Introduction
Liver cancer is a type of malignancy prevalent in
less-developed regions, and was the fifth most common cancer in
males and the ninth in females worldwide in 2012; it is also the
second most common cause of cancer-associated mortality (1). Developing an optimum therapeutic
strategy is one of the major aims of clinical studies at
present.
It has been identified that RAS proteins are
involved in a number of cellular processes, including migration,
proliferation, differentiation and survival (2). RAS protein activator like 1 (RASAL1) is
a member of the RAS GTPase-activating protein (GAP) family, and has
been revealed as downregulated in several solid tumors (3), and also to function as a tumor
suppressor gene that negatively modulates the RAS signaling pathway
by catalyzing RAS inactivation (4).
Previously, evidence has indicated that RASAL1 levels are
correlated with liver injury and hepatic fibrosis (5,6). However,
little is known about the association between RASAL1 and liver
cancer.
Hypoxia-inducible factor (HIF)-1 and HIF-2 are
transcription factors that serve major roles in the cellular
responses to hypoxia, and have recently been considered mediators
of cancer progression and targets for cancer therapy (7). HIF-1α has been identified as a positive
factor for tumor growth, and increased HIF-1α activation was
correlated with the development of more aggressive carcinogenic
phenotypes (8,9). Not only a prognostic marker, high HIF-2α
levels have also been associated with advanced stages or poor
patient outcomes in several types of tumor (10), HIF-2α has also been suggested to serve
an important role in the development of various diseases:
HIF-2α-null embryos have exhibited vascular disorganization
throughout the yolk sac and the embryo itself (11), and can perish due to adrenal
insufficiency, although they may survive with adrenal catecholamine
replacement therapy (12). A prior
study identified that HIF-2α-mediated hypoxic signaling and hepatic
insulin action may modulate glucose metabolism (13,14), and
may participate in the postprandial hepatic glucagon response
(15). Increased metabolic autonomy,
nutrient absorbance and metabolism to support growth and
proliferation has been demonstrated among diverse tumor types
(16), so targeting metabolic
transformation is a promising strategy for cancer therapy.
Therefore, the present study focused on the glucose metabolism
effect to clarify the correlation between RASAL1 and HIF-2α in
liver cancer development.
In the present study, it was identified that RASAL1
was significantly downregulated in liver cancer tissues compared
with the corresponding non-tumor tissues, and may serve as an
independent predictor for the overall survival of patients with
liver cancer. Furthermore, RASAL1 regulated cell proliferation and
invasion through its inhibitory effect on HIF-2α, which may partly
account for HIF-2α-mediated gluconeogenesis via the extracellular
signal-related kinase (ERK)1/2 pathway, thus affecting the
proliferation of liver cancer cells in vitro and in
vivo. This assisted understanding of the tumor suppressive
function of RASAL1. In addition, the present study aimed to reveal
a novel regulatory mechanism of RASAL1 in the development of liver
cancer, and provide a novel direction for its clinical
application.
Materials and methods
Tissue collection and ethics
statement
A total of 16 primary human liver cancer tissues and
adjacent non-tumor tissues were collected from patients who had
undergone surgery at the Linyi People's Hospital (Linyi, China)
between August 2013 and October 2015. All patients had not received
chemotherapy or radiotherapy prior to surgery. The study was
approved by the Linyi People's Hospital Ethics Committee, and was
performed in compliance with the Declaration of Helsinki
Principles. Written informed consent was obtained for all patient
samples. The animal experiments were performed with the approval of
The Institutional Committee for Animal Research of Linyi People's
Hospital and in conformity with National Guidelines for the Care
and Use of Laboratory Animals (17).
Cell culture
Hepatoblastoma HepG2 cells (18) were purchased from the American Type
Culture Collection (Manassas, VA, USA) and cultured at 37°C with 5%
CO2 in Dulbecco's Modified Eagle's Medium (DMEM, Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal calf serum (Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA), 4.5 g/l glucose, 2 mM L-glutamine (Thermo Fisher Scientific,
Inc.,), 100 U/ml penicillin and 100 µg/ml streptomycin.
