Introduction
Aurora kinases (AURKs) are members of the mitotic
serine/threonine kinase family, which includes AURK A, B, and C.
Among these kinases, AURKA is frequently amplified in various
cancer cell types and can act as an oncogene (1). AURKA performs an essential role in
centrosome maturation, chromosome alignment and proper formation of
the mitotic spindle (2,3). Inhibition of AURKA has been demonstrated
to arrest the cell cycle at the G2/M phase and suppress
tumor growth in vivo (4–7). In
prostate cancer cells, knockdown of AURKA expression by shRNA
induces apoptosis through an upregulation of tumor protein p53 and
its downstream target apoptosis regulator Bax (Bax) (8). However, the specific mechanism
underlying the induction of apoptosis via AURKA inhibition remains
unclear.
The endoplasmic reticulum (ER) is an organelle that
has numerous cellular functions, including protein folding,
post-translational modifications and trafficking. Cellular
conditions associated with the accumulation of misfolded or
unfolded proteins in the lumen of the ER induce a series of ER
stress responses, known as the unfolded protein response (UPR)
(9). Thus, this allows for
restoration of protein folding homeostasis by inhibiting further
protein translation, degrading misfolded proteins and increasing
the synthesis of molecular chaperones to augment ER folding
capacity (9). The UPR signaling
pathway is mediated by ER transmembrane proteins, including
double-stranded RNA-activated protein kinase-like ER kinase,
inositol-requiring enzyme 1 (IRE1), and activating transcription
factor 6 (9). However, if the ER
stress is too severe and prolonged for the cell to overcome, the
UPR signaling pathway can activate the caspase cascade and thus
promote the cell death program through apoptosis (10).
The aim of the present study was to investigate the
effects of TCS7010, a specific inhibitor of AURKA, on the induction
of UPR-mediated apoptosis. The results demonstrated that TCS7010
produces reactive oxygen species (ROS) and triggers apoptosis
through ROS-dependent UPR-mediated pathway.
Materials and methods
Cell culture and chemicals
The human colon carcinoma HCT116 cell line was
obtained from the American Type Culture Collection (Manassas, VA,
USA). Cells were grown in Dulbecco's modified Eagle's medium
supplemented with 10% (v/v) heat-inactivated fetal bovine serum
(Cellgro; Corning Incorporated, Corning, NY, USA) at 37°C in a
humidified incubator with an atmosphere containing 5%
CO2. The cells were seeded onto 60-mm culture dishes at
a density of 4×105 cells/dish and were further cultured
until the cells reached up to a subconfluent monolayer (70–80%
confluency). The cells were treated with the stated concentrations
of TSC7010 (Tocris Bioscience, Bristol, UK).
Immunoblot analysis
Cells were harvested and lysed for 30 min at 4°C in
a buffer consisting of 20 mM HEPES (pH 7.2), 1% Triton X-100, 10%
glycerol, 150 mM NaCl, 10 µg/ml leupeptin and 1 mM
phenylmethylsulfonyl fluoride. Protein concentrations were measured
with a Pierce™ Bicinchoninic Protein Assay Kit (#23225; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). The protein extracts
(20 µg each) were separated on a 10% SDS-PAGE gel and transferred
to a nitrocellulose membrane. Following protein transfer, membranes
were washed in Tris Buffered Saline with Tween 20 (TBST; Cell
Signaling Technology, Inc., Danvers, MA, USA) 3 times for 5 min
each at room temperature, followed by blocking in 5% non-fat milk
for 30 min at room temperature. The blots were incubated with
primary antibodies for 2 h at room temperature. Antibodies against
phospho-Aurora A(Thr288)/B(Thr232)/C(Thr198) (#2914; 1:1,000
dilution), Cleaved caspase-7 (#9491; 1:1,000 dilution), IRE1
(#3294; 1:2,000 dilution), CHOP (#2895; 1:1,000 dilution), Bim
(#2819; 1:1,000 dilution), Caspase-2 (#611022; 1:1,000 dilution)
were purchased from Cell Signaling Technology, Inc. Antibodies
against GAPDH (#sc-32233; 1:5,000 dilution), PARP (#sc-7150;
1:4,000 dilution) were obtained from Santa Cruz Biotechnology,
Inc., (Dallas, TX, USA) and antibody against Caspase-2 (#611022;
1:1,000 dilution) was from BD Biosciences (San Jose, CA, USA).
