Introduction
Esophageal cancer is a type of malignant disease
with one of the highest causes of cancer-associated mortality in
developing countries (1).
Chemotherapy and radiotherapy are two indispensable methods for the
treatment of esophageal squamous cell carcinoma (ESCC),
particularly for locally advanced or unresectable cases; however,
therapeutic toxicity and resistance remain two challenges to
overcome. The five-year survival rate for advanced ESCC is ~10%
(2). Thus, the development of novel
therapeutic agents for the treatment of patients with this disease
is warranted.
Advances in understanding of the signaling pathways
involved in carcinogenesis, tumor growth and metastasis may aid in
identifying potential novel molecular targets for esophageal cancer
treatment (3). Angiogenesis serves an
essential role in cancer development and progression, and
determines the rate of tumor cell growth and invasion (4). Thus, targeting angiogenesis is being
considered as a novel therapeutic strategy to overcome tumor growth
(4). Endostar®, a
recombinant human-endostatin (rh-endostatin), is a novel
artificially-synthesized anti-angiogenesis drug, which was approved
by The China Food and Drug Administration (FDA) in 2005 for the
treatment of non-small cell lung cancer when combined with
cisplatin-based chemotherapy (5).
Previous studies have reported that Endostar improves the efficacy
of chemotherapy for esophageal cancer with synergistic effects when
combined with chemoradiotherapy (CRT), and that it is a good
prospect for clinical application (6,7).
Furthermore, it has been demonstrated that CRT combined with
endostatin markedly improves the complete response rate (endostatin
combined with CRT vs. CRT-alone, 44 vs. 30%), and increases the
1-year (72 vs. 50%) and 3-year (32 vs. 22%) overall survival rates
in metastatic ESCC when compared with the CRT-alone treatment group
(8). However, despite the success of
anti-angiogenic therapy regimens for advanced esophageal cancer,
the most advantageous use of this drug remains to be
identified.
Autophagy is a highly conserved catabolic process
that transports cellular macromolecules and organelles to the
lysosome for degradation in eukaryotic cells (9). It has been reported that autophagy is
upregulated in cancer cells in response to antitumor treatment,
including cisplatin, doxorubicin, sorafenib and cetuximab (10–13).
Autophagy is generally considered as a means by which carcinoma
cells develop resistance to therapy, thus the inhibition of
autophagy could represent a promising strategy to improve the
efficacy of cancer treatment.
The potential molecular mechanism underlying the
anti-angiogenic effect of Endostar involves the vascular
endothelial growth factor (VEGF)/VEGF receptor (R) signaling
pathway (14). The AKT
serine/threonine kinase (Akt)/mechanistic target of rapamycin
(mTOR) signaling pathway is one of the essential downstream
pathways of VEGF (15). As inhibition
of the Akt/mTOR signaling pathway is known to activate autophagy,
it is hypothesized that there is an association between autophagy
and Endostar treatment in ESCC therapy.
In the current study, the pro-survival role of
autophagy for ESCC cells was studied. Furthermore, the inhibiting
effects of autophagy on ESCC cells were discussed. The study
evaluated a novel and promising approach for enhancing the clinical
benefits of Endostar for the treatment of ESCC.
Materials and methods
Reagents and antibodies
Endostar was kindly donated by Xiansheng
Pharmaceutical Co., Ltd. (Nanjing China; http://www.simcere.com/). The autophagy inhibitor
chloroquine (CQ) was purchased from J&K Chemical Ltd. (Beijing,
China). The primary antibodies directed against
microtubule-associated protein 1 light chain 3-α (LC3), nucleoporin
p62 (p62), Beclin-1, Akt, phosphorylated Akt, mTOR and
phosphorylated mTOR were obtained from Cell Signaling Technology,
Inc., (Danvers, MA, USA, cat. nos. 4455, 4691, 13038, 2972 and
2971, respectively). Horseradish peroxidase-conjugated anti-rabbit
(cat. no. 2357) and anti-mouse (cat. no. 516190) secondary
antibodies were obtained from Santa Cruz Biotechnology, Inc.,
(Dallas, TX, USA), and a fluorescein isothiocyanate
(FITC)-conjugated anti-rabbit IgG was purchased from Beyotime
Institute of Biotechnology (Nanjing, China).
Cell cultures
Eca-109 and TE-1 cells were purchased from the Type
Culture Collection of the Chinese Academy of Sciences (Shanghai,
China). Cells were maintained in Dulbecco's modified Eagle medium
(DMEM) supplemented with 10% fetal bovine serum (both from Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and were
incubated at 37°C in a humidified incubator with 5% CO2.
Endostar was aliquoted to achieve desired working volumes of 20 µl
and stored at 4°C. The drug was further diluted with DMEM to 25,
50, 100 and 200 µg/ml prior to use.