Plasmids and cell transfection
For RASAL1 overexpression experiments, HepG2 cells
(5×106 per reaction) were transfected with 1 µg
pcDNA3.1-RASAL1 plasmid at 37°C for 48 h, designed and synthesized
by Shanghai GenePharma Co., Ltd (Shanghai, China), using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The RASAL1-overexpressing
HepG2 stable cell line was generated using 10 µg/ml G418
(Invitrogen; Thermo Fisher Scientific, Inc.) in the culture medium
for 4 weeks, and the resulting single clones were expanded to
obtain stably transfected cells. Cells transfected with an empty
vector and un-transfected cells were used as controls. To detect
the function role of ERK1/2 in RASAL1 mediated HIF-2α expression,
100 nM ERK1/2 inhibitor SCH772984 (S7101, Selleck Chemicals,
Houston, TX, USA) was added to the medium 24 h after transfection
to inhibit the activation of ERK1/2.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analyses
The total RNA was extracted from tissues or cultured
cells with TRIzol® reagent (Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Briefly, tissues or
cells were homogenized and RNA was isolated following phase
separation with chloroform, precipitated with 80% isopropanol,
washed twice with 75% ethanol, and finally re-dissolved in water.
RNA concentration was determined by UV spectrophotometry (NanoDrop
2000; Thermo Fisher Scientific, Inc., Wilmington, DE, USA). A
volume of 1 µg total RNA was reverse transcribed to a final volume
of 20 µl, using random primers under standard conditions with the
PrimeScript RT Reagent kit and gDNA Eraser (Takara Biotechnology
Co., Ltd., Dalian, China; cat. no. RR047A). Following the RT
reaction, 1 µl cDNA was used for subsequent RT-qPCR reactions (SYBR
Premix Ex Taq; Takara Biotechnology Co., Ltd., according to the
manufacturer's protocol. Sequences of all primers are summarized in
Table I. The RT-qPCR and data
collection were carried out on an ABI 7500 real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The reaction
was initially denatured (95°C for 15 sec), followed by 40 cycles of
95°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec, with a final
melting curve analysis of the fluorescence performed between 60°C
and 95°C with increments of 0.5°C every 10 sec. The
2−ΔΔCq value was calculated for every sample, finally
the mRNA expression levels were indicated with 2−ΔΔCq
and normalized to GAPDH (19).
 | Table I.Primers sequences, PCR conditions and
products sizes. |
Table I.
Primers sequences, PCR conditions and
products sizes.
| Gene | Sequence | Annealing
Temperature, °C | PCR product size
(bp) |
|---|
| HIF-1α
(NM_181054) | Forward:
5′-GAACGTCGAAAAGAAAAGTCTCG-3′ | 60 | 124 |
|
| Reverse:
5′-CCTTATCAAGATGCGAACTCACA-3′ |
|
|
| HIF-1β
(NM_001197325) | Forward:
5′-TAGTGCCCTGGCTCGAAAAC-3′ | 61 | 239 |
|
| Reverse:
5′-GGTTCAAAACAGGAGTCACGG-3′ |
|
|
| HIF-2α
(NM_001430) | Forward:
5′-CGGAGGTGTTCTATGAGCTGG-3′ | 62 | 115 |
|
| Reverse:
5′-AGCTTGTGTGTTCGCAGGAA-3′ |
|
|
| HIF-3α
(NM_022462.4) | Forward:
5′-CCTGTGGAGTCATCTCACCG-3′ | 60 | 151 |
|
| Reverse:
5′-GACTTTTCCTTGCGCAGCTC-3′ |
|
|
| RASAL1
(NM_004658) | Forward:
5′-CAGCTCCCTGAATGTTCGC-3′ | 61 | 216 |
|
| Reverse:
5′-TCCTCATCCAGCACGTAGAAG-3′ |
|
|
| PEPCK
(NM_001018073) | Forward:
5′-AGTAGAGAGCAAGACGGTGAT-3′ | 60 | 179 |
|
| Reverse:
5′-TGCTGAATGGAAGCACATACAT-3′ |
|
|
| G6Pase
(NM_000151) | Forward:
5′-CTACTACAGCAACACTTCCGTG-3′ | 61 | 160 |
|
| Reverse:
5′-GGTCGGCTTTATCTTTCCCTGA-3′ |
|
|
| GAPDH
(NM_001256799) | Forward:
5′-GGAGCGAGATCCCTCCAAAAT-3′ | 60 | 197 |
|
| Reverse:
5′-GGCTGTTGTCATACTTCTCATGG-3′ |
|
|
MTT assay
A 100 µl suspension of HepG2 cells was seeded into a
96-well plate following transfection for different times (0, 24,
48, 72 and 96 h), at a density of 0.5×105 cells/well at
37°C, Following this, MTT was added to each well at a final
concentration of 0.5 mg/ml for 4 h, and the resulting formazan
crystals were dissolved in dimethyl sulfoxide. Optical density was
measured at 490 nm using a plate microreader (Tecan Austria GmbH,
Grodig, Austria). The growth inhibition ratio was calculated for
three independent repeats.