Following washing 3 times for 5 min each with TBST at room
temperature, horseradish peroxidase-conjugated secondary antibody
was incubated for 1 h at room temperature. Secondary antibody to
anti-mouse IgG (Goat; #BR170-6516; 1:2,000 dilution) and
anti-rabbit IgG (Goat; #BR170-6515; 1:2,000 dilution) were
purchased from Bio-Rad Laboratories, Inc., (Hercules, CA, USA).
Following incubation with secondary antibodies, membranes were
washed 5 times with TBST for 5 min each at room temperature and the
immunoreactivity of protein bands was visualized using an enhanced
chemiluminescence detection system (GE Healthcare Life Sciences,
Chalfont, UK). Each blot is representative of ≥3 separate
experiments.
Cell viability assay
Cell viability was determined using a Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Rockville, MD,
USA), according to the manufacturer's protocol. Briefly,
exponentially growing cells were exposed to 5 or 10 µM TCS7010 for
24 h at 37°C followed by the addition of the CCK-8 solution for an
additional 1 h at 37°C. The absorbance was measured at 450 nm using
an Emax Endpoint ELISA microplate reader (Molecular Devices, LLC,
Sunnyvale, CA, USA). Data were presented as the mean ± standard
deviation of 2 independent experiments performed in triplicate.
Cell cycle analysis
Cellular DNA content was analyzed using flow
cytometry, as previously described (11). Briefly, HCT116 cells were collected
following 18 and 24 h of exposure to 5 µM TCS7010, fixed in 70%
ethanol, and washed twice with phosphate-buffered saline.
Subsequently, cells were stained with a 50 µg/ml propidium iodide
(PI) solution containing 0.1% Triton X-100, 0.1 mM
ethylenediaminetetraacetic acid and 50 µg/ml RNase A. Fluorescence
was measured and analyzed using a NucleoCounter NC-3000 instrument
(ChemoMetec, Allerød, Denmark). Data are representative of ≥3
separate experiments.
Quantification of apoptotic cells
using annexin V staining
HCT116 cells (1×106 cells/sample) were
treated with 5 µM TCS7010 for 24 h at 37°C. The cells were fixed
with 4% paraformaldehyde, followed by incubation with fluorescein
isothiocyanate (FITC)-conjugated annexin V, according to the
manufacturer's protocol (ChemoMetec). PI was used as a counterstain
for dead cells. The fluorescence intensities of FITC-annexin
V-positive cells (green) and PI-positive cells (red) were analyzed
with a NucleoCounter NC-3000 instrument as described previously
(12). Data are representative of ≥3
separate experiments.
Detection of intracellular ROS
levels
Cells were trypsinized and incubated with 10 µM
H2-2′, 7′-dichlorofluorescin diacetate (DCF-DA) (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) for 60 min, followed by incubation
with 5 µM TCS7010 for 3, 6, or 12 h. The fluorescence intensity was
analyzed using a FACSCalibur flow cytometer (BD Biosciences), as
described previously (11). CellQuest
Pro software version 5.2 (BD Biosciences) was used for acquisition
and analysis of the data. Data are representative of ≥3 separate
experiments.
Analysis of X-box binding protein 1
(XBP-1) splicing
Total RNA was isolated using the
phenol/guanidine-based Isol-RNA Lysis reagent (5 PRIME GmbH,
Hamburg, Germany), and the synthesis of cDNA was performed using an
iScript cDNA Synthesis kit (Bio-Rad, Inc.). Reverse
transcription-polymerase chain reaction (RT-PCR) was performed
using XBP-1-specific primers (forward, 5′-AGAGTAGCAGCTCAGACTGC-3′
and reverse, 5′-CATTAATGGCTTCCAGCTTG-3′) that correspond to nt
277–817, encompassing the IRE-1 cleavage site, as previously
described (12). PCR products were
further digested for 2 h at 37°C using PstI, whose
recognition site is located at nt 556 within the 26-nt intron (nt
531–556) that is lost following IRE-1-mediated splicing (13). The PCR product of unspliced XBP-1
(uXBP1) mRNA is expected to be cleaved into two fragments of 279 bp
and 261 bp following PstI digestion, while that of spliced
XBP-1 (sXBP1) mRNA is resistant to PstI digestion, producing
a 540-bp band (11). Data are
representative of ≥3 separate experiments.
