Introduction
Lung cancer is one of the most common types of
tumors and the leading cause of cancer-associated mortality in the
world (1). Non-small cell lung cancer
(NSCLC) accounts for ~80% of all lung cancer cases (2). Although many advances have been achieved
in diagnosis and treatment and progress has been made in lung
cancer therapy, there is ~5% improvement in 5-year survival rates
for the previous 20 years (3).
Epithelial-mesenchymal transition (EMT) is an
important cell biological process, which is associated with cancer
migration, metastasis, asthma and fibrosis in the lung (4). When EMT occurs, epithelial cells
gradually transform into mesenchymal-like cells by losing their
epithelial-associated functions and characteristics (5). Epigenetic downregulation of E-cadherin
expression was observed in advanced NSCLC and restoration of
E-cadherin expression strongly suppressed the invasion/migration of
tumor cells (6,7). Several signaling pathways are involved
in EMT in NSCLC, including TGF-β (8),
Slug (9), Wnt/β-catenin/zinc finger
E-box binding homeobox 1 signaling (10) and c-Jun N-terminal kinase (JNK)
signaling (11).
MicroRNAs (miRNAs) are small non-coding RNAs, which
regulate gene expression post-transcriptionally. miRNAs are
involved in the pathogenesis of lung diseases, including lung
cancer, cystic fibrosis, chronic obstructive pulmonary disease,
asthma and idiopathic pulmonary fibrosis (12). miRNAs contribute to the development of
novel diagnostic biomarkers and personalized therapeutic tools in
NSCLC (13).
miR-145 is silenced by DNA methylation and acts as a
prognostic biomarker in NSCLC (14).
miR-145 regulates chemoresistance in hepatocellular carcinoma
(15) and bladder cancer (16) via EMT. miR-145 inhibits migration and
invasion by down-regulating fascin actin-bundling protein 1 (FSCN1)
(17) and mucin 1 (18).
The aim of the present study was to identify the
function of miR-145-5p in EMT in NSCLC cells. In addition, it was
investigated whether the target of miR-145-5p mediated this
process. The findings of the present study will provide new
insights into the functions of miR-145-5p as well as its molecular
mechanisms in NSCLC.
Materials and methods
Cell culture and treatment
The NSCLC cell lines A549 and H520, and the normal
lung bronchus epithelial cell line 16HBE were obtained from
American Type Culture Collection (Manassas, VA, USA). The cells
were grown in Dulbecco's modified Eagle's medium supplemented with
10% fetal bovine serum, 2 µM glutamine, 100 IU/ml penicillin, and
100 µg/ml streptomycin sulfate and were cultured in a humidified
chamber with 5% CO2 at 37°C. All transfections were
performed using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). In order to interrupt the
MAP3K1 mediated signaling pathway, cells were treated with 30 µm
SP100625 (Enzo Life Sciences, Inc., Farmingdale, NY, USA) for 24
h.
Target prediction of miR-145-5p by
Targetscan
Targetscan (http://www.targetscan.org/vert_71/) is an online
software for the prediction of the binding of miRNAs to target
genes. miR-145-5p investigated using the software to search for its
target genes.
Transfection and apoptosis assay
The cells were placed in six-well plates at 70–90%
confluence 24 h prior to transfection in serum-free media. The
cells were transfected with plasmids, pcDNA3.1 or pcDNA3.1-MAP3K1
(2 µg/well; Fulengen, Guangzhou, China; http://www.fulengen.com/) or miRNA/small-interfering
RNA (100 pmol/well) using Lipofectamine 2000, according to the
manufacture's protocol. After 48 h, the cells were collected by
trypsinization, washed with phosphate-buffered saline and used for
subsequent experiments.
Luciferase reporter assay
The entire 3′-untranslated region (UTR) of human
mitogen-activated protein kinase kinase kinase 1 (MAP3K1) was
cloned by Genscript (Nanjing, China; http://www.genscript.com.cn/). The entire 3′-UTR of
MAP3K1 were inserted into the pMIR-reporter plasmid (Ambion; Thermo
Fisher Scientific, Inc.). The insertion was confirmed to be correct
by DNA sequencing. The sequences that interact with the seed
sequences of miR-145-5p were mutated. Luciferase assays were
performed using A549 cells. The cells were transfected with:
miR-145-5p mimics, 5′-GUCCAGUUUUCCCAGGAAUCCCU-3′ or scrambled
sequences, 5′-UUCUCCGAACGUGUCACGUTT-3′; GenePharm, Inc. (Sunnyvale,
CA, USA) and Renilla luciferase plasmids (Ambion; Thermo
Fisher Scientific, Inc.) in 24-well plates. The cells were lysed
for luciferase assay 48 h following transfection. Luciferase assays
were performed using dual luciferase assay kit (Promega
Corporation, Madison, WI, USA) according to the manufacturer's
protocol.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from NSCLC cell lines and
16HBE cells using TRIzol (Thermo Fisher Scientific, Inc.).
