Introduction
Nasopharyngeal carcinoma (NPC), a malignant tumor
arising from the epithelium of the nasopharynx, is the most
prevalent in southern China (1). In
Hong Kong, the incidence of NPC is as high as 0.02–0.03% in males
and 0.01–0.02% in females (2).
Although the classic treatment of high-dose radiotherapy plus
adjunctive chemotherapy is able to achieve a 5-year survival rate
of 80%, recurrence and metastasis may occur, which are the primary
causes of mortality (3). Therefore,
it is necessary to identify molecular biomarkers for NPC prognosis
and targeted therapy.
Aberrant DNA methylation usually leads to the
occurrence of tumors. CpG island promoter hypermethylation and
global DNA hypomethylation are the characteristics of the cancer
epigenome (4,5). In NPC, aberrant methylation has been
considered as the most frequent event for gene silencing: For
example, a previous study identified that abnormal methylation on
chromosome 6p occurs in 76.9% of patients with early-stage NPC,
based on the comparative methylome analysis (6). Furthermore, tumor-suppressor genes such
as protocadherin 8 (PCDH8), FEZ family zinc finger 2
(FEZF2) and argininosuccinate synthetase (ASS1) have
been previously demonstrated to be frequently methylated in NPC,
which promotes NPC cell migration, and are associated with poorer
clinical outcomes (7–9).
With the exception of DNA methylation, the
differential expression of genes is also frequently detected in
NPC: A previous study demonstrated that the overexpression of
branched-chain-amino-acid aminotransferase cytosolic (BCAT1)
protein in NPC at different pathological stages and BCAT1
deficiency reduced tumor cell proliferation and decreased cell
migration and invasion abilities (10). Recently, upregulation of Kelch domain
containing 4 (KLHDC4) was demonstrated to result in poor
overall and metastasis-free survival rates, and the deletion of
KLHDC4 significantly induced the spontaneous apoptosis of
NPC cells (11). However, the
molecular mechanisms of NPC remain incompletely understood. There
have been no studies that have comprehensively analyzed
differentially expressed methylated genes using array data from
multiple platforms.
In the present study, methylation profiling data in
the dataset GSE62336 (6), sourced
from the Gene Expression Omnibus (GEO) database, were used to
identify differentially methylated regions (DMRs) and
differentially methylated CpG islands (DMIs). Concurrently,
compared with differentially expressed genes (DEGs) that were
identified using a meta-analysis of three gene expression datasets
(GSE53819, GSE13597 and GSE12452) (12–14),
differentially methylated genes were identified. Additionally, a
functional network consisting of the differentially methylated
genes was constructed to reveal the potential functional
associations of those genes. These results may provide novel
information for the study of molecular mechanisms underlying NPC
and provide potential therapeutic targets for NPC.
Materials and methods
Data acquisition
Methylation profiling data from the dataset GSE62336
(6) were downloaded from GEO
(http://www.ncbi.nlm.nih.gov/geo/)
(15). A total of 25 primary NPC
tumors and non-tumor counterparts were included in this dataset,
and the data were produced on the platform of Illumina
HumanMethylation450 BeadChip (GPL13534,
HumanMethylation450_15017482) (Illumina, Inc., San Diego, CA,
USA).
Three gene expression profiling datasets (GSE53819,
GSE13597 and GSE12452) were obtained from GEO. In the GSE53819
dataset (13), 18 primary NPC tumors
and 18 non-cancerous nasopharyngeal tissues were included; the
median ages were 46 years (range, 19–77 years) for patients with
NPC, and 45 years (range, 18–78 years) for the non-cancerous
cohort; almost one third of patients were females; all samples were
collected prior to any anticancer treatment. Data in the GSE53819
dataset were produced on the platform of Agilent-014850 Whole Human
Genome Microarray 4×44 K G4112F (Probe Name version; GPL6480;
Agilent Technologies, Inc., Santa Clara CA, USA). The GSE13597
dataset (14) contained data of 25
histologically confirmed undifferentiated NPC tissues and 3
non-malignant nasopharyngeal controls, which were produced on the
platform of [HG-U133A] Affymetrix Human Genome U133A Array (GPL96)
(Affymetrix, Inc., Santa Clara, CA, USA). Additionally, the
GSE12452 dataset (12) contained 31
NPC tumor samples and 10 normal healthy nasopharyngeal tissues,
data of which were produced on the platform of [HG-U133_Plus_2]
Affymetrix Human Genome U133 Plus 2.0 Array (GPL570) (Affymetrix,
Inc.).
Data preprocessing
The downloaded raw data were preprocessed. For the
methylation data in GSE62336, documents of normalized average
β-value were downloaded. The β-mixture quantile normalization
method (BMIQ) (16) was utilized to
preprocess β-values.
Due to different platforms being used for the three
gene expression profiling datasets, two different methods were
utilized for data preprocessing. For the gene expression data in
the GSE13597 and GSE12452 datasets, raw gene expression data were
preprocessed using the method of robust microarray analysis in Affy
package (version 1.46.1; https://www.bioconductor.org/packages/3.1/bioc/html/affy.html)
in R (17). The preprocessing steps
included background correction, quantile normalization and
calculation of expression. By contrast, Limma (version 3.24.15;
https://www.bioconductor.org/packages/3.1/bioc/html/limma.html)
package (18) in R was applied to
preprocess raw data in the GSE53819 dataset. The preprocessing
steps included background correction, normalization between arrays
and concentration of microarray data. Following this, an annotation
file of the platform corresponding to each dataset was used for the
transformation of probe identities into gene symbols. If one probe
corresponded to multiple genes, the expression value of this probe
would be removed. However, if multiple probes corresponded to a
certain gene, the mean expression value was defined as the final
expression value of the gene.
Prediction of DMRs and DMIs
DMRs between NPC and normal samples were predicted
using COHCAP package (version 1.6.0; http://www.bioconductor.org/pacages/3.1/bioc/html/COHCAP.html)
(19) in R. Briefly, based on the
β-value file of CpG site probes, Δβ, P-value and adjusted P-value
of NPC and normal samples were calculated by COHCAP. Only regions
with |Δβ|>0.1 and adjusted P<0.05 were identified as
DMRs.