Statistical analysis
Statistical analysis was performed using one-way
analysis of variance followed by Sidak's multiple comparisons test
using GraphPad Prism software (version 7.0; GraphPad Software Inc.,
La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results and discussion
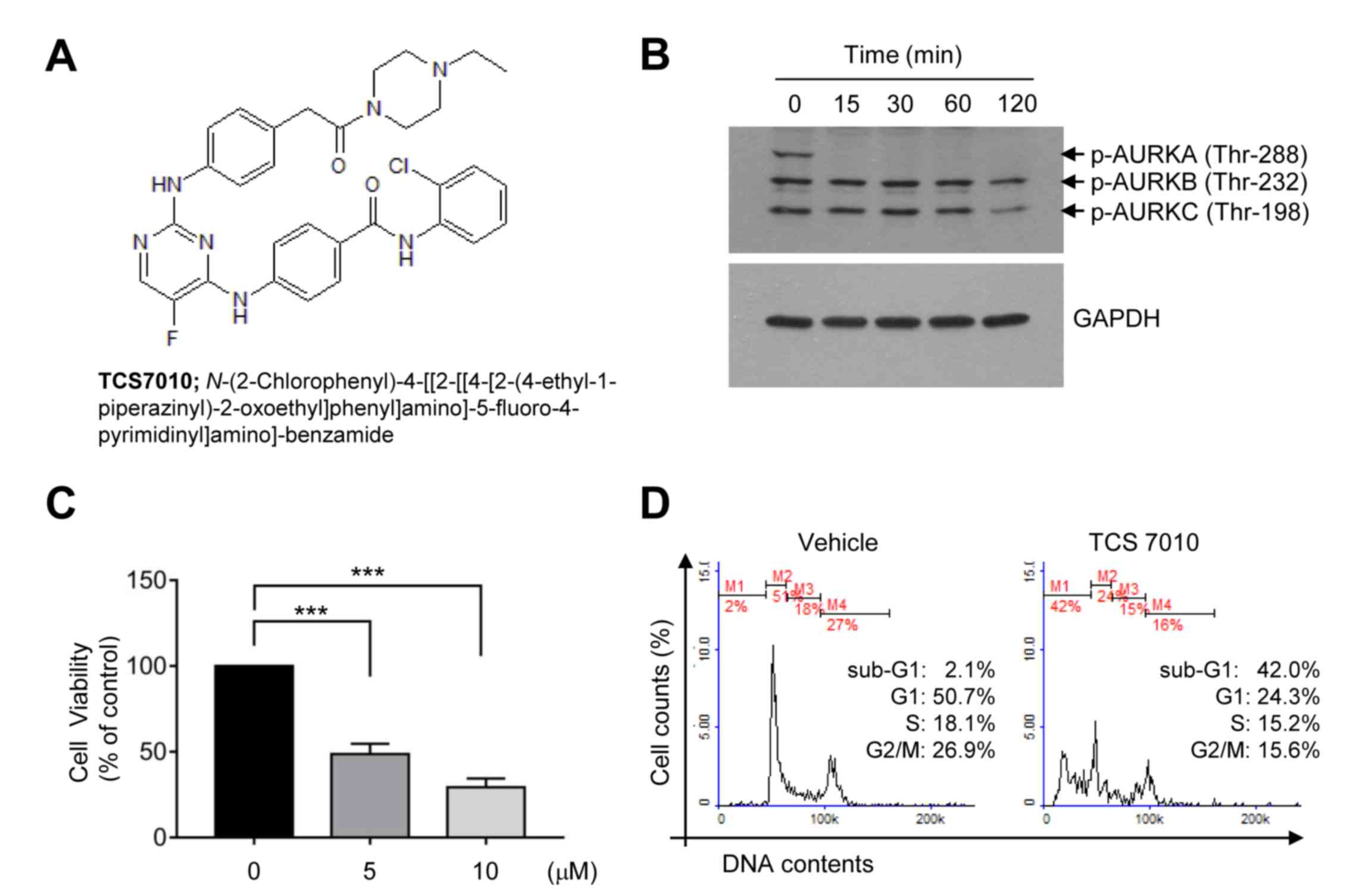
Effect of TCS7010 on the
phosphorylation of AURKA in HCT116 cells
AURKA is overexpressed in a broad range of tumor
cells (14). Immunoblot analysis was
performed to evaluate whether TCS7010 inhibits AURKA in HCT116
colon cancer cells. The phosphorylation of all AURK subtypes (A, B
and C) was highly elevated at the basal status (Fig. 1B). It was demonstrated that TCS7010
selectively inhibited AURKA phosphorylation on threonine-288 within
15 min of treatment (Fig. 1B).