Complementary DNAs were synthesized by using specific-miRNA primers
(GeneCopoeia Inc., Guangzhou, China). The primers of miR-145-5p
(HmiRQP0192) were obtained from Fulengen. U6 snRNA was used as an
internal control. Gene expression levels were measured by RT-qPCR
using BioRad CFX (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and SYBR-Green kit (Takara Biotechnology Co., Ltd., Dalian, China).
Reactions were incubated for 10 min at 95°C, followed by 40 cycles
of 95°C for 10 sec; 60°C for 20 sec and 72°C for 10 sec. The
primers of were as follows: MAP3K1 forward,
5′-TAGGGCCTAACTCTTTCCTGAT-3′ and reverse,
5′-GTTCTAGTTGAAACACCCGGA-3′; GAPDH forward,
5′-CAATGACCCCTTCATTGACC-3′ and reverse, 5′-GACAAGCTTCCCGTTCTCAG-3′.
GAPDH was used as endogenous control to normalize the expression
data. Reactions were incubated for 10 min at 95°C, followed by 40
cycles of 95°C for 10 sec; 60°C for 20 sec and 72°C for 15 sec.
Each sample was analyzed in quadruplicate. The relative amount of
miRNA was normalized to U6: Forward, 5′-CTCGCTTCGGCAGCACA-3′ and
reverse, 5′-AACGCTTCACGAATTTGCGT-3′. The data was calculated with
the equation 2−ΔΔCq, where ΔΔCq = (CqmiRNA -
CqU6) target - (CqmiRNA - CqU6)
control. Gene expression was calculated with the equation
2−ΔΔCq, where ΔΔCq = (Cqtarget -
CqGAPDH) - (Cqcontrol - CqGAPDH)
(19).
Western blotting
The cells were suspended in radioimmunoprecipitation
assay protein lysis buffer (pH 7.4), containing 20 mM sodium
phosphate, 150 mM sodium chloride, 5 mM EDTA, 5 mM
phenylmethylsulfonyl fluoride, 1% aprotinin, 1 µg/ml leupeptin, and
500 µM Na3VO4. Protein concentration was
quantified using Pierce BCA protein assay kit (Thermo Fisher
Scientific, Inc.). Total protein (50 µg) was resolved with 6%
SDS-PAGE, and transferred to a polyvinylidene difluoride membrane.
Membranes were blocked in 5% non-fat milk diluted in TBST for 1 h
at room temperature. The blots were probed with primary antibodies
against MAP3K1 (cat. no. PA5-15085; Invitrogen; Thermo Fisher
Scientific, Inc.), E-cadherin (cat. no. 14472), Vimentin (cat. no.
5741), JNK (cat. no. 9252), p-JNK (cat. no. 9255), GAPDH (cat. no.
5174) all purchased from Cell Signaling Technology, Inc., Danvers,
MA, USA. All primary antibodies were diluted at 1:1,000 and
incubated at 4°C overnight. After washing, membranes were probed
with appropriate secondary antibodies, horseradish peroxidase
conjugated-goat anti mouse IgG (cat. no. BA1051; Boster Biological
Technology, Pleasanton, CA, USA) or horseradish peroxidase
conjugated-goat anti rabbit IgG (cat. no. BA1054; Boster Biological
Technology). Secondary antibodies were diluted at 1:5,000 and
incubated at room temperature for 1 h. All membranes were stripped
by incubating in Restore PLUS Western blot stripping buffer (Thermo
Fisher Scientific, Inc.) for 15 min at room temperature and
reprobed with anti-GAPDH antibody for loading control. The blots
were visualized using enhanced chemiluminescence (GE Healthcare,
Chicago, IL, USA).
Statistical analysis
Statistical analysis was performed with SPSS17.0
(SPSS, Inc., Chicago, IL, USA). The Student's t-test was used to
compare the different groups. P<0.05 was considered to indicate
a statistically significant difference, and P-values are
represented by an asterisk on the bars in the figures.