Furthermore, the COHCAP package was also utilized to
predict DMIs. CpG island statistics were calculated by averaging
β-values among samples per site and comparing the average β-values
across groups. If the number of DMRs in the CpG island was >4,
this CpG island was identified as a DMI.
Identification of DEGs using
meta-analysis
MetaDE package (version 1.1.6; http://www.bioconductor.org/packages/2.11/bioc/html/metahdep.html)
(20) in R was applied to integrate
DEGs in the three gene expression profiling datasets. Briefly, a
heterogeneity test for each gene under different platforms was
firstly performed to evaluate whether each gene was homogeneous and
unbiased. If the parameter tau2=0 and Qpval>0.05 (tau2 is used
to estimate amount of heterogeneity; Qpval represents P-value of
Qval test statistics; and Q is a statistical magnitude in
statistics), the gene was homogeneous and unbiased. Then, the
differential expression of the genes was analyzed. Only genes with
P<0.05 were considered significant. Finally, the relative
fold-change (FC) in expression of each gene between the NPC tissues
samples and normal control samples was calculated. Collectively,
genes with P<0.05 were identified as DEGs.
Gene Ontology (GO) functional and
pathway enrichment analyses
The gene functional analysis tool Database for
Annotation, Visualization and Integrated Discovery (http://david.abcc.ncifcrf.gov/) (21) was used to perform GO [including
biological process (BP), cell component (CC) and molecular function
(MF)] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway
enrichment analyses of DEGs. Only the GO and pathway terms with
gene count ≥2 and P<0.05 were considered significant.
Selection of DEGs with DMRs
Based on the gene symbols corresponding to the DMRs
and identified DEGs, the overlapped genes between genes with
hypermethylated DMRs and downregulated DEGs, and the genes with
hypomethylated DMRs and upregulated DEGs were selected.
Functional association analysis of the
DEGs with DMRs
The GeneMANIA prediction server (22,23)
(http://apps.cytoscape.org/download/stats/genemania/),
a plugin in Cytoscape software (version 3.2.1; National Institute
of General Medical Sciences, Seattle, WA, USA), was utilized to
analyze the correlations among the identified DEGs that had DMRs,
based on a large set of functional association data, including
protein and genetic interactions, co-expression, co-localization
pathways, and protein domain similarity.
Results
Statistics of DMRs, DMIs and DEGs
In total, 2,262 probes of DMRs were obtained,
including 2,234 hypermethylated CpG site probes corresponding to
1,676 gene symbols and 28 hypomethylated CpG site probes
corresponding to 28 gene symbols. Furthermore, 2,983 DEGs were
identified, including 1,655 upregulated and 1,328 downregulated
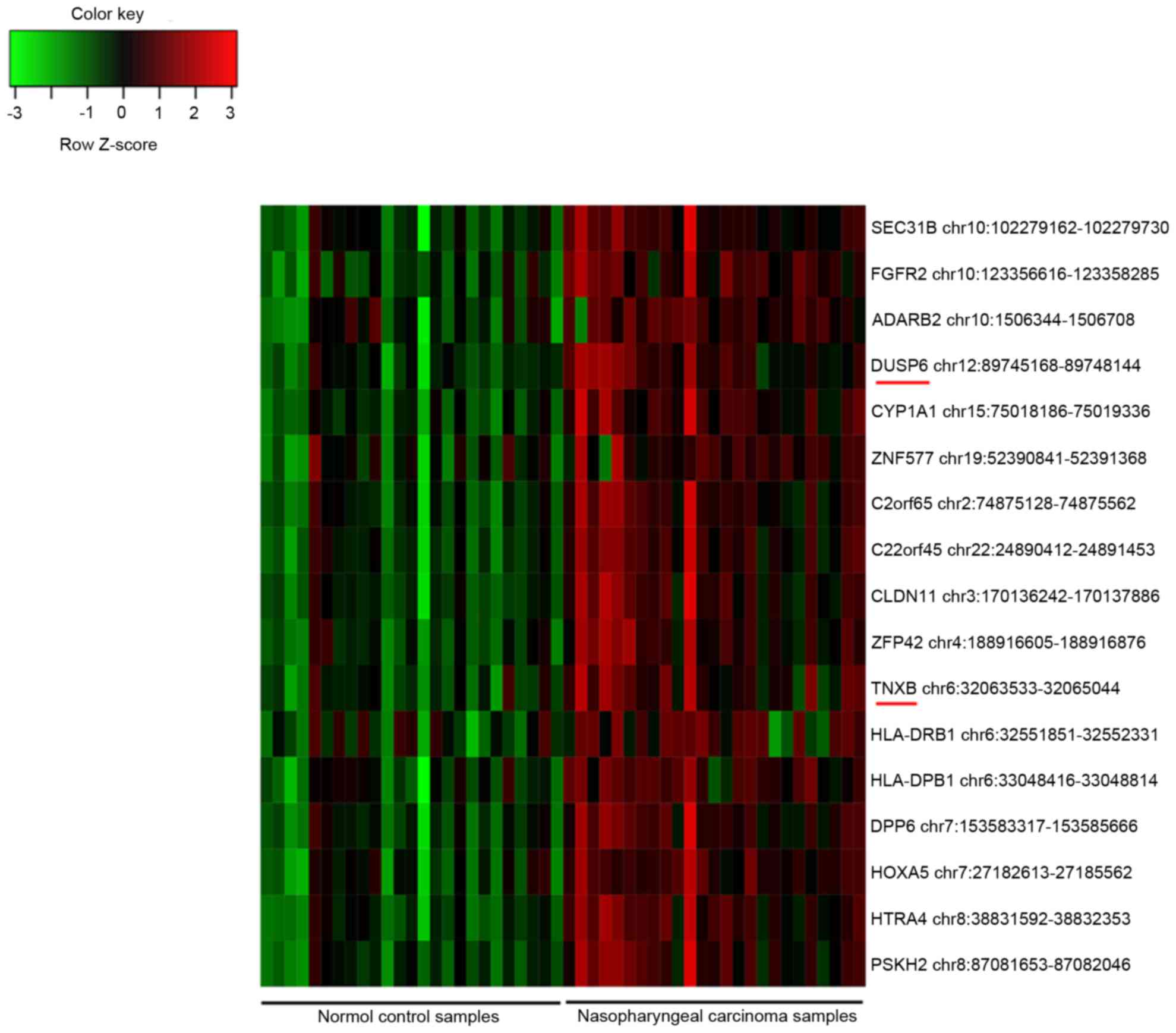
genes. Additionally, 17 DMIs were identified, and all of them were
hypermethylated in the NPC samples compared with their methylation
status in the controls (Fig. 1).