Effect of TCS7010 on the cell
viability and cell cycle progression of HCT116 cells
To assess the cytotoxic effect of TCS7010,
exponentially growing HCT116 cells were exposed to different
TCS7010 concentrations for 24 h, and cell viability was measured
using CCK-8. Treatment with TCS7010 significantly reduced HCT116
cell viability in a dose-dependent manner (Fig. 1C). The effective dose causing 50% of
maximal cytotoxicity (ED50) was ~5 µM. To investigate
the mechanism underlying TCS7010-induced cytotoxicity, the effect
of TCS7010 on cell cycle progression was evaluated. HCT116 cells
were treated with 5 µM TCS7010 for 24 h and cell cycle progression
was monitored using flow cytometry. The number of G1
phase cells decreased (50.7 to 24.3%), with a concomitant increase
in the number of sub-G1 phase cells (2.10 to 42.0%),
which is indicative of a hypodiploid DNA cell population undergoing
apoptosis (Fig. 1D). These results
suggest that TCS7010 has a cytotoxic effect against HCT116 colon
cancer cells, possibly via the induction of apoptosis.
Effect of TCS7010 on the induction of
apoptosis
To determine whether TCS7010 triggers apoptosis,
annexin V staining was performed. In the early stages of apoptosis,
phosphatidylserine (PS) in the inner side of the plasma membrane
translocates to the outer surface. As annexin V preferentially
binds to PS (15), detection of PS
using annexin V is widely used to identify apoptotic cells in
response to cytotoxic agents. PI, which cannot enter live cells,
was used to counterstain dead cells. HCT116 cells were treated with
5 µM TCS7010 for 24 h and then incubated with FITC-conjugated
annexin V and PI. In Fig. 2A, the
lower left quadrant of each cytogram (PI and FITC double-negative)
represents viable cells, the lower right quadrant (PI negative/FITC
positive) represents early apoptotic cells, and the upper right
quadrant (PI and FITC double-positive) represents late apoptotic
and dead cells. The PI-negative/FITC-positive population was
increased in the cells treated with TCS7010 compared with the
untreated control group (16 vs. 48%) following, suggesting that
TCS7010 triggers apoptosis in HCT116 cells.
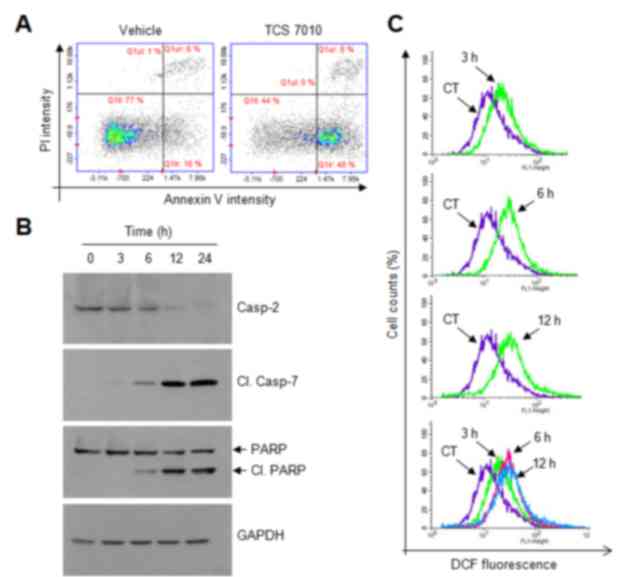 | Figure 2.Effect of TCS7010 on the activation of
caspases and produces reactive oxygen species. (A) Annexin V
staining assay in which HCT116 cells were treated with vehicle
(DMSO) or 5 µM TCS7010 for 24 h and then stained with FITC-annexin
V and PI. Cells were harvested, washed and analyzed for
fluorescence intensity. Scatter plots demonstrating the intensity
of FITC-annexin V vs. that of PI. x-axis, Annexin V intensity;
y-axis, PI intensity. (B) HCT116 cells were treated with 5 µM
TCS7010 for various time periods (0–24 h). Whole cell lysates were
prepared and analyzed using immunoblotting. GAPDH was used as an
internal control. (C) HCT116 cells were incubated with 10 µM DCF-DA
for 1 h, followed by the addition of 5 µM TCS7010 for 3, 6, and 12
h. CT, control treated with DCF-DA alone. Fluorescence was assessed
using a FACSCalibur instrument. PI, propidium iodide; DCF-DA, H2-2′
7′-dichlorofluorescin diacetate; casp, caspase; Cl., cleaved; PARP,
poly(ADP-ribose) polymerase. |
Caspases are a family of cysteine proteases that
have an essential role in the progression of apoptosis (16). The effect of TCS7010 on the induction
of caspase activation was determined in the present study. Upon
TCS7010 treatment, the pro-caspase-2 level gradually decreased in a
time-dependent manner and the amount of cleaved active caspase-7,
and cleaved poly (ADP-ribose) polymerase (PARP), a well-known
substrate for caspase-7, increased in a time-dependent manner
(Fig. 2B). These data suggest that
caspase activation is involved in TCS7010-induced apoptosis.