Results
Ectopic expression of miR-145-5p
inhibits EMT in NSCLC cells
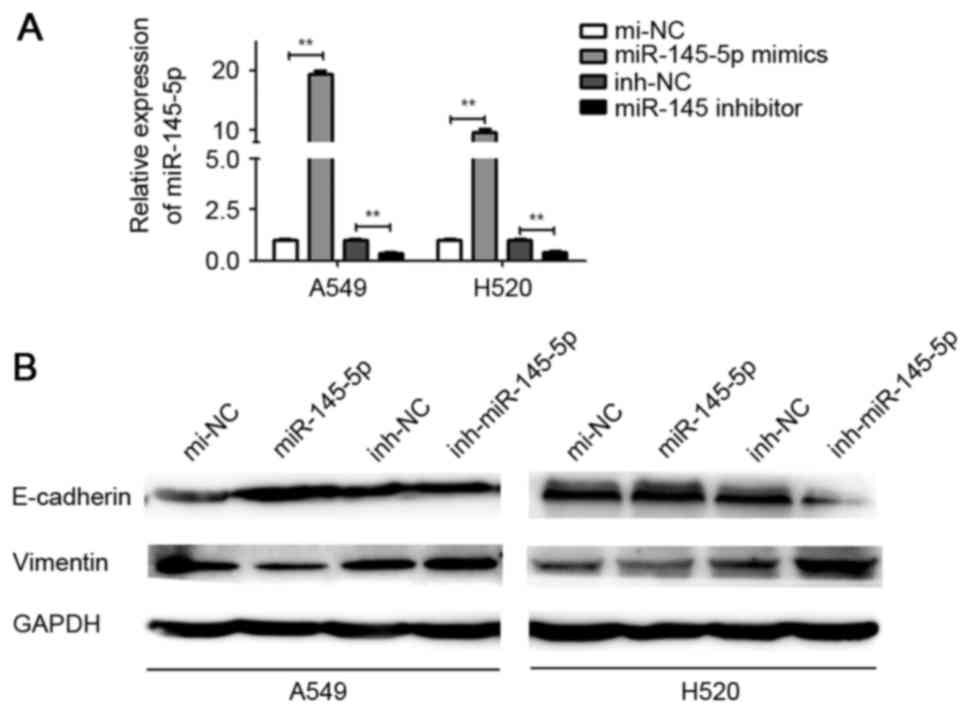
The effect of miR-145-5p mimics and inhibitor
transfection was detected (Fig. 1A).
It was demonstrated that ectopic expression of miR-145-5p markedly
increased the expression of E-cadherin and suppressed the
expression of vimentin in A549 and H520 cells, compared with mi-NC.
By contrast, depletion of miR-145-5p induced E-cadherin
downregulation and vimentin upregulation in A549 and H520 cells
(Fig. 1B).
Involvement of JNK signaling in
miR-145-5p-induced EMT suppression in NSCLC
JNK is not required for initiation of NSCLC, but
associated with EMT. JNK drives transcription of numerous key EMT
genes (20). It has been reported
that TGF-β1 induced JNK activation during EMT in idiopathic
pulmonary fibrosis (21). In the
present study, it was examined whether JNK signaling pathway has a
critical role in the regulation of miR-145-5p-mediated EMT in NSCLC
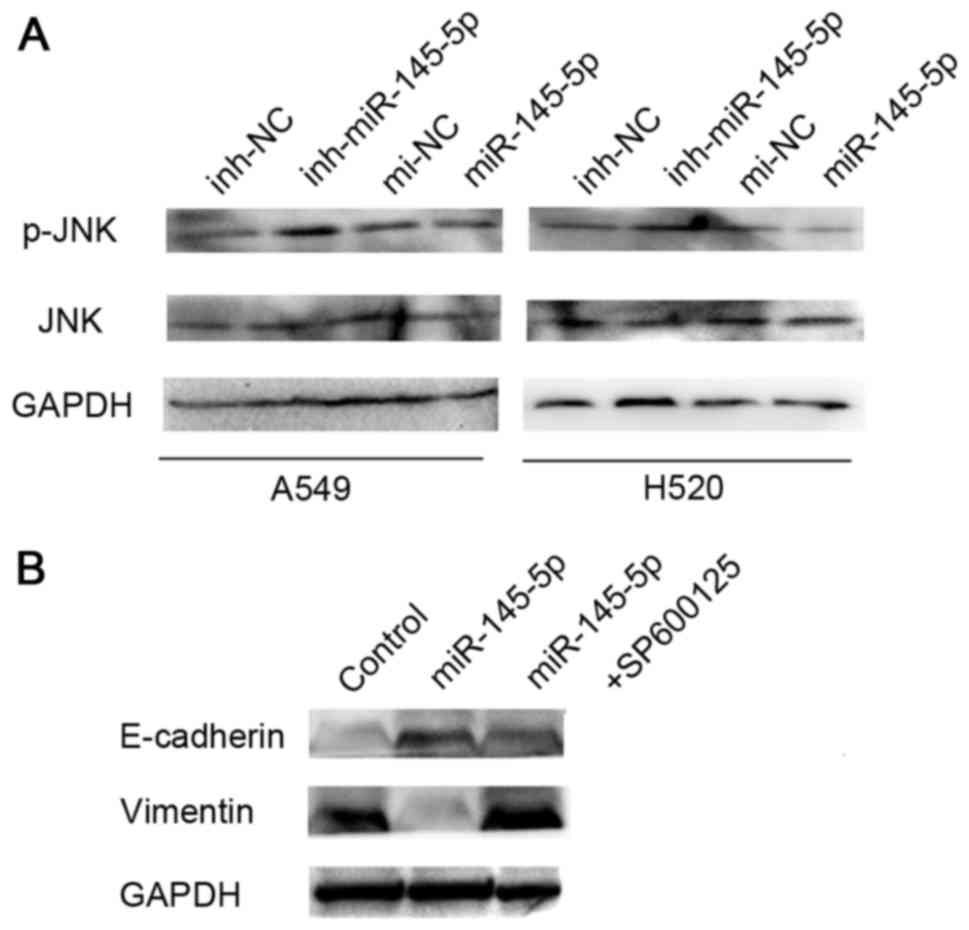
cells. The results revealed that overexpression of miR-145-5p was
able to inhibit the expression of phosphorylated (p)-JNK, but has
no effect on the level of total JNK protein (Fig. 2A). The cells were also treated with a
combination of a specific JNK inhibitor (SP600125) and miR-145-5p.