Among them, dual specificity phosphatase 6 (DUSP6) and
tenascin XB (TNXB) were also identified as DEGs.
Significant pathways enriched by
DEGs
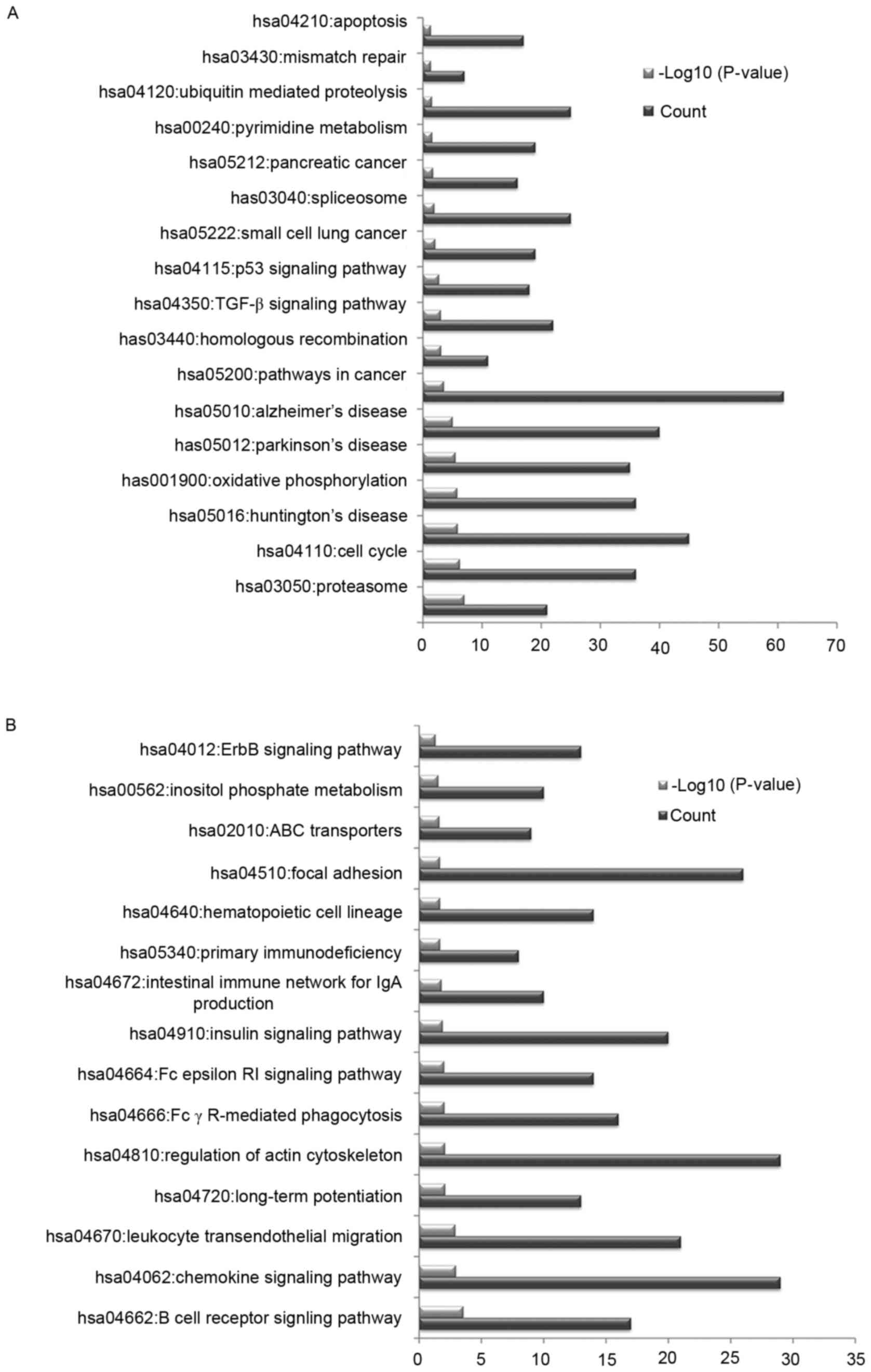
In order to investigate the potential pathways
associated with the identified DEGs, KEGG pathway enrichment
analysis was conducted. The upregulated DEGs were primarily
associated with pathways in cancer, oxidative phosphorylation and
the cell cycle (Fig. 2A). The
downregulated DEGs were primarily associated with the regulation of
actin cytoskeleton, chemokine signaling pathway and focal adhesion
(Fig. 2B).
Overlapped genes between genes with
DMRs and DEGs
In order to reveal whether the genes with DMRs were
DEGs, genes with DMRs were compared with DEGs. It was identified
that 135 genes with hypermethylated DMRs were downregulated in the
NPC samples compared with their expression levels in the normal
controls, including prostaglandin D2 synthase, elongation factor
for RNA polymerase II 3, ATPase sarcoplasmic/endoplasmic reticulum
Ca2+ transporting 3 and claudin 3 (Table I).
 | Table I.Information regarding the
downregulated genes containing hypermethylated regions. |
Table I.
