33342 staining demonstrated that cell apoptosis was
enhanced in 5637 cells where HMGN5 was depleted compared to cells
transfected with the shRNA control vector.
Cell apoptosis is executed by members of the caspase
family, which can be activated by the mitochondrial apoptotic
pathway (33). Inactivation of
PI3K/Akt signaling leads to the release of cytochrome c from the
mitochondria to the cytoplasm, which leads to the activation of
caspase-3 and PARP, and subsequently triggers apoptosis (34). In the present study, increased
expression of cytochrome c, cleaved-caspase-3 and cleaved-PARP was
observed in 5637 cells where HMGN5 was knocked down and treated
with CDDP, which confirms the activation of the mitochondrial
apoptotic pathway. The changes caused by the inhibition of HMGN5 in
5637 cells were able to be reversed by treatment with the PI3K/Akt
signaling activator IGF-1, indicating a mechanism in which HMGN5
positively regulates the activity of the PI3K/Akt signaling
pathway.
Notably, CDDP was previously observed to impair the
expression of HMGN5 and therefore able to affect its downstream
signal molecules. A potential explanation may be that HMGN5 can
serve as a molecular target for CDDP, and therefore CDDP is able to
directly inhibit HMGN5 in UBC cells (29). Another explanation may be that CDDP is
able to kill the UBC cells, which then leads to a decrease in HMGN5
expression. However, the mechanism remains unclear and required to
be further investigated.
Although certain limitations were inevitable,
including the lack of in vivo experiments and the
experimental data from HMGN5-overexpressing bladder cancer cells, a
number of primary findings were reported. Based on results of the
present study, it was concluded that HMGN5 regulates the PI3K/Akt
signaling pathway in UBC cell lines and determines the response of
UBC cell lines to CDDP. Through inhibiting PI3K/Akt signaling,
knock down of HMGN5 increases the chemosensitivity of UBC cells to
CDDP. Therefore, HMGN5 may be a promising therapeutic target for
the treatment of UBC and hinder its progression.
Acknowledgements
The present study was supported by the Hunan
Provincial Natural Science Foundation of China (grant no. 14JJ3044)
and the China Scholarship Council (grant no. 201606370204).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar
|
|
2
|
Burger M, Catto JW, Dalbagni G, Grossman
HB, Herr H, Karakiewicz P, Kassouf W, Kiemeney LA, La Vecchia C,
Shariat S, et al: Epidemiology and risk factors of urothelial
bladder cancer. Eur Urol. 63:234–241. 2013. View Article : Google Scholar
|
|
3
|
Kaufman DS, Shipley WU and Feldman AS:
Bladder cancer. Lancet. 374:239–249. 2009. View Article : Google Scholar
|
|
4
|
Jacobs BL, Lee CT and Montie JE: Bladder
cancer in 2010: How far have we come? CA Cancer J Clin. 60:244–272.
2010. View Article : Google Scholar
|
|
5
|
von der Maase H, Sengelov L, Roberts JT,
Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A and Arning
M: Long-term survival results of a randomized trial comparing
gemcitabine plus cisplatin, with methotrexate, vinblastine,
doxorubicin, plus cisplatin in patients with bladder cancer. J Clin
Oncol. 23:4602–4608. 2005. View Article : Google Scholar
|
|
6
|
Vaishampayan U: Systemic therapy of
advanced urothelial cancer. Curr Treat Options Oncol. 10:256–266.
2009. View Article : Google Scholar
|
|
7
|
King LM and Francomano CA:
Characterization of a human gene encoding nucleosomal binding
protein NSBP1. Genomics. 71:163–173. 2001. View Article : Google Scholar
|
|
8
|
Rochman M, Malicet C and Bustin M:
HMGN5/NSBP1: A new member of the HMGN protein family that affects
chromatin structure and function. Biochim Biophys Acta. 1799:86–92.
2010. View Article : Google Scholar
|
|
9
|
Shi Z, Tang R, Wu D and Sun X: Research
advances in HMGN5 and cancer. Tumour Biol. 37:1531–1539. 2016.
View Article : Google Scholar
|
|
10
|
Gan Y, Tan J, Yang J, Zhou Y, Dai Y, He L,
Yao K and Tang Y: Knockdown of HMGN5 suppresses the viability and
invasion of human urothelial bladder cancer 5637 cells in vitro and
in vivo. Med Oncol. 32:1362015. View Article : Google Scholar
|
|
11
|
Yao K, He L, Gan Y, Zeng Q, Dai Y and Tan
J: MiR-186 suppresses the growth and metastasis of bladder cancer
by targeting NSBP1. Diagn Pathol. 10:1462015. View Article : Google Scholar
|
|
12
|
Haslehurst AM, Koti M, Dharsee M, Nuin P,
Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, et al: EMT
transcription factors snail and slug directly contribute to
cisplatin resistance in ovarian cancer. BMC Cancer. 12:912012.
View Article : Google Scholar
|
|
13
|
Zhao J, Dong D and Sun L, Zhang G and Sun
L: Prognostic significance of the epithelial-to-mesenchymal
transition markers e-cadherin, vimentin and twist in bladder
cancer. Int Braz J Urol. 40:179–189. 2014. View Article : Google Scholar
|
|
14
|
Zhu H, Yun F, Shi X and Wang D: VEGF-C
inhibition reverses resistance of bladder cancer cells to cisplatin
via upregulating maspin. Mol Med Rep. 12:3163–3169. 2015.