Cell viability assays
The viability of Eca-109 and TE-1 cells was
determined using an MTS assay kit (Jiangsu Kaiji Bio-Technology
Co., Nanjing, China). Cells were seeded into 96-well plates at a
density of 3×103 cells/well in 100 µl DMEM (n=6) and
treated with Endostar (25, 50, 100 and 200 µg/ml), CQ (5 mM),
combination therapy or vehicle control (DMSO, 0.1%) for 24, 48, or
72 h when cells were in the logarithmic phase. After 24, 48 or 72 h
incubation at 37°C, 10 µl MTS was added into each well and
incubated for 4 h at 37°C. Subsequently, the absorbance was
measured using an ELX800 microplate reader (Omega Bio-Tek, Inc.,
Norcross, GA, USA) at a wave length of 490 nm and used to calculate
the cell viability rates. The half maximal inhibitory concentration
(IC50) for Endostar was calculated for esophageal cancer
cell lines using standard algorithms (16).
Transmission electron microscopy
(TEM)
For TEM, treated Eca-109 cells were washed with PBS
and fixed at room temperature for 30 min in 2.5% glutaraldehyde.
Then, the samples were treated with 1.5% osmium tetroxide at room
temperature for 30 min, dehydrated with acetone and embedded in
Durcupan resin. The sections were stained at room temperature for 2
h with 0.2% lead citrate and 1% uranyl acetate, and examined using
a Tecnai 10 electron microscope (FEI; Thermo Fisher Scientific,
Inc.; ×4,000-50,000) at 60 kV in five fields of view. For
quantification of autophagic vesicles, cells with >3-4
double-membrane vesicles were scored as positive for
autophagosomes.
Immunofluorescence
Cells seeded at a density of 3×105
cells/well into six-wells plates were treated with a specific dose
(25, 50, 100 and 200 µg/ml) of Endostar for 24 h at 37°C, then
incubated with LysoTracker (Invitrogen; Thermo Fisher Scientific,
Inc.,) for 90 min at 37°C. Subsequently, cells were washed twice
with PBS, fixed with 4% paraformaldehyde at 37°C for 2 h and
permeabilized with 1% CHAPS buffer (150 mM NaCl, 10 mM HEPES, 1.0%
CHAPS) at room temperature for 15 min. Then, cells were incubated
with an anti-LC3 antibody (dilution, 1:1,000; cat. no. 4455) for 2
h at 37°C, washed with PBS three times and incubated with
FITC-conjugated goat anti-rabbit IgG (dilution, 1:200; cat. no.
A0562) for 1 h at 37°C. Next, the cell nuclei were stained using
DAPI (Invitrogen; Thermo Fisher Scientific, Inc.) for 15 min at
37°C. Samples were examined under a Zeiss LSM 710 fluorescence
microscope system (Carl Zeiss AG, Oberkochen, Germany;
magnification, ×1,000). Images were processed using ZEN LE software
(version 2.0; Carl Zeiss AG).
For the quantification of LC3-positive cells,
150–200 cells were randomly selected from the acquired image and
counted. Cells with >5 dots of specific red signals were
considered to be autophagic (LC3-positive).
Western blot analysis
Cells were lysed by cell lysis buffer (Beyotime
Institute of Biotechnology, Nanjing, China; cat. no. P0013) and
immune blotted as previously described (15). Briefly, proteins from a total cell
extract (20 µg/lane) were separated using 10% SDS-PAGE, transferred
to a polyvinylidene difluoride membrane (EMD Millipore, Billerica,
MA, USA) and blocked with non fat milk powder (5%) for 2 h at room
temperature, then detected using the aforementioned primary
antibodies (dilution, 1:200, 4°C, overnight) and secondary
antibodies (dilution, 1:1,000, room temperature, 2 h), prior to
visualization using an enhanced chemiluminescence kit (Beyotime
Institute of Biotechnology). Visualization was performed using the
ImageQuant LAS-4000 imager with Multi Gauge software (version 1.03;
both from Fujifilm, Tokyo, Japan).
Cell apoptosis assay
Cells weretreated with Endostar (25 µg/ml), CQ (5
mM) or vehicle control (DMSO, 0.1%) for 24 h when cells were in the
logarithmic phase. After 24 h, cells were collected by
trypsinization, washed twice with cold PBS and resuspended in 1X
binding buffer. The final concentration of cells was
1×106/ml. Then, 100 µl cells were mixed with 5 µl
Annexin V/FITC and 5 µl propidium iodide, then gently vortexed and
incubated for 15 min at room temperature in the dark according to
the manufacturer's protocol (Nanjing KeyGen Biotech Co., Ltd.,
Nanjing, China). Cell apoptosis was determined using flow
cytometric analysis with the Annexin V/FITC Apoptosis Detection kit
(Nanjing KeyGen Biotech Co., Ltd.). The samples were then detected
by flow cytometric analysis within 1 h. The results were analyzed
using the BD FACSCalibur™ system and FACStation™
software version 6.1 (BD Biosciences, Franklin Lakes, NJ, USA), as
suggested by the manufacturer.