Information regarding the
downregulated genes containing hypermethylated regions.
| Site ID | Chr | Loc | Gene | Island | Mean
log2FCa | Mean β-value in NPC
samples | Mean β-value in
normal samples | Δβ (NPC vs.
normal) | FDR (NPC vs.
normal) |
|---|
| cg20450318 | 11 | 65415260 | SIPA1 |
chr11:65413778-65415203 | −0.72 |
6.57×10−1 |
4.65×10−1 |
1.92×10−1 |
1.30×10−4 |
| cg17953300 | 11 | 65418265 |
|
chr11:65419853-65420527 |
|
5.91×10−1 |
4.57×10−1 |
1.34×10−1 |
2.33×10−3 |
| cg16915828 | 11 | 73371940 | PLEKHB1 |
chr11:73371800-73372632 | −0.59 |
6.19×10−1 |
4.31×10−1 |
1.88×10−1 |
2.93×10−5 |
| cg07223180 | 13 | 20989142 | CRYL1 |
chr13:20989007-20989836 | −0.62 |
5.98×10−1 |
4.63×10−1 |
1.35×10−1 |
5.69×10−5 |
| cg21177426 | 15 | 37386586 | MEIS2 |
chr15:37387386-37387614 | −0.82 |
6.28×10−1 |
4.68×10−1 |
1.60×10−1 |
1.70×10−5 |
| cg24361265 | 15 | 44068668 | ELL3 |
chr15:44068586-44069792 | −1.24 |
6.36×10−1 |
4.32×10−1 |
2.04×10−1 |
2.51×10−5 |
| cg06786050 | 16 | 84401247 | ATP2C2 |
chr16:84401957-84402497 | −0.82 |
5.58×10−1 |
4.36×10−1 |
1.22×10−1 |
1.21×10−4 |
| cg26824780 | 16 | 89004908 | CBFA2T3 |
chr16:89006334-89008600 | −0.87 |
5.95×10−1 |
4.73×10−1 |
1.23×10−1 |
6.23×10−6 |
| cg09013975 | 17 | 3847872 | ATP2A3 |
chr17:3847999-3848570 | −1.06 |
6.68×10−1 |
4.90×10−1 |
1.78×10−1 |
4.51×10−5 |
| cg05247914 | 19 | 35629701 | FXYD1 |
chr19:35632356-35632572 | −0.62 |
6.76×10−1 |
4.83×10−1 |
1.93×10−1 |
3.71×10−5 |
| cg03078169 | 19 | 35629791 |
|
chr19:35632356-35632572 |
|
6.31×10−1 |
4.79×10−1 |
1.52×10−1 |
4.40×10−5 |
| cg27461196 | 19 | 35630106 |
|
chr19:35632356-35632572 |
|
5.59×10−1 |
4.51×10−1 |
1.08×10−1 |
1.81×10−4 |
| cg16334795 | 21 | 42538894 | BACE2 |
chr21:42539367-42540872 | −0.86 |
6.06×10−1 |
4.51×10−1 |
1.55×10−1 |
1.58×10−5 |
| cg08481491 | 3 | 125900108 | ALDH1L1 |
chr3:125898662-125899568 | −0.80 |
5.85×10−1 |
4.46×10−1 |
1.38×10−1 |
1.04×10−4 |
| cg04161526 | 6 | 31696519 | DDAH2 |
chr6:31695894-31698245 | −0.63 |
5.65×10−1 |
4.33×10−1 |
1.32×10−1 |
2.25×10−5 |
| cg21286967 | 6 | 31696710 |
|
chr6:31695894-31698245 |
|
6.01×10−1 |
4.71×10−1 |
1.30×10−1 |
3.27×10−5 |
| cg25526039 | 6 | 107813291 | SOBP |
chr6:107810066-107812733 | −0.81 |
5.53×10−1 |
4.36×10−1 |
1.17×10−1 |
4.45×10−4 |
| cg24419391 | 7 | 73183516 | CLDN3 |
chr7:73183379-73185115 | −1.02 |
5.89×10−1 |
4.67×10−1 |
1.21×10−1 |
1.64×10−3 |
| cg08580268 | 7 | 150038502 | RARRES2 |
chr7:150037459-150039031 | −0.94 |
5.66×10−1 |
3.95×10−1 |
1.71×10−1 |
9.63×10−5 |
| cg27494647 | 7 | 150038898 |
|
chr7:150037459-150039031 |
|
6.14×10−1 |
4.15×10−1 |
1.99×10−1 |
7.82×10−5 |
| cg11714502 | 9 | 130640212 | AK1 |
chr9:130639738-130640143 | −0.61 |
5.71×10−1 |
4.47×10−1 |
1.24×10−1 |
5.00×10−4 |
| cg02156769 | 9 | 139872246 | PTGDS |
chr9:139872237-139873143 | −1.81 |
5.58×10−1 |
4.56×10−1 |
1.01×10−1 |
1.95×10−5 |
| cg07390373 | X | 43741933 | MAOB |
chrX:43741299-43741827 | −0.78 |
5.96×10−1 |
4.88×10−1 |
1.08×10−1 |
3.94×10−5 |
| cg05605944 | X | 43741945 |
|
chrX:43741299-43741827 |
|
5.84×10−1 |
4.62×10−1 |
1.22×10−1 |
1.72×10−5 |
However, the 28 genes with hypomethylated DMRs were
not differentially expressed between the two group samples.