Western blotting analysis showed SP600125 was able to reverse the
effect of miR-145-5p on EMT in A549 and H520 cells (Fig. 2B).
MAP3K1 is a target of miR-145-5p
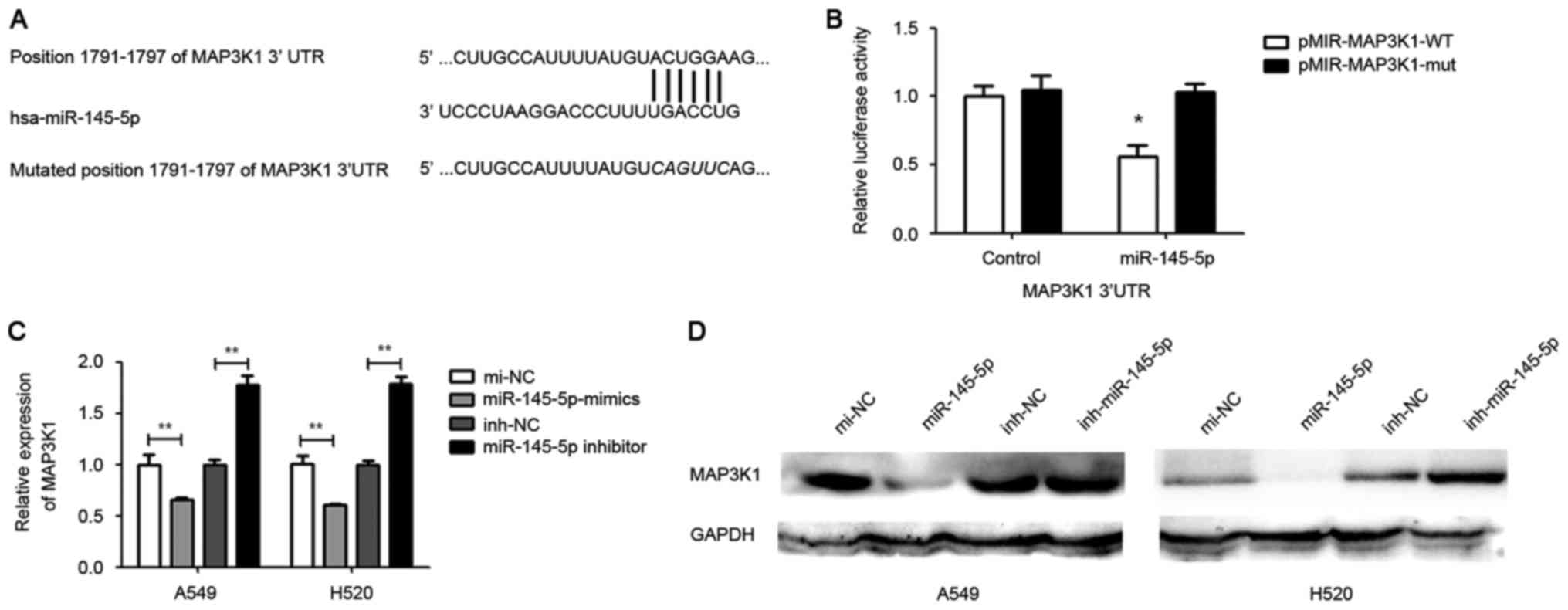
TargetScan online software was used to search for
putative protein-coding gene targets of miR-145-5p. The results
indicated that MAP3K1 is a putative target of miR-145-5p (Fig. 3A). The MAP3K1 3′UTR was cloned into a
luciferase reporter vector and binding sites of the mutant vector.