Statistical analysis
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis. Data are presented as the mean ±
standard deviation. Comparisons between two groups were performed
by unpaired t-test. Multiple comparisons were performed by one-way
analysis of variance with Scheffe's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Endostar reduces the viability of
Eca-109 and TE-1 cells
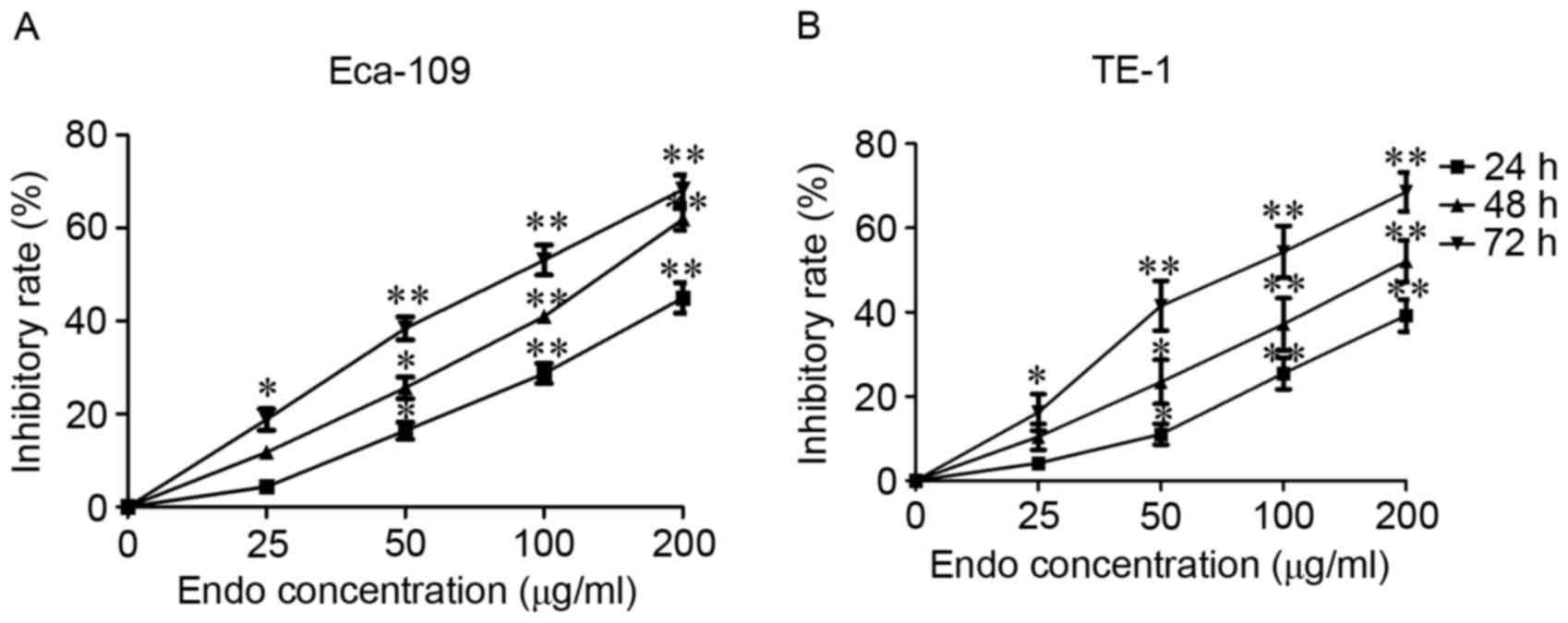
Human ESCC Eca-109 and TE-1 cells were selected to
evaluate the cytotoxic activity of Endostar. Cell viability was
measured using an MTS assay. Time- and dose-dependent inhibitory
effects on cell viability were observed following 72 h of Endostar
treatment in Eca-109 and TE-1 cells, and the IC50 was
calculated as being 67.23±8.42, and 75.39±11.56 µg/ml, respectively
(Fig. 1). These results suggest that
Endostar can significantly inhibit ESCC cell viability (Fig. 1).
Endostar induces autophagy in Eca-109
and TE-1 cells
To evaluate the significance of autophagy in
esophageal cancer therapy, the occurrence of autophagy in
Endostar-treated human ESCC Eca-109 and TE-1 cells was detected.
Autophagosomes were observed using TEM, a standard approach for
detecting autophagy. As presented in Fig.
2A and B, typical double-membrane autophagosomes emerged
following exposure to Endostar for 24 h in Eca-109 and in TE-1
cells. The control cells exhibited normal nuclei with a
significantly lower number of autophagosomes compared with in the
Endostar-treated group (Fig. 2A).