Transwell chamber assay
HepG2 cells that stably expressed RASAL1 and the
control cells were trypsinized with 0.25% phenol red trypsin (cat
no. 25200056; Thermo Fisher Scientific, Inc.), centrifuged at room
temperature for 3 min at 100 × g and resuspended in serum-free
DMEM. A total of ×105 HepG2 cell suspension was added to
the upper wells of Transwell chambers (Corning Incorporated,
Corning, NY, USA) pre-coated with matrigel (to observe migration
ability, upper Transwell chamber wells were not coated with
Matrigel). The medium was added to the lower chamber. Subsequent to
culturing at 37°C, the cells for 48 h, the cells remaining in the
upper chamber were removed with a cotton swab. The wells were
washed twice with PBS and stained at room temperature for 10 min
with 2 mg/ml crystal violet. The migrated/invaded cells were
counted under a light microscope at magnification, ×200 in at least
6 fields of view. The experiments were repeated three times.
Cell proliferation assay
To measure the effect of RASAL1 on proliferation
activity, 3×103 cells/well HepG2 cells were plated onto
96-well plates. Following overnight culture at 37°C, HepG2 cells
were transfected with 100 ng/well pcDNA3.1 or RASAL1 using
Lipofectamine® 2000 at 37°C, and after 24 h of
incubation at 37°C, cell proliferation was measured with a BrdU
assay kit (Roche Applied Science, Penzberg, Germany) in accordance
with the manufacturer's protocol. All experiments were repeated
three times independently.
Western blot analysis
Liver cancer tissues and HepG2 cells were collected,
lysed with radioimmunoprecipitation assay lysis buffer (50 mM
Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium
deoxycholate, 0.1% SDS, 1 mM EGTA, 1 mM EDTA) containing complete
protease inhibitor (Roche Applied Science) on ice for 30 min and
subjected to protein extraction. A total of 25 µg total lysate per
sample was separated on 10% SDS-PAGE gels and then transferred onto
nitrocellulose membranes. Specific monoclonal anti-RASAL1 (cat. no.
ab170711; 1:1,000 dilution), anti-HIF-2α (cat. no. ab73895; 1:1,000
dilution) and anti-glucose 6-phosphatase (G6Pase; cat. no. ab83690;
1:1,000 dilution) primary antibodies (all Abcam, Cambridge, MA,
USA), and anti-ERK1/2 (cat. no. 4695; 1:1,000 dilution),
anti-phospho-ERK1/2 (cat. no. 4370; 1:1,000 dilution) and
anti-phosphoenolpyruvate carboxy kinase (PEPCK; cat. no. 8565;
1:1,000 dilution) (all Cell Signaling Technology, Inc., Danvers,
MA, USA) were used. An anti-rabbit horseradish
peroxidase-conjugated secondary antibody (1:5,000 dilution, cat.
no. 7074; Cell Signaling Technology, Inc.,) was also used. West
Pico Chemiluminescent substrate kit (Pierce; Thermo Fisher
Scientific, Inc.) was used as a substrate to visualize the protein
bands, which were quantified using densitometry image analysis
software version 3.0 (Image Master VDS; Pharmacia Biotech; GE
Healthcare, Chicago, IL, USA). Normalization was performed using
β-actin (cat. no. ab6276; 1:3,000 dilution, Abcam) expression.
Glucose production
A total of 48 h after HepG2 cells were transfected
with pcDNA3.1 or RASAL1, cells were washed twice with PBS and then
incubated at 37°C with KRB buffer for 2 h. Following incubation,
0.5 mM pyruvate and 1 mM lactate were added to the KRB buffer and
incubated at 37°C for an additional 4 h. Glucose release was
measured using a glucose LiquiColor® diagnostic kit
(Stanbio Laboratories; EKF Diagnostics, Inc., Boerne, TX, USA).