The A549 cells were co-transfected with either mutated
pmiR-MAP3K1-3′UTR and miR-145-5p mimics or wild-type
pmiR-MAP3K1-3′UTR and miR-145-5p mimics. The cells were also
co-transfected with mutated pmiR-MAP3K1 and negative control or
wild-type pmiR-MAP3K1 and negative control. Luciferase activity was
measured 48 h following transfection.
The results showed that the cells co-transfected
with wild-type pmiR-MAP3K1-3′UTR and miR-145-5p mimic exhibited
significant reduction in luciferase activity compared with cells
cotransfected with the control. By contrast, the luciferase
activity in cells transfected with mutated pmiR-MAP3K1-3′UTR and
miR-145-5p mimic was not significantly altered (Fig. 3B). Furthermore, RT-qPCR and western
blotting analysis was used to investigate the expression of MAP3K1
regulated by miR-145-5p in A549 and H520 cells. The results showed
that a marked reduction in mRNA and protein level of MAP3K1 in
miR-145-5p-overexpressed A549 and H520 cells compared with the
control (Fig. 3C and D).
MAP3K1 reverses miR-145-5p- induced
inhibition of EMT in NSCLC
Since MAP3K1 was a novel target of miR-145-5p in
NSCLC cells, it was investigated whether the effect of miR-145 on
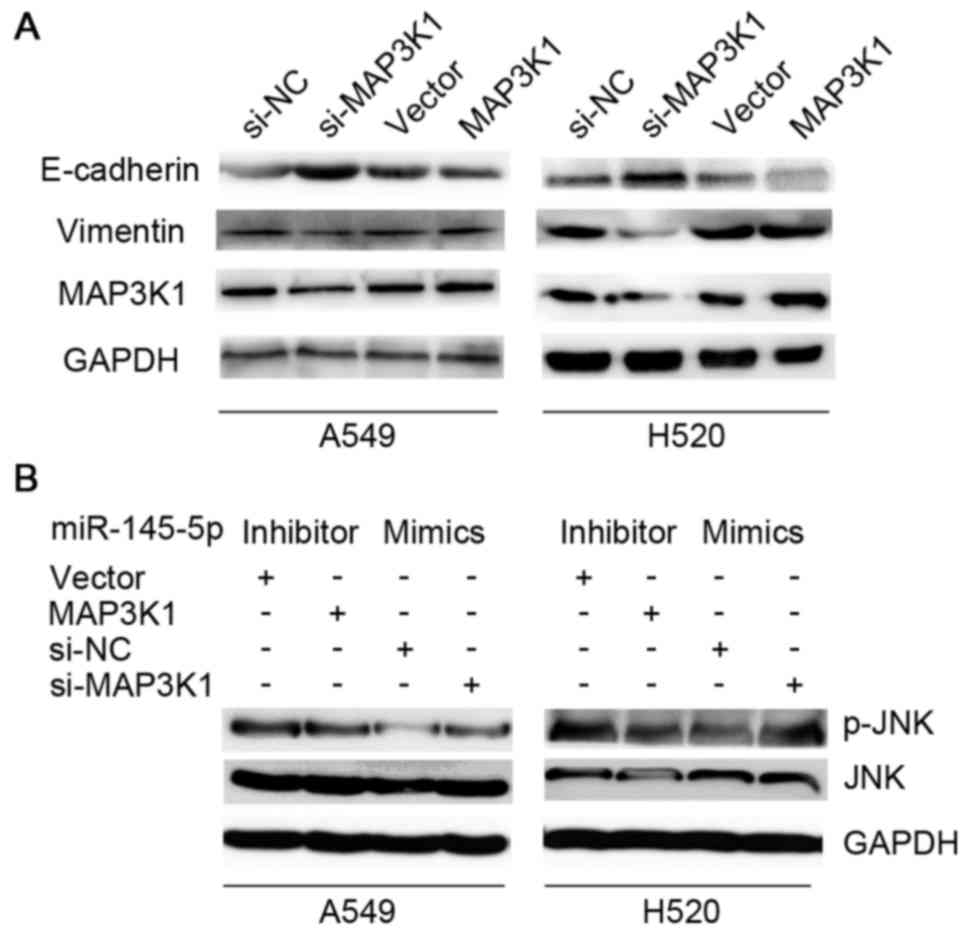
EMT was mediated by MAP3K1. Fig. 4A
showed that the knockdown of MAP3K1 markedly induced the expression
of E-cadherin and inhibited the expression of vimentin in A549 and
H520 cells, while overexpression of MAP3K1 treatment reversed this
effect. The ectopic expression of MAP3K1 was able to partly reverse
the downregulation of p-JNK induced by the miR-145-5p inhibitor
compared to the vector group. While, the suppression of MAP3K1 was
able to interrupt the upregulation of p-JNK induced by miR-145-5p
mimics compared to the siRNA negative control group (Fig. 4B).