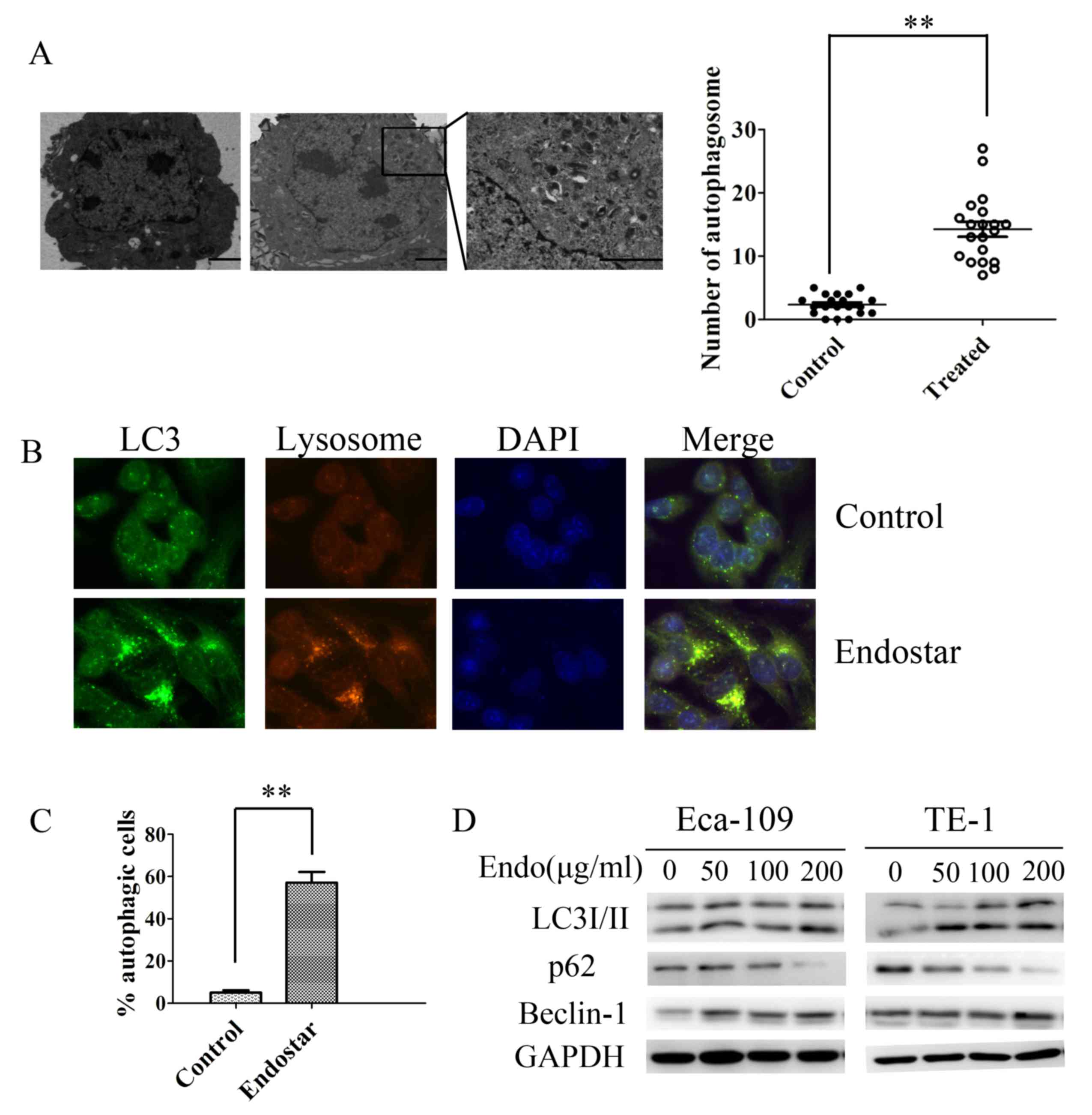 | Figure 2.Endostar promotes autophagy activation
in human esophageal cancer cells. (A) Representative transmission
electron microscopy images demonstrating autophagic vacuoles,
including autophagosomes, in Eca-109 cells treated with Endostar or
0.1% DMSO. Magnification, ×4,000 (×50,000 in the highlighted area).
(B) Representative immunofluorescence microscopy images of
endogenous punctae fluorescence of LC3 in Eca-109 cells after 24 h
treatment with Endostar. Magnification, ×1,000. Green, FITC-labeled
LC3; Red, lyso-tracker-labeled lysosome; Blue, DAPI-labeled
nucleus. (C) The percentage of green or yellow punctae-positive
cells was quantified and statistically analyzed using a threshold
of >5 dots/cell. Results are presented as the mean ± standard
deviation. (D) Western blot analysis indicating the transition of
LC3-I to LC3-II, p62 and Beclin-1 protein expression in Eca-109 and
TE-1 cells treated with Endostar (0–200 µg/ml). **P<0.01. LC3,
microtubule-associated protein 1 light chain 3 α; p62, nucleoporin
p62; Endo, Endostar; DMSO, dimethyl sulfoxide. |
These results were further confirmed by analyzing
the endogenous punctae fluorescence of LC3 in cells following
Endostar treatment using immunofluorescence staining. Eca-109 and
TE-1 cells demonstrated overlapping of LC3 punctae, and lysosomes
in the Endostar-treated group, indicating the formation of
autolysosomes (Fig. 2C). Autophagic
punctae were scarce in the untreated cell group compared with
Endostar-treated group (Fig. 2C).