View Article : Google Scholar
|
|
15
|
Gan Y, Wang Y, Tan Z, Zhou J, Kitazawa R,
Jiang X, Tang Y and Yang J: TDRG1 regulates chemosensitivity of
seminoma TCam-2 cells to cisplatin via PI3K/Akt/mTOR signaling
pathway and mitochondria-mediated apoptotic pathway. Cancer Biol
Ther. 17:741–750. 2016. View Article : Google Scholar
|
|
16
|
West KA, Castillo SS and Dennis PA:
Activation of the PI3K/Akt pathway and chemotherapeutic resistance.
Drug Resist Updat. 5:234–248. 2002. View Article : Google Scholar
|
|
17
|
Zhou X, Yuan B, Yuan W, Wang C, Gao R and
Wang J: The expression and clinical significance of high mobility
group nucleosome binding domain 5 in human osteosarcoma. Tumour
Biol. 35:6539–6547. 2014. View Article : Google Scholar
|
|
18
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar
|
|
19
|
Liang W, Hao Z, Han JL, Zhu DJ, Jin ZF and
Xie WL: CAV-1 contributes to bladder cancer progression by inducing
epithelial-to-mesenchymal transition. Urol Oncol. 32:855–863. 2014.
View Article : Google Scholar
|
|
20
|
Wissmann C and Detmar M: Pathways
targeting tumor lymphangiogenesis. Clin Cancer Res. 12:6865–6868.
2006. View Article : Google Scholar
|
|
21
|
Chen H, Guan R, Lei Y, Chen J, Ge Q, Zhang
X, Dou R, Chen H, Liu H, Qi X, et al: Lymphangiogenesis in gastric
cancer regulated through Akt/mTOR-VEGF-C/VEGF-D axis. BMC Cancer.
15:1032015. View Article : Google Scholar
|
|
22
|
Gerlitz G: HMGNs, DNA repair and cancer.
Biochim Biophys Acta. 1799:80–85. 2010. View Article : Google Scholar
|
|
23
|
Rochman M, Postnikov Y, Correll S, Malicet
C, Wincovitch S, Karpova TS, McNally JG, Wu X, Bubunenko NA,
Grigoryev S, et al: The interaction of NSBP1/HMGN5 with nucleosomes
in euchromatin counteracts linker histone-mediated chromatin
compaction and modulates transcription. Mol Cell. 35:642–656. 2009.
View Article : Google Scholar
|
|
24
|
Wahafu W, He ZS, Zhang XY, Zhang CJ, Yao
K, Hao H, Song G, He Q, Li XS and Zhou LQ: The nucleosome binding
protein NSBP1 is highly expressed in human bladder cancer and
promotes the proliferation and invasion of bladder cancer cells.
Tumour Biol. 32:931–939. 2011. View Article : Google Scholar
|
|
25
|
Burgering BM and Coffer PJ: Protein kinase
B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction.
Nature. 376:599–602. 1995. View
Article : Google Scholar
|
|
26
|
Nordentoft I, Dyrskjot L, Bodker JS, Wild
PJ, Hartmann A, Bertz S, Lehmann J, Orntoft TF and
Birkenkamp-Demtroder K: Increased expression of transcription
factor TFAP2alpha correlates with chemosensitivity in advanced
bladder cancer. BMC Cancer. 11:1352011. View Article : Google Scholar
|
|
27
|
Yang L, Shi T, Liu F, Ren C, Wang Z, Li Y,
Tu X, Yang G and Cheng X: REV3L, a promising target in regulating
the chemosensitivity of cervical cancer cells. PloS One.
10:e01203342015. View Article : Google Scholar
|
|
28
|
He J, Liu C, Wang B, Li N, Zuo G and Gao
D: HMGN5 blockade by siRNA enhances apoptosis, suppresses invasion
and increases chemosensitivity to temozolomide in meningiomas. Int
J Oncol. 47:1503–1511. 2015. View Article : Google Scholar
|
|
29
|
Guo Z, Zhang X, Li X, Xie F, Su B, Zhang M
and Zhou L: Expression of oncogenic HMGN5 increases the sensitivity
of prostate cancer cells to gemcitabine. Oncol Rep. 33:1519–1525.
2015. View Article : Google Scholar
|
|
30
|
Zhang HY, Zhang PN and Sun H: Aberration
of the PI3K/AKT/mTOR signaling in epithelial ovarian cancer and its
implication in cisplatin-based chemotherapy. Eur J Obstet Gynecol
Reprod Biol. 146:81–86. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun D, Sawada A, Nakashima M, Kobayashi T,
Ogawa O and Matsui Y: MK2206 potentiates cisplatin-induced
cytotoxicity and apoptosis through an interaction of inactivated
Akt signaling pathway. Urol Oncol. 33:111.e17–111.e26. 2015.
View Article : Google Scholar
|
|
32
|
Chang F, Lee JT, Navolanic PM, Steelman
LS, Shelton JG, Blalock WL, Franklin RA and McCubrey JA:
Involvement of PI3K/Akt pathway in cell cycle progression,
apoptosis, and neoplastic transformation: A target for cancer
chemotherapy. Leukemia. 17:590–603. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nicholson DW and Thornberry NA: Apoptosis.
Life and death decisions. Science. 299:214–215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Estaquier J, Vallette F, Vayssiere JL and
Mignotte B: The mitochondrial pathways of apoptosis. Adv Exp Med
Biol. 942:157–183. 2012. View Article : Google Scholar : PubMed/NCBI
|