Discussion
EMT is a characteristic feature of the majority of
metastatic cells (22). Epithelial
cells are highly differentiated, polarized and organized. When EMT
occurs, these cells are transformed into undifferentiated, isolated
and mesenchymal-like cells (23).
E-cadherin and vimentin are known to be closely associated with EMT
(24). E-cadherin has important
functions in regulating cell-cell interactions (25). Epigenetic silencing of E-cadherin
expression was observed in advanced NSCLC, and restoration of
E-cadherin expression strongly suppressed the metastasis of cancer
cells (7). Vimentin is specifically
expressed in normal mesenchymal cells and overexpressed in lung
cancer cells (26). In previous
studies, vimentin has been considered as a marker for EMT (26), and EMT is considered to be involved in
the pathogenesis of lung cancer (5).
Insulin like growth factor 1 receptor-induced resistance to
epidermal growth factor receptor-tyrosine kinase inhibitors in
advanced NSCLC is mediated by EMT (27). MARVELD1 suppresses EMT in NSCLC cells
(28). Inactivation of M2
acetylcholine receptor/nuclear factor-κB signaling axis reverses
EMT and suppresses migration and invasion in NSCLC cells (29).
miR-145 was reported to inhibit metastasis in
multiple tumors, including hepatocellular carcinoma (30), osteosarcoma (31) and nasopharyngeal carcinoma (32). miR-145 inhibits migration and invasion
by downregulating FSCN1 (17) and
mucin1 (18). A previous study by the
present authors revealed that miR-145 acts as a metastasis
suppressor by targeting metadherin in lung cancer (33). miR-145 regulates chemoresistance in
hepatocellular carcinoma via EMT (15). However, the underlying mechanisms of
miR-145-5p in regulating EMT in NSCLC are unclear. In the present
investigation, it was demonstrated that miR-145-5p increased the
expression of E-cadherin and suppressed the expression of vimentin.
Therefore, it was hypothesized that miR-145-5p may inhibit
metastasis via suppressing EMT in NSCLC cells.
The JNK signaling pathway exert an important role in
the function of NSCLC cells. It was previously demonstrated that
protein 4.1N, a member of the protein 4.1 family, was able to
suppress metastasis by inactivating JNK-c-Jun signaling pathway in
NSCLC (34). Celastrol induces
cisplatin re-sensitization through inhibition of the JNK/activating
transcription factor 2 signaling pathway in NSCLC (35). JNK signaling mediates EPH receptor
A2-dependent tumor cell proliferation, motility and cancer stem
cell-like properties in NSCLC cells (36). miR-145 induces apoptosis in
ethanol-induced acute gastric mucosal injury via the JNK signaling
pathway (37). However, no studies
have yet shown the association between miR-145-5p and the JNK
signaling pathway in NSCLC.
In the present study, the effect of miR-145-5p on
JNK signaling pathway in NSCLC cells was detected. The results
showed that miR-145-5p was able to inactivate the JNK signaling
pathway in NSCLC cells. The results of the present study are in
accordance with previous research from another group (38).
MAP3K1 is a component of an evolutionarily conserved
signal transduction cascade, which can activate mitogen-activated
protein kinases (MAPKs) (39). It has
been previously revealed that the single nucleotide polymorphism
MAP3K1 rs889312 is closely associated with the risk of breast
cancer and outcome among patients with diffuse-type gastric cancer
(40).
In the present study, it was predicted and confirmed
that MAP3K1 is a potential target of miR-145-5p. In order to
clarify the function of MAP3K1 on EMT in NSCLC cells, the
expression of E-cadherin and vimentin regulated by MAP3K1 was
detected. The data illustrated that ectopic expression of MAP3K1
was able to inhibit E-cadherin protein levels and increase vimentin
protein levels in NSCLC cells. Subsequently, whether MAP3K1
regulates ETM via a miR-145-5p mediated-JNK signaling pathway was
investigated. The results revealed that overexpression of
miR-145-5p and inhibition of MAP3K1 was able to reverse the
expression of p-JNK in NSCLC cells. To conclude, miR-145-5p was
able to inhibit EMT via JNK signaling pathway by targeting MAP3K1
in NSCLC cells.
Acknowledgements
The present study was supported by the Natural
Science Foundation of Guangdong Province (grant no. 2016zc0160) and
the Science and Technology Planning Project of Guangdong Province,
China (grant no. 2014A020212447).
References
|
1
|
Hu J, Qiu M, Jiang F, Zhang S, Yang X,
Wang J, Xu L and Yin R: MiR-145 regulates cancer stem-like
properties and epithelial-to-mesenchymal transition in lung
adenocarcinoma-initiating cells. Tumour Biol. 35:8953–8961. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mariotto AB, Noone AM, Howlader N, Cho H,
Keel GE, Garshell J, Woloshin S and Schwartz LM: Cancer survival:
An overview of measures, uses, and interpretation. J Natl Cancer
Inst Monogr. 2014:145–186. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Johnson DH, Schiller JH and Bunn PA Jr:
Recent clinical advances in lung cancer management. J Clin Oncol.