Although increased autophagosome numbers and
autophagic punctae were observed following Endostar incubation, it
remains unclear whether these changes are the result of autophagy
activation or inhibition of the process at the late degradation
stage. Thus, the expression of autophagy-associated proteins LC3,
Beclin-1 and p62 were assessed using western blotting. LC3 is used
as a marker of autophagosomes and expressed by two forms: LC3-I and
LC3-II. The ratio of LC-II to LC3-I is increased when autophagy
activated. Beclin-1 performed an important role in the regulation
of autophagy activation. P62 was a selective substrate of
autophagy. P62 facilitated binding of LC3 to ubiquitinated proteins
and degraded ubiquitinated proteins in autolysosomes (9). The transition of LC3-I to LC3-II was
induced after 24 h of Endostar treatment in a dose-dependent manner
(Fig. 2D). Similarly, under various
doses of Endostar treatment (0–200 µg/ml), the expression of
Beclin-1 increased in Eca-109 and TE-1 cell lines in a
dose-dependent manner. In addition, when ESCC cells were treated
with Endostar the p62 protein expression was markedly inhibited,
which indicates that autophagy was activated by Endostar (Fig. 2D). These findings provide evidence
that Endostar favors autophagy activation in ESCC cells.
Endostar-induced autophagy is
regulated through suppression of the Akt/mTOR signaling
pathway
TheAkt/mTOR signaling pathway serves an essential
role in the regulation of autophagy. As a multi target inhibitor of
VEGF and platelet derived growth factor receptor family members,
linifanib-induced autophagy has been reported to be associated with
the inhibition of Akt/mTOR phosphorylation in hepatocellular
carcinoma (17). It has been
demonstrated that Endostar suppresses VEGF and its receptor, thus
inhibiting the downstream Akt/mTOR signaling pathway (18). However, the precise effect of Endostar
on the Akt/mTOR signaling pathway in ESCC cell lines has not been
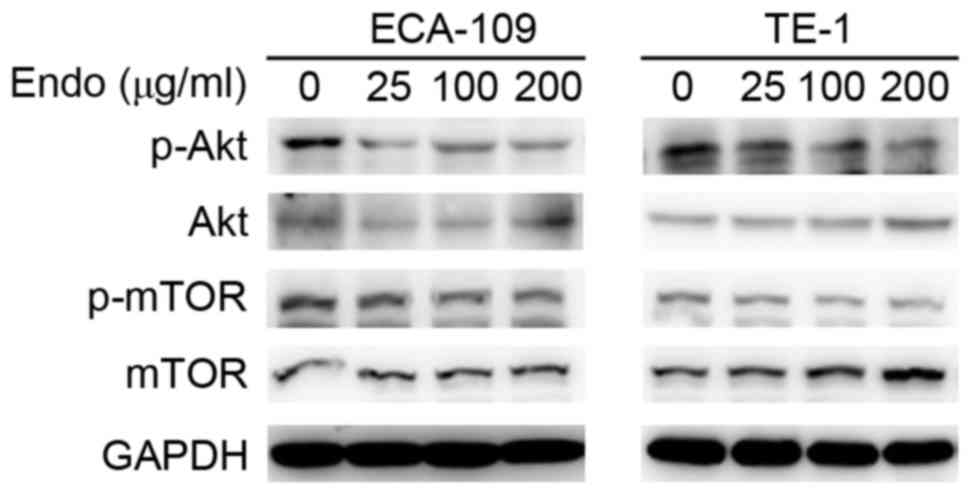
clearly established. To investigate the effect on Akt/mTOR
signaling, the activity of Akt/mTOR was detected in ESCCs following
treatment with Endostar using western blot analysis. The
phosphorylation of Akt/mTOR in ESCC cell lines was markedly reduced
following Endostar treatment in a dose-dependent manner, whereas
the total levels were unaffected (Fig.
3). This suggests that the activation of autophagy following
Endostar treatment is regulated through inhibition of the Akt/mTOR
signaling pathway.