Plasmid construction and luciferase
assay
The entire human HIF-2α 3′-untranslated region
segment was amplified by PCR using homo genomic DNA extracted from
HepG2 cell as a template using Prime STAR® HS DNA
Polymerase (R045Q, Takara Biotechnology Co., Ltd.). Primers were as
follows: Forward: 5′-CCGCTCGAGGCCAGGCCTTCTACCTGGGCAGCACC-3′,
Reverse: 5′-GAATGCGGCCGCTAGGATCAGAATACTTTAATAAGATACC-3′, and the
thermocycling conditions were as follows: 95°C initial denaturation
for 5 min; 95°C degeneration for 1 min, 56.1°C annealing for 1 min,
72°C for 1 min in 35 cycles; 72°C extension for 10 min with MluI
and XhoI overhangs. The PCR products were inserted into MluI and
XhoI sites in the pGL3-Basic (Ambion; Thermo Fisher Scientific,
Inc.) vector to obtain the reporter pGL3-HIF-2a construct. MluI
(cat no. 1071B) and XhoI (cat no. 1094B) restriction enzymes were
obtained from Takara Biotechnology Co., Ltd., (Dalian, China). For
the luciferase reporter assays, 0.3 µg pGL3-HIF-2α plasmid, 0.3 µg
Renilla luciferase (Ambion; Thermo Fisher Scientific, Inc.)
and 0.3 µg pcDNA3.1 or RASAL1 were transfected into HepG2 cells
using Lipofectamine® 2000 (Thermo Fisher Scientific,
Inc.) in 6-well plates at 37°C for 48 h. Renilla was used as
the transfection control. A total of 48 h after transfection, cells
were assayed using Dual-Luciferase Reporter Assay kits (E1910,
Promega Corporation, Madison, WI, USA).
Oxygen consumption rate (OCR)
Measurement of the OCR was performed using a
Seahorse XF96 analyzer (Seahorse Bioscience; Agilent Technologies,
Inc., North Billerica, MA, USA). HepG2 cells were transfected with
pcDNA3.1 or RASAL1 using Lipofectamine® 2000 at 37°C for
48 h, and then resuspended with un-buffered medium and seeded at
1×105 cells/well in XF96 plates. Cells were equilibrated
in the un-buffered medium for 45 min at 37°C in a
CO2-free incubator, prior to being transferred to the
XF96 analyzer. Basal OCR and the change in oxygen consumption were
measured upon treatment with oligomycin and carbonyl cyanide
p-trifluoromethoxyphenylhydrazone in succession, according to the
manufacturer's protocol (cat no. 103344-100, Seahorse Bioscience;
Agilent Technologies, Inc., Santa Clara, CA, USA).
In vivo tumor study
A total of 20 male nude mice (4–6 weeks; 18–20 g)
were purchased from the Model Animal Research Center of Shandong
University (Jinan, China). The animals were housed in a
temperature- (20–26°C) and humidity- (40–70%) controlled room with
a 12:12 light: dark cycle, and provided free access to food and
water. After 1week adaptive feeding, 20 mice were randomly divided
into two groups: The control group was injected with control HepG2
cells and the RASAL1 group, which was injected with
RASAL1-overexpressing HepG2 cells. A total of 5×106
RASAL1-overexpressing HepG2 stable or control cells were injected
subcutaneously into the right flank of each mouse. Tumor volumes
were determined every 5 days after injection and calculated as
described previously (20). Mice were
sacrificed by CO2 asphyxiation in a 1.5 l cage with 1.8
m3/min CO2 flow rate, approximately 5 min
later, the mice died the death was confirmed by observing no
spontaneous breathing for 2–3 min and no blink reflex, the final
concentration of CO2 in the cage reach approximately
80%. Tumors were dissected for RT-qPCR and western blot analysis.
All animal studies were performed in strict accordance with the
recommendations in the Guide for the Care and Use of Laboratory
Animals of the Linyi People's Hospital and the Research Institute
Animal Care and Use Committee. All protocols were approved by the
Shandong Cancer Hospital and Research Institute Animal Care and Use
Committee (approval number, 1040608). All surgery was performed
under sodium pentobarbital anesthesia (60 mg/kg, i.p.), and all
efforts were made to minimize suffering.