Effect of TCS7010 on the production of
ROS
ROS are chemically reactive molecules containing
oxygen, including superoxide anion radical, hydrogen peroxide and
the highly reactive hydroxyl radical, all of which are formed as a
byproduct of normal cellular metabolism. Abnormally excessive ROS
levels may result in cellular damage, known as oxidative stress.
Thus, under normal physiological conditions, various antioxidant
enzymes, including catalase and superoxide dismutase maintain the
proper cellular redox states. However, there is growing evidence
for the beneficial effects of ROS on chemotherapy (17,18). As
numerous cancer cells are more sensitive to ROS compared with
normal cells, ROS production could be used for the selective
targeting of cancer cells (12,19–21).
As ROS activates the caspase signaling pathway
(22), ROS production levels were
analyzed using a fluorescent DCF-DA probe. Upon TCS7010 treatment,
ROS accumulation was detectable at 3 h and maintained up to 12 h
following TCS7010 treatment (Fig.
2C). These data suggested that ROS may act as an upstream
signal in TCS7010-induced apoptosis.
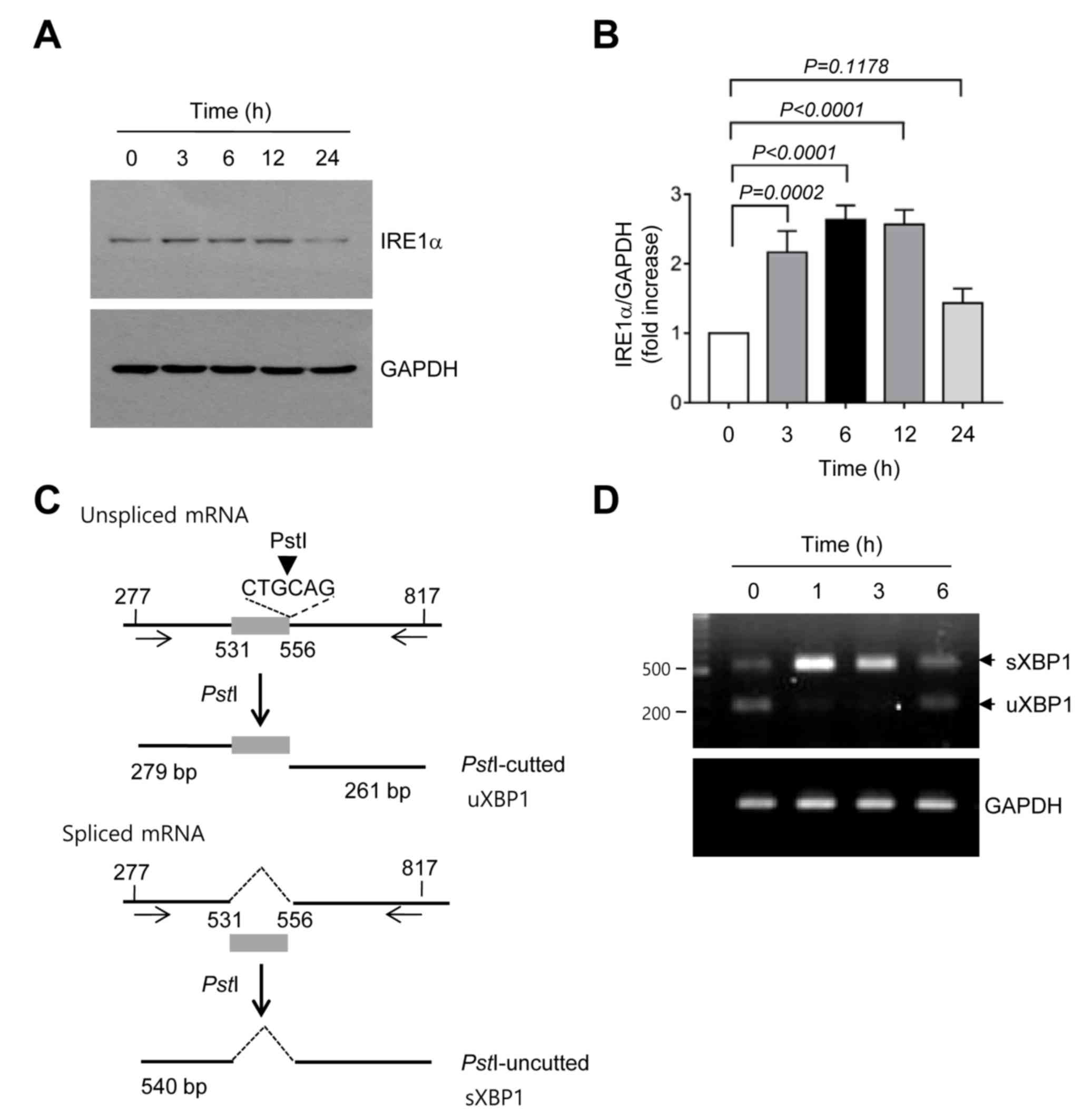
Effect of TCS7010 on the activation of
the UPR signaling pathway
ROS can induce ER stress (23), and ER stress can trigger apoptosis