32:973–982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tian L, Shen D, Li X, Shan X, Wang X, Yan
Q and Liu J: Ginsenoside Rg3 inhibits epithelial-mesenchymal
transition (EMT) and invasion of lung cancer by down-regulating
FUT4. Oncotarget. 7:1619–1632. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartis D, Mise N, Mahida RY, Eickelberg O
and Thickett DR: Epithelial-mesenchymal transition in lung
development and disease: Does it exist and is it important? Thorax.
69:760–765. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S,
Chen Y and Wu K: Notch signaling and EMT in non-small cell lung
cancer: Biological significance and therapeutic application. J
Hematol Oncol. 7:872014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mateen S, Raina K, Agarwal C, Chan D and
Agarwal R: Silibinin synergizes with histone deacetylase and DNA
methyltransferase inhibitors in upregulating E-cadherin expression
together with inhibition of migration and invasion of human
non-small cell lung cancer cells. J Pharmacol Exp Ther.
345:206–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qu MH, Han C, Srivastava AK, Cui T, Zou N,
Gao ZQ and Wang QE: miR-93 promotes TGF-β-induced
epithelial-to-mesenchymal transition through downregulation of
NEDD4L in lung cancer cells. Tumour Biol. 37:5645–5651. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang LK, Pan SH, Chang YL, Hung PF, Kao
SH, Wang WL, Lin CW, Yang SC, Liang CH, Wu CT, et al:
MDA-9/Syntenin-Slug transcriptional complex promote
epithelial-mesenchymal transition and invasion/metastasis in lung
adenocarcinoma. Oncotarget. 7:386–401. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qu J, Li M, An J, Zhao B, Zhong W, Gu Q,
Cao L, Yang H and Hu C: MicroRNA-33b inhibits lung adenocarcinoma
cell growth, invasion, and epithelial-mesenchymal transition by
suppressing Wnt/β-catenin/ZEB1 signaling. Int J Oncol.
47:2141–2152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim J, Moon SH, Kim BT, Chae CH, Lee JY
and Kim SH: A novel aminothiazole KY-05009 with potential to
inhibit Traf2- and Nck-interacting kinase (TNIK) attenuates
TGF-β1-mediated epithelial-to-mesenchymal transition in human lung
adenocarcinoma A549 cells. PLoS One. 9:e1101802014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ebrahimi A and Sadroddiny E: MicroRNAs in
lung diseases: Recent findings and their pathophysiological
implications. Pulm Pharmacol Ther. 34:55–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Feng B, Zhang K, Wang R and Chen L:
Non-small-cell lung cancer and miRNAs: Novel biomarkers and
promising tools for treatment. Clin Sci (Lond). 128:619–634. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xia W, Chen Q, Wang J, Mao Q, Dong G, Shi
R, Zheng Y, Xu L and Jiang F: DNA methylation mediated silencing of
microRNA-145 is a potential prognostic marker in patients with lung
adenocarcinoma. Sci Rep. 5:169012015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ju BL, Chen YB, Zhang WY, Yu CH, Zhu DQ
and Jin J: miR-145 regulates chemoresistance in hepatocellular
carcinoma via epithelial mesenchymal transition. Cell Mol Biol
(Noisy-le-grand). 61:12–16. 2015.PubMed/NCBI
|
|
16
|
Tan J, Qiu K, Li M and Liang Y:
Double-negative feedback loop between long non-coding RNA TUG1 and
miR-145 promotes epithelial to mesenchymal transition and
radioresistance in human bladder cancer cells. FEBS Lett.
589:3175–3181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Yang X, Wu H, Zhou W and Liu Z:
MicroRNA-145 inhibits migration and invasion via inhibition of
fascin 1 protein expression in non-small-cell lung cancer cells.
Mol Med Rep. 12:6193–6198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ye Z, Shen N, Weng Y, Li K, Hu L, Liao H,
An J, Liu L, Lao S and Cai S: Low miR-145 silenced by DNA
methylation promotes NSCLC cell proliferation, migration and
invasion by targeting mucin 1. Cancer Biol Ther. 16:1071–1079.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sahu SK, Garding A, Tiwari N, Thakurela S,
Toedling J, Gebhard S, Ortega F, Schmarowski N, Berninger B, Nitsch
R, et al: JNK-dependent gene regulatory circuitry governs
mesenchymal fate. EMBO J. 34:2162–2181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ding H, Ji X, Chen R, Ma T, Tang Z, Fen Y
and Cai H: Antifibrotic properties of receptor for advanced
glycation end products in idiopathic pulmonary fibrosis. Pulm
Pharmacol Ther. 35:34–41. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alizadeh AM, Shiri S and Farsinejad S:
Metastasis review: From bench to bedside. Tumour Biol.