Inhibition autophagy sensitizes
esophageal cancer to Endostar
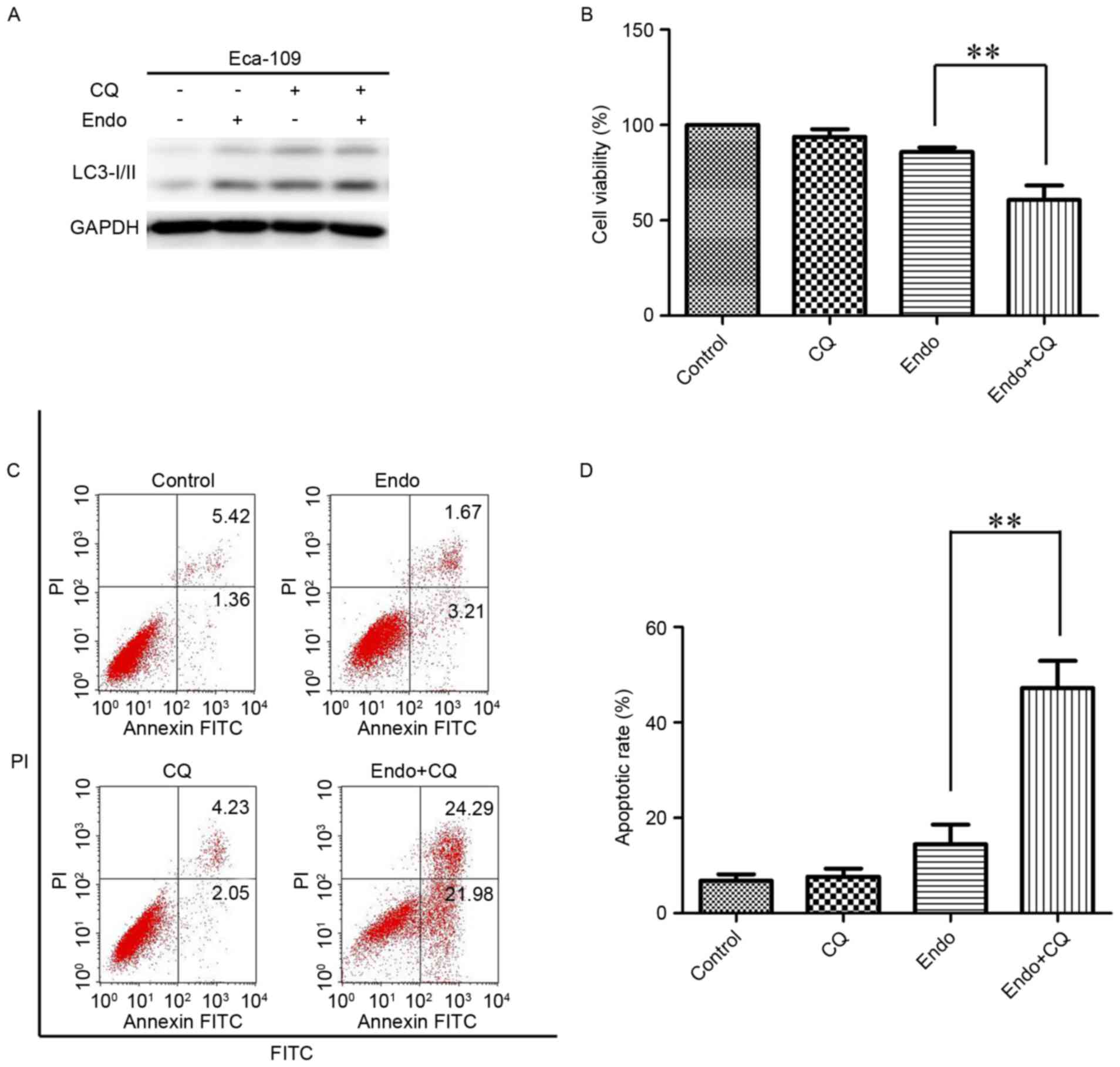
To investigate the biological effect of autophagy
activation following Endostar treatment, ESCC cells were incubated
with Endostar combined with the autophagy inhibitor CQ for 24 h to
detect cell viability. The conversion of LC3-I to LC3-II was
markedly increased in the combined group compared with the
Endostar-alone treatment group (Fig.
4A). Next, the viability of ESCC cells was detected. In
combination with 5 mM CQ, which inhibits the fusion of
autophagosomes and lysosomes, Endostar treatment significantly
suppressed the viability of ESCC cells compared with in the
Endostar-alone treatment group (Fig.
4B). As presented in Fig. 4B, the
cell viability of the untreated, CQ-alone, Endostar-alone and
combined groups was 99.7±0.75, 93.8±3.21, 85.8±1.21 and 62.6±8.42%,
respectively. Furthermore, a significant increase in the number of
apoptotic cells was observed in the Endostar combined with CQ
group, as compared with in the Endostar-alone treatment group
(46.27±5.61 vs. 14.78±3.42%; Fig. 4C and
D). Together, this data indicates that autophagy activation
promotes cell growth and survival in response to Endostar
treatment.
Discussion
Endostatin is a 22 kD a polypeptide and was obtained
from the C-terminal fragment of collagen type XVIII. Endostatin has
an anti angiogenic effect on cancer cells (19). Endostar is a novel rh-endostatin that
has an additional nine-amino acid sequence and an attached
six-histidine tag at the N-terminal end of the protein (20). It has been reported that Endostar
suppresses the proliferation and migration of endothelial cells by
inhibiting VEGFR signaling (14). In
tumor cells, this results in anti-proliferative and -invasive
effects. Treatment with Endostar has been demonstrated to suppress
the growth of lung cancer cells (21,22).
Furthermore, Endostar significantly enhanced the inhibitory effect
of chemotherapy on an ESCC Eca-109 xenograft model (23). In the present study, it was
demonstrated that Endostar significantly reduced the viability of
Eca-109 and TE-1 ESCC cells in a dose- and time-dependent manner
in vitro.
Autophagy is considered to be a pro-survival
mechanism in cancer therapy (17).