through the UPR signaling pathway (24,25). Thus,
whether TCS7010 activates the UPR signaling pathway was determined
in the present study. A typical feature of the ER stress response
is the accumulation of UPR sensor proteins. It was demonstrated
that the amount of IRE1α protein significantly increased within 3 h
compared with no treatment and then returned to the basal level at
24 h following TCS7010 treatment (3 h, P=0.0002; Sidak's multiple
comparisons test; n=3; Fig. 3A and
B). Activated IRE1 functions as a site-specific
endoribonuclease that catalyzes the excision of an unconventional
26-nt intron from uXBP1 to generate sXBP1 mRNA (26). To investigate whether TCS7010 induces
XBP1 mRNA splicing, RT-PCR was performed using primers that amplify
the region spanning the 26-nt intron, followed by digestion with
PstI. As the PstI restriction site is within the
intron site, the uXBP1 PCR product should be cleaved into two
fragments (279 and 261 bp) by PstI digestion, whereas the
sXBP1 PCR product lacking the PstI site should produce a
540-bp band (Fig. 3C). As illustrated
in Fig. 3D, TCS7010 caused the
accumulation of a shifted PCR band (540 bp) between 1 and 6 h,
reflecting the splicing of XBP1 mRNA. These data suggest that
TCS7010 activates the ER stress-induced UPR signaling pathway.