Functional associations of the DEGs
with DMRs
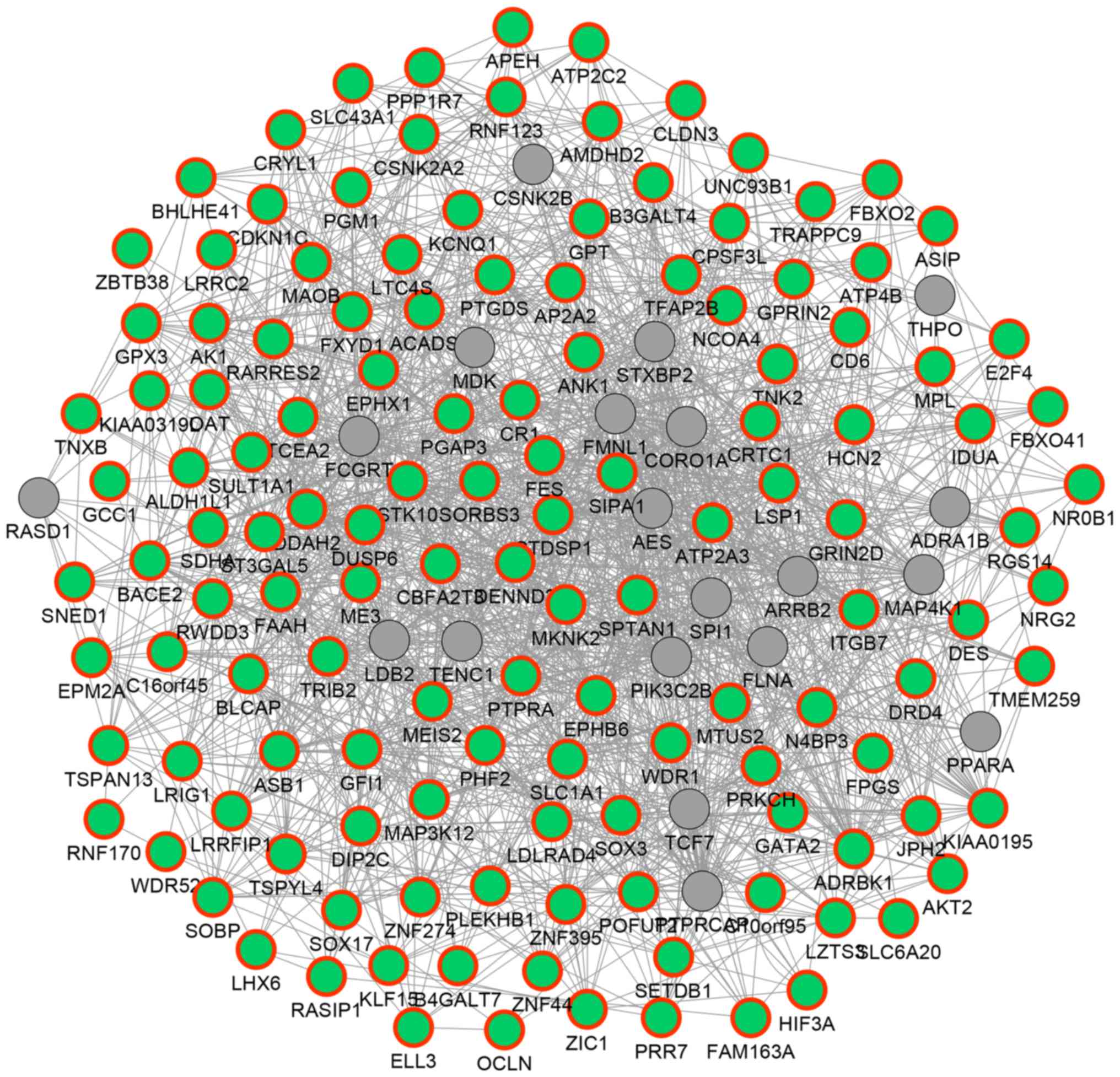
In order to reveal the potential functional
associations among the above 135 hypermethylated DEGs, a functional
network was constructed (Fig. 3). In
the network, 154 genes and 1,651 association pairs were included.
The association pairs included 1,155 co-expression associations, 24
physical interactions, 351 genetic interactions, 83 co-localization
associations and 38 associations between shared protein domains.
For example, DUSP6 was predicted to exhibit genetic
interactions with other hypermethylated DEGs such as malic enzyme 3
(ME3) and ST3 β-galactoside α-2,3-sialytansferase 5
(ST3GAL5); TNXB was predicted to be co-expressed with
genes such as EPH receptor B6 (EPHB6), aldehyde
dehydrogenase 1 family, member L1 (ALDH1L1) and glutathione
peroxidase 3 (GPX3).
According to the enrichment analysis, the 135
hypermethylated DEGs were significantly associated with the GO
functions of protein amino acid phosphorylation and phosphate
metabolic process, and the tight junction pathway (Table II).
| Table II.Results of GO and Kyoto Encyclopedia
of Genes and Genomes pathway enrichment analyses of the 135
downregulated genes containing hypermethylated regions. |
Table II.
Results of GO and Kyoto Encyclopedia
of Genes and Genomes pathway enrichment analyses of the 135
downregulated genes containing hypermethylated regions.