Prior studies have revealed that angiogenesis inhibitor-induced
autophagy is initiated in the presence of VEGF (24). Furthermore, Selvakumaran et al
(25) reported that bevacizumab, the
first anti-angiogenic medicine approved by the FDA, induces
autophagy in colon cancer cells, and effect that was also observed
in hepatocellular carcinoma cells (26). Thus, it was hypothesized that Endostar
may induce autophagy in the treatment of esophageal cancer. In the
present study, it was demonstrated that the number of
autophagosomes significantly increased following Endostar exposure,
compared with that in the vehicle-treated cells. In addition, the
punctae fluorescence of LC3 and the LC3-1 to LC3-II transition were
enhanced in the Endostar-treated group, compared with in the
vehicle-treated control group. To elucidate whether the changes
observed were due to increases in autophagy activation or to
inhibition of the late stage of degradation, the expression
patterns of Beclin-1 and p62 were determined. Following exposure to
Endostar, the expression of Beclin-1 markedly increased and p62
decreased in a dose-dependent manner, suggesting that Endostar
initiates autophagy in ESCC cells.
The precise biological significance of autophagy
activation in cancer treatment is controversial and
context-dependent. It has been reported that endostatin causes
autophagic cell death in human EAhy927 endothelial cells (27). However, in the present study, it was
demonstrated that autophagy activation promotes the survival of
ESCC cells, contributing to the occurrence of innate or acquired
resistance. CQ, an inhibitor of autophagy, sensitized the ESCC
cells to Endostar and significantly enhanced the growth inhibitory
effect of Endostar. Thus, autophagy may serve a protective role in
ESCC treated with Endostar, and inhibiting autophagy provides a
potential approach for improving the clinical efficacy of Endostar
for ESCC therapy.
The underlying mechanisms by which Endostar
activates autophagy remain to be elucidated. Previous studies have
revealed that the Akt/mTOR signaling pathway serves a significant
role in cell survival and migration (28). It is well known that the Akt/mTOR
signaling pathway is significantly involved in autophagy (29). In endothelial and gastric cancer,
Endostar has been demonstrated to inhibit the interaction between
VEGF, and its receptor, which consequently inhibits downstream
biological activity, including Akt/mTOR signaling (30,31). In
the current study, the expression of phosphorylated Akt and mTOR
were markedly decreased, suggesting their involvement in
Endostar-mediated autophagy.
In conclusion, it was demonstrated that Endostar
induces autophagy activation and significantly reduces the
viability of ESCC cells. A potential reason for the autophagy
initiation was identified to be the inhibition of Akt/mTOR
signaling, as suggested by the decrease in phosphorylated Akt and
mTOR protein levels. Furthermore, autophagy initiation was revealed
to serve a pro-survival role in cancer cells; thus, autophagy
inhibition may confer clinical benefits in the treatment of
patients with ESCC.
Acknowledgements
The present study was supported by the Anhui
Provincial Key Science and Technology Project (grant no.
1401042007).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sant M, Aareleid T, Berrino F, Lasota M
Bielska, Carli PM, Faivre J, Grosclaude P, Hedelin G, Matsuda T,
Moller H, et al: EUROCARE-3: Survival of cancer patients diagnosed
1990-94-results and commentary. Ann Oncol. 14 Suppl 5:v61–v118.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pande AU, Iyer RV, Rani A, Maddipatla S,
Yang GY, Nwogu CE, Black JD, Levea CM and Javle MM: Epidermal
growth factor receptor-directed therapy in esophageal cancer.
Oncology. 73:281–289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Folkman J: Angiogenesis: An organizing
principle for drug discovery? Nat Rev Drug Discov. 6:273–286. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang J, Sun Y, Liu Y, Yu Q, Zhang Y, Li K,
Zhu Y, Zhou Q, Hou M, Guan Z, et al: Results of randomized,
multicenter, double-blind phase III trial of rh-endostatin (YH-16)
in treatment of advanced non-small cell lung cancer patients.