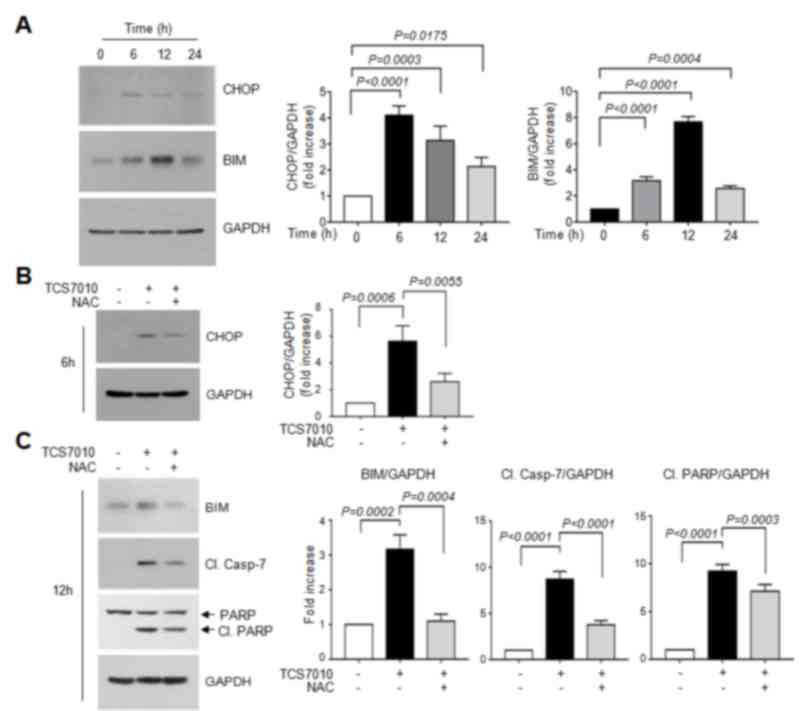
Effect of TCS7010 on the accumulation
of CHOP and BIM proteins
Accumulating evidence has demonstrated that CHOP
performs an essential role in ER-stress-induced apoptosis via
upregulation of the pro-apoptotic protein BIM (24,25,27,28).
BIM is a Bcl-2 homology domain 3 (BH3)-containing pro-apoptotic
Bcl-2 family member. In general, BIM cooperates with other BH3-only
proteins, including truncated BH3 interacting domain death agonist
and BAX, to trigger mitochondria-mediated apoptosis (29). To determine whether CHOP expression is
upregulated by TCS7010, HCT116 cells were treated with TCS7010 for
different periods of time. Immunoblot analysis demonstrated that
CHOP protein expression was maximal at 6 h following TCS7010
treatment and were maintained up to 24 h (Fig. 4A). The amount of BIM protein, a CHOP
target, peaked at 12 h and then decreased (Fig. 4A). Densitometric analysis revealed
that the increases of CHOP and BIM levels were significant (all
P<0.0001; Sidak's multiple comparisons test; n=3) upon TCS7010
treatment. These results suggest that the UPR signaling pathway may
be involved in TCS7010-induced apoptosis.
Effect of N-acetylcystein (NAC) on
TCS7010-induced CHOP accumulation
The association between ROS accumulation by TCS7010
and the UPR response was determined. Fig.
4B demonstrates that pretreatment of HCT116 cells with 2 mM
NAC, a thiol-containing ROS scavenger, significantly abrogated
TCS7010-induced CHOP accumulation compared with cells treated with
TCS7010 alone (P=0.0055; Sidak's multiple comparisons test; n=3).
Furthermore, pretreatment with NAC significantly reduced the
TCS7010-induced accumulation of BIM and cleaved caspase-7 compared
with cells treated with TCS7010 alone (P=0.0004 and P<0.0001,
respectively; Sidak's multiple comparisons test; n=3; Fig. 4C). In addition, the TCS7010-induced
cleavage of PARP was significantly reduced by pretreatment with NAC
compared with cells treated with TCS7010 alone (P=0.0003; Sidak's
multiple comparisons test; n=3). These results suggest that ROS
production by TCS7010 is upstream of UPR-mediated apoptosis.
In conclusion, the results of the present study
demonstrated that TCS7010 induces ROS-mediated apoptosis.
Upregulation of BIM through the UPR signaling pathway may have an
important role in TCS7010-induced apoptosis. Currently, the
mechanism underlying AURKA regulation of the UPR signaling pathway
remains unclear. Additional studies are required to investigate the
role of AURKA in the control of ER stress responses.
Acknowledgements
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Science and Future Planning (grant no.
2016R1A2B4008570). This paper was supported by the KU Research
Professor Program of Konkuk University.
References
|
1
|
Mountzios G, Terpos E and Dimopoulos MA:
Aurora kinases as targets for cancer therapy. Cancer Treat Rev.
34:175–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fu J, Bian M, Jiang Q and Zhang C: Roles
of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res.
5:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Barr AR and Gergely F: Aurora-A: The maker
and breaker of spindle poles. J Cell Sci. 120:2987–2996. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qi W, Cooke LS, Liu X, Rimsza L, Roe DJ,
Manziolli A, Persky DO, Miller TP and Mahadevan D: Aurora inhibitor
MLN8237 in combination with docetaxel enhances apoptosis and
anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol.