| Category | Term | P-value | Gene count | Genes |
|---|
| BP | GO:0006468~protein
amino acid phosphorylation | 0.0021 | 14 | STK10, DRD4,
PTPRA, MKNK2, PRKCH, ADRBK1, FES, TRIB2, CSNK2A2, EPHB6, TNK2, CD6,
MAP3K12, AKT2 |
|
|
GO:0006796~phosphate metabolic
process | 0.0087 | 16 | STK10, DRD4,
EPM2A, PTPRA, MKNK2, PRKCH, ADRBK1, FES, TRIB2, CSNK2A2, EPHB6,
TNK2, CD6, MAP3K12, AKT2, DUSP6 |
|
|
GO:0006793~phosphorus metabolic
process | 0.0087 | 16 | STK10, DRD4,
EPM2A, PTPRA, MKNK2, PRKCH, ADRBK1, FES, TRIB2, CSNK2A2, EPHB6,
TNK2, CD6, MAP3K12, AKT2, DUSP6 |
|
|
GO:0016310~phosphorylation | 0.0096 | 14 | STK10, DRD4,
PTPRA, MKNK2, PRKCH, ADRBK1, FES, TRIB2, CSNK2A2, EPHB6, TNK2, CD6,
MAP3K12, AKT2 |
|
|
GO:0006357~regulation of transcription
from RNA polymerase II promoter | 0.0113 | 13 | CDKN1C, GATA2,
SORBS3, MEIS2, E2F4, CRTC1, TFAP2B, GFI1, TCEA2, LRRFIP1, ELL3,
NtnxbR0B1, ZBTB38 |
| CC | GO:0044459~plasma
membrane part | 0.0118 | 27 | FXYD1, OCLN,
CLDN3, SLC6A20, DRD4, ADRBK1, PRR7, SORBS3, EPHB6, ANK1, ST3GAL5,
ITGB7, GRIN2D, CD6, SLC43A1, KCNQ1, SLC1A1, HCN2, CR1, PTPRA, MAOB,
TSPAN13, ACTN3, LSP1, AP2A2, MPL, SPTAN1 |
|
| GO:0005887~integral
to plasma membrane | 0.0166 | 17 | FXYD1, HCN2,
CR1, CLDN3, SLC6A20, DRD4, PTPRA, TSPAN13, EPHB6, ST3GAL5, GRIN2D,
ITGB7, MPL, CD6, KCNQ1, SLC1A1, SLC43A1 |
|
|
GO:0031226~intrinsic to plasma
membrane | 0.0201 | 17 | FXYD1, HCN2,
CR1, CLDN3, SLC6A20, DRD4, PTPRA, TSPAN13, EPHB6, ST3GAL5, GRIN2D,
ITGB7, MPL, CD6, KCNQ1, SLC1A1, SLC43A1 |
|
|
GO:0005626~insoluble fraction | 0.0247 | 13 | DES, JPH2,
BACE2, GRIN2D, EPHX1, TSPAN13, ADRBK1, LTC4S, NR0B1, SLC1A1,
MAP3K12, SPTAN1, AKT2 |
|
| GO:0000267~cell
fraction | 0.0336 | 15 | JPH2, TSPAN13,
EPHX1, ADRBK1, LTC4S, NR0B1, DES, BACE2, GRIN2D, GPX3, SLC1A1,
MAP3K12, AKT2, SPTAN1, DUSP6 |
| MF |
GO:0008134~transcription factor
binding | 0.0009 | 13 | ZNF274, E2F4,
CRTC1, NR0B1, GATA2, SORBS3, MEIS2, DIP2C, NCOA4, GPX3, TFAP2B,
SOX17, DDAH2 |
|
| GO:0048037~cofactor
binding | 0.0156 | 7 | SDHA, CRYL1,
ME3, ALDH1L1, ACADS, GPT, OAT |
|
|
GO:0005200~structural constituent of
cytoskeleton | 0.0220 | 4 | SORBS3, ANK1,
DES, SPTAN1 |
|
| GO:0004672~protein
kinase activity | 0.0247 | 11 | CSNK2A2, EPHB6,
STK10, MKNK2, PRKCH, ADRBK1, TNK2, FES, MAP3K12, TRIB2,
AKT2 |
| Pathway | hsa04530: Tight
junction | 0.0036 | 7 | CSNK2A2, OCLN,
CLDN3, PRKCH, ACTN3, SPTAN1, AKT2 |
Discussion
In the present study, 2,234 hypermethylated CpG site
probes corresponding to 1,676 gene symbols, 28 hypomethylated CpG
site probes corresponding to 28 gene symbols and 17 DMIs were
identified based on analysis of the methylation profiling dataset.
Furthermore, 2,983 DEGs (1,655 upregulated and 1,328 downregulated)
were identified based on the three gene expression profiling
datasets. Among these DEGs, 135 downregulated genes were
hypermethylated, including DUSP6 and TNXB, which were
also among the 17 DMIs identified.
DUSP6 encodes dual specificity phosphatase 6,
also termed mitogen-activated protein kinase phosphatase 3, which
belongs to the dual specificity protein phosphatase subfamily
(24). Phosphatases in this family
inactivate their target kinases, such as members of the
mitogen-activated protein kinase superfamily, which are involved in
cellular proliferation and differentiation (25). In the present study, DUSP6 was
identified to be hypermethylated and downregulated in NPC samples
compared with its methylation status in the normal controls, which
was consistent with other studies (14,26).
DUSP6 has been identified as a tumor suppressor, and it is
able to inhibit epithelial-mesenchymal transition (EMT) and cell
invasion by negatively modulating the activity of
extracellular-signal-regulated kinase in NPC (26). In the present study, DUSP6 was
predicted to exhibit genetic interactions with other
hypermethylated DEGs such as ME3 and ST3GAL5.
ME3 is a mitochondrial nicotinamide adenine dinucleotide
phosphate(+) -dependent enzyme (27), and it serves a unique role in tumor
mitochondria (28). The protein
encoded by ST3GAL5 is a sialyltransferase, a type II
membrane protein that catalyzes the formation of
α-2,3-sialyltransferase (GM3), a protein participating in cell
differentiation and cell adhesion (29). There is no other evidence to indicate
the associations of ME3 and ST3GAL5 with NPC at
present. Therefore, ME3 and ST3GAL5 may be potential
novel biomarker molecules in the progression of NPC.
TNXB was also identified to be
hypermethylated and downregulated in NPC samples compared with its
methylation status in the normal controls, which was consistent
with previous studies (6,12). In Epstein-Barr virus-positive gastric
cancer and pancreatic cancer, TNXB was also hypermethylated
(30,31). TNXB encodes a tenascin, which
exhibits an anti-adhesive effect (32). It is able to promote EMT by activating
latent transforming growth factor-β (33). In malignancy, TNXB is usually
suppressed, and it has been identified as a marker for malignant
mesothelioma (34). Furthermore, in
the present study, TNXB was predicted to be co-expressed
with a set of other hypermethylated DEGs, including EPHB6,
ALDH1L1 and GPX3. EPH receptor B6 (EPHB6) encodes
a transmembrane protein, which may affect cell adhesion and
migration. In tumor progression, EPHB6 is usually
downregulated due to promoter DNA hypermethylation (35,36). It
has been suggested that EPHB6 alters invasiveness, and is
associated with the prognosis and/or diagnosis of breast carcinoma
(35,37). ALDH1L1 and GPX3 have
been previously identified to be silenced by methylation, which is
associated with tumorigenesis (38,39).
Although there is no experimental evidence to confirm the
involvement of EPHB6, ALDH1L1 and GPX3 in NPC, the
present study hypothesizes that these molecules may also serve a
significant role in the progression of NPC, along with
TNXB.