Zhongguo Fei Ai Za Zhi. 8:283–290. 2005.(In Chinese). PubMed/NCBI
|
|
6
|
Xu M, Huang H, Xiong Y, Peng B, Zhou Z,
Wang D and Yang X: Combined chemotherapy plus Endostar with
sequential stereotactic radiotherapy as salvage treatment for
recurrent esophageal cancer with severe dyspnea: A case report and
review of the literature. Oncol Lett. 8:291–294. 2014.PubMed/NCBI
|
|
7
|
Deng WY, Song T, Li N, Luo SX and Li X:
Clinical observation and therapeutic evaluation of Rh-endostatin
combined with DP regimen in treating patients with advanced
esophageal cancer. Asian Pac J Cancer Prev. 15:6565–6570. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong Z, Gu X, Zhang Z, Wang D, Qing Y, Li
M and Dai N: Recombinant human endostatin combined with definitive
chemoradiotherapy as primary treatment for patients with
unresectable but without systemic metastatic squamous cell
carcinoma of the oesophagus. Br J Radiol. 85:e1104–e1109. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J,
Su J, Li H and Sun L: Inhibition of autophagy enhances cisplatin
cytotoxicity through endoplasmic reticulum stress in human cervical
cancer cells. Cancer Lett. 314:232–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fong MY, Jin S, Rane M, Singh RK, Gupta R
and Kakar SS: Withaferin A synergizes the therapeutic effect of
doxorubicin through ROS-mediated autophagy in ovarian cancer. PLoS
One. 7:e422652012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shimizu S, Takehara T, Hikita H, Kodama T,
Tsunematsu H, Miyagi T, Hosui A, Ishida H, Tatsumi T, Kanto T, et
al: Inhibition of autophagy potentiates the antitumor effect of the
multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J
Cancer. 131:548–557. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X and Fan Z: The epidermal growth
factor receptor antibody cetuximab induces autophagy in cancer
cells by downregulating HIF-1alpha and Bcl-2 and activating the
beclin 1/hVps34 complex. Cancer Res. 70:5942–5952. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ling Y, Yang Y, Lu N, You QD, Wang S, Gao
Y, Chen Y and Guo QL: Endostar, a novel recombinant human
endostatin, exerts antiangiogenic effect via blocking VEGF-induced
tyrosine phosphorylation of KDR/Flk-1 of endothelial cells. Biochem
Biophys Res Commun. 361:79–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bernatchez PN, Allen BG, Gelinas DS,
Guillemette G and Sirois MG: Regulation of VEGF-induced endothelial
cell PAF synthesis: Role of p42/44 MAPK, p38 MAPK and PI3K
pathways. Br J Pharmacol. 134:1253–1262. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li N, Fan LL, Sun GP, Wan XA, Wang ZG, Wu
Q and Wang H: Paeonol inhibits tumor growth in gastric cancer in
vitro and in vivo. World J Gastroenterol. 16:4483–4490. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pan H, Wang Z, Jiang L, Sui X, You L, Shou
J, Jing Z, Xie J, Ge W, Cai X, et al: Autophagy inhibition
sensitizes hepatocellular carcinoma to the multikinase inhibitor
linifanib. Sci Rep. 4:66832014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang L, Ge W, Hu K, Zhang Y, Li C, Xu X,
He D, Zhao Z, Zhang J, Jie F, et al: Endostar down-regulates HIF-1
and VEGF expression and enhances the radio response to human lung
adenocarcinoma cancer cells. Mol Biol Rep. 39:89–95. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harris AL: Are angiostatin and endostatin
cures for cancer? Lancet. 351:1598–1599. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Reilly MS, Boehm T, Shing Y, Fukai N,
Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR and Folkman J:
Endostatin: An endogenous inhibitor of angiogenesis and tumor
growth. Cell. 88:277–285. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
You ZY, Zhao Y, Liu F, Zhang YD and Wang
JJ: The radiosensitization effects of Endostar on human lung
squamous cancer cells H-520. Cancer Cell Int. 10:172010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ni Q, Ji H, Zhao Z, Fan X and Xu C:
Endostar, a modified endostatin inhibits non small cell lung cancer
cell in vitro invasion through osteopontin-related mechanism. Eur J
Pharmacol. 614:1–6. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chang L, Guo F, Lv Y, Wang Y, Huo B, Wang
L and Liu W: The inhibitory effects of Endostar combined with
chemotherapy on human esophageal squamous cell carcinoma xenograft
in mice. Mol Biol Rep. 40:669–673. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bansode RR, Ahmedna M, Svoboda KR and
Losso JN: Coupling in vitro and in vivo paradigm reveals a dose
dependent inhibition of angiogenesis followed by initiation of
autophagy by C6-ceramide. Int J Biol Sci. 7:629–644. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Selvakumaran M, Amaravadi RK, Vasilevskaya
IA and O'Dwyer PJ: Autophagy inhibition sensitizes colon cancer
cells to anti angiogenic and cytotoxic therapy. Clin Cancer Res.
19:2995–3007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo XL, Li D, Sun K, Wang J, Liu Y, Song
JR, Zhao QD, Zhang SS, Deng WJ, Zhao X, et al: Inhibition of
autophagy enhances anticancer effects of bevacizumab in
hepatocarcinoma. J Mol Med (Berl). 91:473–483. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chau YP, Lin SY, Chen JH and Tai MH:
Endostatin induces autophagic cell death in EAhy926 human
endothelial cells. Histol Histopathol. 18:715–726. 2003.PubMed/NCBI
|
|
28
|
Porta C, Paglino C and Mosca A: Targeting
PI3K/Akt/mTOR signaling in cancer. Front Oncol. 4:642014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12 Suppl 2:S1509–S1518. 2005. View Article : Google Scholar
|
|
30
|
Xu X, Mao W, Chen Q, Zhuang Q, Wang L, Dai
J, Wang H and Huang Z: Endostar, a modified recombinant human
endostatin, suppresses angiogenesis through inhibition of
Wnt/beta-catenin signaling pathway. PloS One. 9:e1074632014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang YB, Liu JH and Song ZM: Effects of
recombinant human endostatin on the expression of vascular
endothelial growth factor in human gastric cancer cell line
MGC-803. Biomed Rep. 1:77–79. 2013. View Article : Google Scholar : PubMed/NCBI
|