35:8483–8523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guan X: Cancer metastases: Challenges and
opportunities. Acta Pharm Sin B. 5:402–418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qi D, Gill Kaur N, Santiskulvong C,
Sifuentes J, Dorigo O, Rao J, Taylor-Harding B, Wiedemeyer Ruprecht
W and Rowat AC: Screening cell mechanotype by parallel
microfiltration. Sci Rep. 5:175952015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng H and Kang Y: Multilayer control of
the EMT master regulators. Oncogene. 33:1755–1763. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Satelli A and Li S: Vimentin in cancer and
its potential as a molecular target for cancer therapy. Cell Mol
Life Sci. 68:3033–3046. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou J, Wang J, Zeng Y, Zhang X, Hu Q,
Zheng J, Chen B, Xie B and Zhang WM: Implication of
epithelial-mesenchymal transition in IGF1R-induced resistance to
EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget.
29:44332–44345. 2015. View Article : Google Scholar
|
|
28
|
Yao Y, Shi M, Liu S and Li Y, Guo K, Ci Y,
Liu W and Li Y: MARVELD1 modulates cell surface morphology and
suppresses epithelial-mesenchymal transition in non-small cell lung
cancer. Mol Carcinog. 55:1714–1727. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao Q, Yue J, Zhang C, Gu X, Chen H and
Xu L: Inactivation of M2 AChR/NF-κB signaling axis reverses
epithelial-mesenchymal transition (EMT) and suppresses migration
and invasion in non-small cell lung cancer (NSCLC). Oncotarget.
6:29335–29346. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding W, Tan H, Zhao C, Li X, Li Z, Jiang
C, Zhang Y and Wang L: MiR-145 suppresses cell proliferation and
motility by inhibiting ROCK1 in hepatocellular carcinoma. Tumour
Biol. 37:6255–6260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen B, Huang Z, Zhang Y, Chen Y and Li Z:
MicroRNA-145 suppresses osteosarcoma metastasis via targeting
MMP16. Cell Physiol Biochem. 37:2183–2193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li YQ, He QM, Ren XY, Tang XR, Xu YF, Wen
X, Yang XJ, Ma J and Liu N: MiR-145 inhibits metastasis by
targeting fascin actin-bundling protein 1 in nasopharyngeal
carcinoma. PLoS One. 10:e01222282015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang M, Wang J, Deng J, Li X, Long W and
Chang Y: MiR-145 acts as a metastasis suppressor by targeting
metadherin in lung cancer. Med Oncol. 32:3442015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Z, Ma B, Li H, Xiao X, Zhou W, Liu F,
Zhang B, Zhu M, Yang Q and Zeng Y: Protein 4.1 acts as a potential
tumor suppressor linking PP1 to JNK-c-Jun pathway regulation in
NSCLC. Oncotarget. 7:509–523. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lo Iacono M, Monica V, Vavalà T, Gisabella
M, Saviozzi S, Bracco E, Novello S, Papotti M and Scagliotti GV:
ATF2 contributes to cisplatin resistance in non-small cell lung
cancer and celastrol induces cisplatin resensitization through
inhibition of JNK/ATF2 pathway. Int J Cancer. 136:2598–2609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Song W, Ma Y, Wang J, Brantley-Sieders D
and Chen J: JNK signaling mediates EPHA2-dependent tumor cell
proliferation, motility, and cancer stem cell-like properties in
non-small cell lung cancer. Cancer Res. 74:2444–2454. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Luo XJ, Liu B, Dai Z, Li TB, Li NS, Zhang
XJ, Yang ZC, Li YJ and Peng J: Expression of apoptosis-associated
microRNAs in ethanol-induced acute gastric mucosal injury via JNK
pathway. Alcohol. 47:481–493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen R, Chen S, Liao J, Chen X and Xu X:
MiR-145 facilitates proliferation and migration of endothelial
progenitor cells and recanalization of arterial thrombosis in
cerebral infarction mice via JNK signal pathway. Int J Clin Exp
Pathol. 8:13770–13776. 2015.PubMed/NCBI
|
|
39
|
Hu P, Huang Q, Li Z, Wu X, Ouyang Q, Chen
J and Cao Y: Silencing MAP3K1 expression through RNA interference
enhances paclitaxel-induced cell cycle arrest in human breast
cancer cells. Mol Biol Rep. 41:19–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wei X, Zhang E, Wang C, Gu D, Shen L, Wang
M, Xu Z, Gong W, Tang C, Gao J, et al: A MAP3k1 SNP predicts
survival of gastric cancer in a Chinese population. PLoS One.
9:e960832014. View Article : Google Scholar : PubMed/NCBI
|