Additionally, there are several limitations in the
present study. The expression levels and methylation status of the
aforementioned genes are required to be validated using
experiments. Functional associations of these genes also need to be
confirmed. These validations will be performed and presented
separately. Despite the absence of experiments, several genes such
as ME3, ST3GAL5, EPHB6, ALDH1L1 and GPX3 were
primarily identified to be potentially associated with NPC, and
they may become novel therapeutic targets for NPC, once
validated.
In conclusion, based on the comprehensive analysis
of methylation profiling and gene expression profiling, 135
downregulated genes were identified to be hypermethylated in NPC
compared with its methylation status in the controls in the present
study. Among them, DUSP6 and TNXB contained DMIs.
Hypermethylated DEGs that exhibited genetic interactions with
DUSP6, including ME3 and ST3GAL5, and genes
that co-expressed with TNXB, including EPHB6, ALDH1L1
and GPX3, may be potential novel molecules involved in the
progression of NPC, and they may become novel therapeutic targets
for NPC.
Acknowledgements
The present study was supported by a grant from the
Natural Science Foundation of Liaoning Province of China (grant no.
201202287).
Glossary
Abbreviations
Abbreviations:
|
NPC
|
nasopharyngeal carcinoma
|
|
DMRs
|
differentially methylated regions
|
|
DMIs
|
differentially methylated CpG
islands
|
|
DEGs
|
differentially expressed genes
|
|
PCDH8
|
protocadherin 8
|
|
FEZF2
|
FEZ family zinc finger 2
|
|
ASS1
|
argininosuccinate synthetase
|
|
KLHDC4
|
Kelch domain containing 4
|
|
GEO
|
Gene Expression Omnibus
|
|
BMIQ
|
β-mixture quantile normalization
method
|
|
FC
|
fold-change
|
|
BP
|
biological process
|
|
CC
|
cell component
|
|
MF
|
molecular function
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
MAP
|
mitogen-activated protein
|
|
EMT
|
epithelial-mesenchymal transition
|
|
ME3
|
malic enzyme 3
|
|
GM3
|
α-2,3-sialyltransferase
|
|
EPHB6
|
EPH Receptor B6
|
|
ALDH1L1
|
aldehyde dehydrogenase 1 family,
member L1
|
|
GPX3
|
glutathione peroxidase 3
|
References
|
1
|
Hildesheim A and Wang CP: Genetic
predisposition factors and nasopharyngeal carcinoma risk: A review
of epidemiological association studies, 2000–2011: Rosetta Stone
for NPC: Genetics, viral infection, and other environmental
factors. Semin Cancer Biol. 22:107–116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wei WI and Sham JS: Nasopharyngeal
carcinoma. Lancet. 365:2041–2054. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang TJ, Riaz N, Cheng SK, Lu JJ and Lee
NY: Intensity-modulated radiation therapy for nasopharyngeal
carcinoma: A review. J Radiat Oncol. 1:129–146. 2012. View Article : Google Scholar
|
|
4
|
Meissner A, Mikkelsen TS, Gu H, Wernig M,
Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB,
et al: Genome-scale DNA methylation maps of pluripotent and
differentiated cells. Nature. 454:766–770. 2008.PubMed/NCBI
|
|
5
|
Irizarry RA, Ladd-Acosta C, Wen B, Wu Z,
Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al:
The human colon cancer methylome shows similar hypo- and
hypermethylation at conserved tissue-specific CpG island shores.
Nat Genet. 41:178–186. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dai W, Cheung AK, Ko JM, Cheng Y, Zheng H,
Ngan RK, Ng WT, Lee AW, Yau CC, Lee VH and Lung ML: Comparative
methylome analysis in solid tumors reveals aberrant methylation at
chromosome 6p in nasopharyngeal carcinoma. Cancer Med. 4:1079–1090.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
He D, Zeng Q, Ren G, Xiang T, Qian Y, Hu
Q, Zhu J, Hong S and Hu G: Protocadherin8 is a functional tumor
suppressor frequently inactivated by promoter methylation in
nasopharyngeal carcinoma. Eur J Cancer Prev. 21:569–575. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shu XS, Li L, Ji M, Cheng Y, Ying J, Fan
Y, Zhong L, Liu X, Tsao SW, Chan AT and Tao Q: FEZF2, a novel 3p14
tumor suppressor gene, represses oncogene EZH2 and MDM2 expression
and is frequently methylated in nasopharyngeal carcinoma.
Carcinogenesis. 34:1984–1993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lan J, Tai HC, Lee SW, Chen TJ, Huang HY
and Li CF: Deficiency in expression and epigenetic DNA Methylation
of ASS1 gene in nasopharyngeal carcinoma: Negative prognostic
impact and therapeutic relevance. Tumour Biol. 35:161–169. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou W, Feng X, Ren C, Jiang X, Liu W,
Huang W, Liu Z, Li Z, Zeng L, Wang L, et al: Over-expression of
BCAT1, a c-Myc target gene, induces cell proliferation, migration
and invasion in nasopharyngeal carcinoma. Mol Cancer. 12:532013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lian YF, Yuan J, Cui Q, Feng QS, Xu M, Bei
JX, Zeng YX and Feng L: Upregulation of KLHDC4 predicts a poor
prognosis in human nasopharyngeal carcinoma. PLoS One.
11:e01528202016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sengupta S, den Boon JA, Chen IH, Newton
MA, Dahl DB, Chen M, Cheng YJ, Westra WH, Chen CJ, Hildesheim A, et
al: Genome-wide expression profiling reveals EBV-associated
inhibition of MHC class I expression in nasopharyngeal carcinoma.
Cancer Res. 66:7999–8006. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bao YN, Cao X, Luo DH, Sun R, Peng LX,
Wang L, Yan YP, Zheng LS, Xie P, Cao Y, et al: Urokinase-type
plasminogen activator receptor signaling is critical in
nasopharyngeal carcinoma cell growth and metastasis. Cell Cycle.