81:881–890. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harrington EA, Bebbington D, Moore J,
Rasmussen RK, Ajose-Adeogun AO, Nakayama T, Graham JA, Demur C,
Hercend T, Diu-Hercend A, et al: VX-680, a potent and selective
small-molecule inhibitor of the Aurora kinases, suppresses tumor
growth in vivo. Nat Med. 10:262–267. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Keen N and Taylor S: Aurora-kinase
inhibitors as anticancer agents. Nat Rev Cancer. 4:927–936. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Katayama H, Zhou H, Li Q, Tatsuka M and
Sen S: Interaction and feedback regulation between
STK15/BTAK/Aurora-A kinase and protein phosphatase 1 through
mitotic cell division cycle. J Biol Chem. 276:46219–46224. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He W, Zhang MG, Wang XJ, Zhong S, Shao Y,
Zhu Y and Shen ZJ: AURKA suppression induces DU145 apoptosis and
sensitizes DU145 to docetaxel treatment. Am J Transl Res.
5:359–367. 2013.PubMed/NCBI
|
|
9
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: Coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu J and Kaufman RJ: From acute ER stress
to physiological roles of the unfolded protein response. Cell Death
Differ. 13:374–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee da H, Jung Y Jung, Koh D, Lim Y, Lee
YH and Shin SY: A synthetic chalcone,
2′-hydroxy-2,3,5′-trimethoxychalcone triggers unfolded protein
response-mediated apoptosis in breast cancer cells. Cancer Lett.
372:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shin SY, Lee JM, Lee MS, Koh D, Jung H,
Lim Y and Lee YH: Targeting cancer cells via the reactive oxygen
species-mediated unfolded protein response with a novel synthetic
polyphenol conjugate. Clin Cancer Res. 20:4302–4313. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calfon M, Zeng H, Urano F, Till JH,
Hubbard SR, Harding HP, Clark SG and Ron D: IRE1 couples
endoplasmic reticulum load to secretory capacity by processing the
XBP-1 mRNA. Nature. 415:92–96. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang Y, Zhang Y, Lees E and Seghezzi W:
AuroraA overexpression overrides the mitotic spindle checkpoint
triggered by nocodazole, a microtubule destabilizer. Oncogene.
22:8293–8301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Andree HA, Reutelingsperger CP, Hauptmann
R, Hemker HC, Hermens WT and Willems GM: Binding of vascular
anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J
Biol Chem. 265:4923–4928. 1990.PubMed/NCBI
|
|
16
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 5:a0086562013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rigas B and Sun Y: Induction of oxidative
stress as a mechanism of action of chemopreventive agents against
cancer. Br J Cancer. 98:1157–1160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schumacker PT: Reactive oxygen species in
cancer cells: Live by the sword, die by the sword. Cancer Cell.
10:175–176. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raj L, Ide T, Gurkar AU, Foley M, Schenone
M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al:
Selective killing of cancer cells by a small molecule targeting the
stress response to ROS. Nature. 475:231–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adams DJ, Dai M, Pellegrino G, Wagner BK,
Stern AM, Shamji AF and Schreiber SL: Synthesis, cellular
evaluation, and mechanism of action of piperlongumine analogs. Proc
Natl Acad Sci USA. 109:pp. 15115–15120. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui X: Reactive oxygen species: The
Achilles' heel of cancer cells? Antioxid Redox Signal.
16:1212–1214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim BM, Rode AB, Han EJ, Hong IS and Hong
SH: 5-Phenylselenyl- and 5-methylselenyl-methyl-2′-deoxyuridine
induce oxidative stress, DNA damage, and caspase-2-dependent
apoptosis in cancer cells. Apoptosis. 17:200–216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kitamura M: The unfolded protein response
triggered by environmental factors. Semin Immunopathol. 35:259–275.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Puthalakath H, O'Reilly LA, Gunn P, Lee L,
Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin
J, Motoyama N, et al: ER stress triggers apoptosis by activating
BH3-only protein Bim. Cell. 129:1337–1349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sidrauski C and Walter P: The
transmembrane kinase Ire1p is a site-specific endonuclease that
initiates mRNA splicing in the unfolded protein response. Cell.
90:1031–1039. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Happo L, Strasser A and Cory S: BH3-only
proteins in apoptosis at a glance. J Cell Sci. 125:1081–1087. 2012.
View Article : Google Scholar : PubMed/NCBI
|