13:1958–1969. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bose S, Yap LF, Fung M, Starzcynski J,
Saleh A, Morgan S, Dawson C, Chukwuma MB, Maina E, Buettner M, et
al: The ATM tumour suppressor gene is down-regulated in
EBV-associated nasopharyngeal carcinoma. J Pathol. 217:345–352.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Norfo R, Zini R, Pennucci V, Bianchi E,
Salati S, Guglielmelli P, Bogani C, Fanelli T, Mannarelli C, Rosti
V, et al: miRNA-mRNA integrative analysis in primary myelofibrosis
CD34+ cells: Role of miR-155/JARID2 axis in abnormal
megakaryopoiesis. Blood. 124:e21–e32. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Teschendorff AE, Marabita F, Lechner M,
Bartlett T, Tegner J, Gomez-Cabrero D and Beck S: A beta-mixture
quantile normalization method for correcting probe design bias in
Illumina Infinium 450 k DNA methylation data. Bioinformatics.
29:189–196. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smyth GK: Limma: Linear models for
microarray dataBioinformatics and computational biology solutions
using R and Bioconductor. Springer; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
19
|
Warden CD, Lee H, Tompkins JD, Li X, Wang
C, Riggs AD, Yu H, Jove R and Yuan YC: COHCAP: An integrative
genomic pipeline for single-nucleotide resolution DNA methylation
analysis. Nucleic Acids Res. 41:e1172013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi C, Hong L, Cheng Z and Yin Q:
Identification of metastasis-associated genes in colorectal cancer
using metaDE and survival analysis. Oncol Lett. 11:568–574.
2016.PubMed/NCBI
|
|
21
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Warde-Farley D, Donaldson SL, Comes O,
Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT,
et al: The GeneMANIA prediction server: Biological network
integration for gene prioritization and predicting gene function.
Nucleic Acids Res. 38:W214–W220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zuberi K, Franz M, Rodriguez H, Montojo J,
Lopes CT, Bader GD and Morris Q: GeneMANIA prediction server 2013
update. Nucleic Acids Res. 41:W115–W122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smith A, Price C, Cullen M, Muda M, King
A, Ozanne B, Arkinstall S and Ashworth A: Chromosomal localization
of three human dual specificity phosphatase genes (DUSP4, DUSP6 and
DUSP7). Genomics. 42:524–527. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jurek A, Amagasaki K, Gembarska A, Heldin
CH and Lennartsson J: Negative and positive regulation of MAPK
phosphatase 3 controls platelet-derived growth factor-induced Erk
activation. J Biol Chem. 284:4626–4634. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wong VC, Chen H, Ko JM, Chan KW, Chan YP,
Law S, Chua D, Kwong DL, Lung HL, Srivastava G, et al: Tumor
suppressor dual-specificity phosphatase 6 (DUSP6) impairs cell
invasion and epithelial-mesenchymal transition (EMT)-associated
phenotype. Int J Cancer. 130:83–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Loeber G, Maurer-Fogy I and Schwendenwein
R: Purification, cDNA cloning and heterologous expression of the
human mitochondrial NADP(+)-dependent malic enzyme. Biochem J.
304:687–692. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wise DR and Thompson CB: Glutamine
addiction: A new therapeutic target in cancer. Trends Biochem Sci.
35:427–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishii A, Ohta M, Watanabe Y, Matsuda K,
Ishiyama K, Sakoe K, Nakamura M, Inokuchi J, Sanai Y and Saito M:
Expression cloning and functional characterization of human cDNA
for ganglioside GM3 synthase. J Biol Chem. 273:31652–31655. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao J, Liang Q, Cheung KF, Kang W, Lung
RW, Tong JH, To KF, Sung JJ and Yu J: Genome-wide identification of
Epstein-Barr virus-driven promoter methylation profiles of human
genes in gastric cancer cells. Cancer. 119:304–312. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimizu H, Horii A, Sunamura M, Motoi F,
Egawa S, Unno M and Fukushige S: Identification of epigenetically
silenced genes in human pancreatic cancer by a novel method
‘microarray coupled with methyl-CpG targeted transcriptional
activation’ (MeTA-array). Biochem Biophys Res Commun. 411:162–167.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chiovaro F, Chiquet-Ehrismann R and
Chiquet M: Transcriptional regulation of tenascin genes. Cell Adh
Migr. 9:34–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alcaraz LB, Exposito JY, Chuvin N, Pommier
RM, Cluzel C, Martel S, Sentis S, Bartholin L, Lethias C and
Valcourt U: Tenascin-X promotes epithelial-to-mesenchymal
transition by activating latent TGF-β. J Cell Biol. 205:409–428.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan Y, Nymoen DA, Stavnes HT, Rosnes AK,
Bjørang O, Wu C, Nesland JM and Davidson B: Tenascin-X is a novel
diagnostic marker of malignant mesothelioma. Am J Surg Pathol.
33:1673–1682. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fox BP and Kandpal RP: DNA-based assay for
EPHB6 expression in breast carcinoma cells as a potential
diagnostic test for detecting tumor cells in circulation. Cancer
Genomics Proteomics. 7:9–16. 2010.PubMed/NCBI
|
|
36
|
Yu J, Bulk E, Ji P, Hascher A, Tang M,
Metzger R, Marra A, Serve H, Berdel WE, Wiewroth R, et al: The
EPHB6 receptor tyrosine kinase is a metastasis suppressor that is
frequently silenced by promoter DNA hypermethylation in non-small
cell lung cancer. Clin Cancer Res. 16:2275–2283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fox BP and Kandpal RP: EphB6 receptor
significantly alters invasiveness and other phenotypic
characteristics of human breast carcinoma cells. Oncogene.
28:1706–1713. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
He Y, Wang Y, Li P, Zhu S, Wang J and
Zhang S: Identification of GPX3 epigenetically silenced by CpG
methylation in human esophageal squamous cell carcinoma. Dig Dis
Sci. 56:681–688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Oleinik NV, Krupenko NI and Krupenko SA:
Epigenetic silencing of ALDH1L1, a metabolic regulator of cellular
proliferation, in cancers. Genes Cancer. 2:130–139. 2011.
View Article : Google Scholar : PubMed/NCBI
|