Statistical analysis
The results are expressed as mean ± standard error
of the mean from ≥3 independent experiments. Data between the
groups were analyzed using the Student's t-test or one-way analysis
of variance, followed by the Bonferroni-Dunn multiple comparisons
test with SPSS statistical software program (v 20.0; IBM Corp.,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
RASAL1 is downregulated in human liver
cancer tissues and is associated with poor prognosis
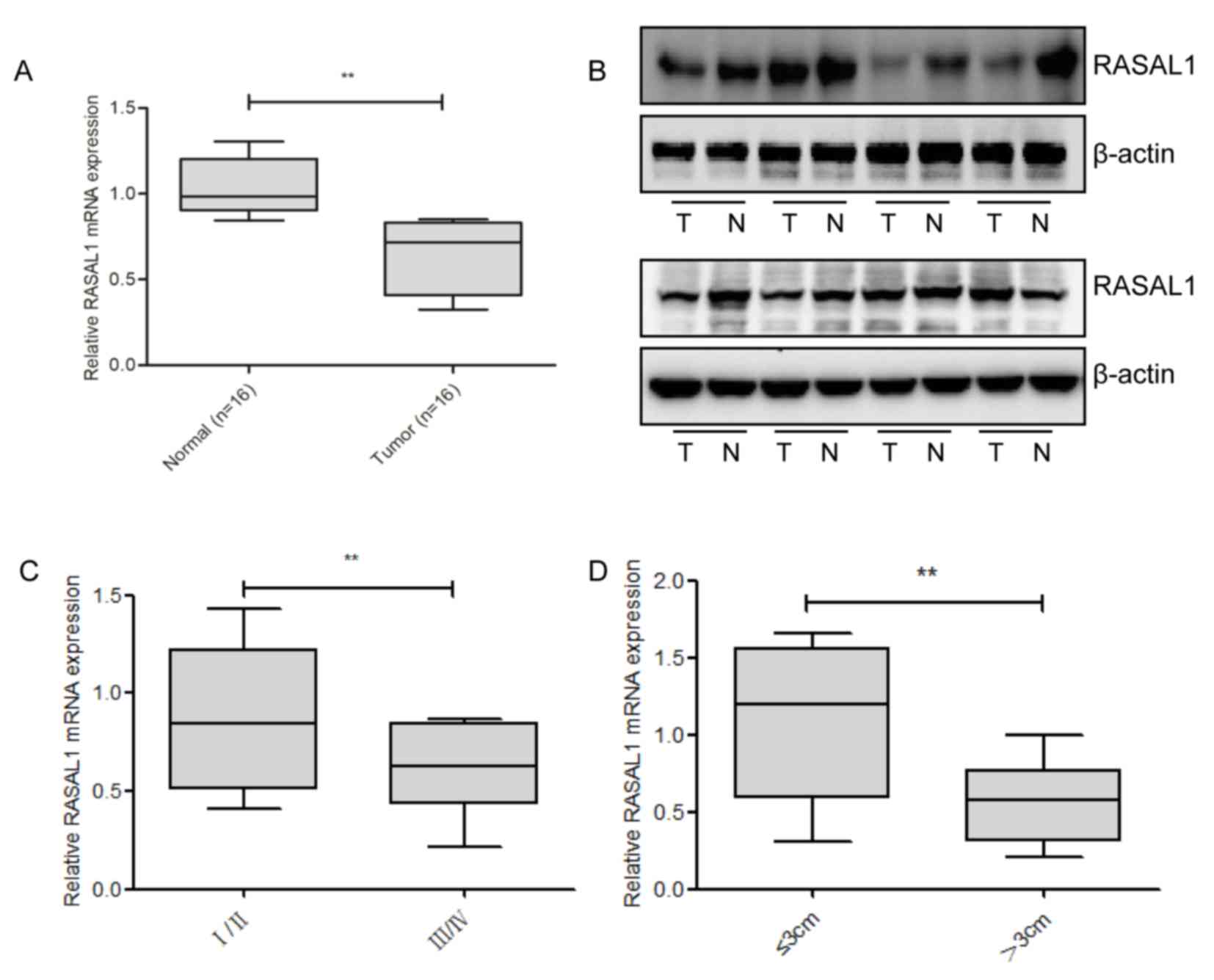
To detect the levels of RASAL1 expression, RT-qPCR
and western blotting were used in 16 pairs of liver cancer tissues,
and compared with the corresponding non-tumor tissues; it was
identified that RASAL1 was significantly downregulated at the mRNA
and protein level in the cancerous tissues (Fig. 1A and B). Subsequently, the
associationbetweenRASAL1 expression levels and the clinical
parameters of liver cancer was examined. As presented in Fig. 1C and D, RASAL1 downregulation was
correlated with advanced pathological stage (P=0.009) and increased
tumor size (P=0.008). Combined, these results suggest that the
downregulation of RASAL1 may serve an important role in liver
cancer development and progression.
RASAL1 inhibits proliferation and
invasion in HepG2 cells
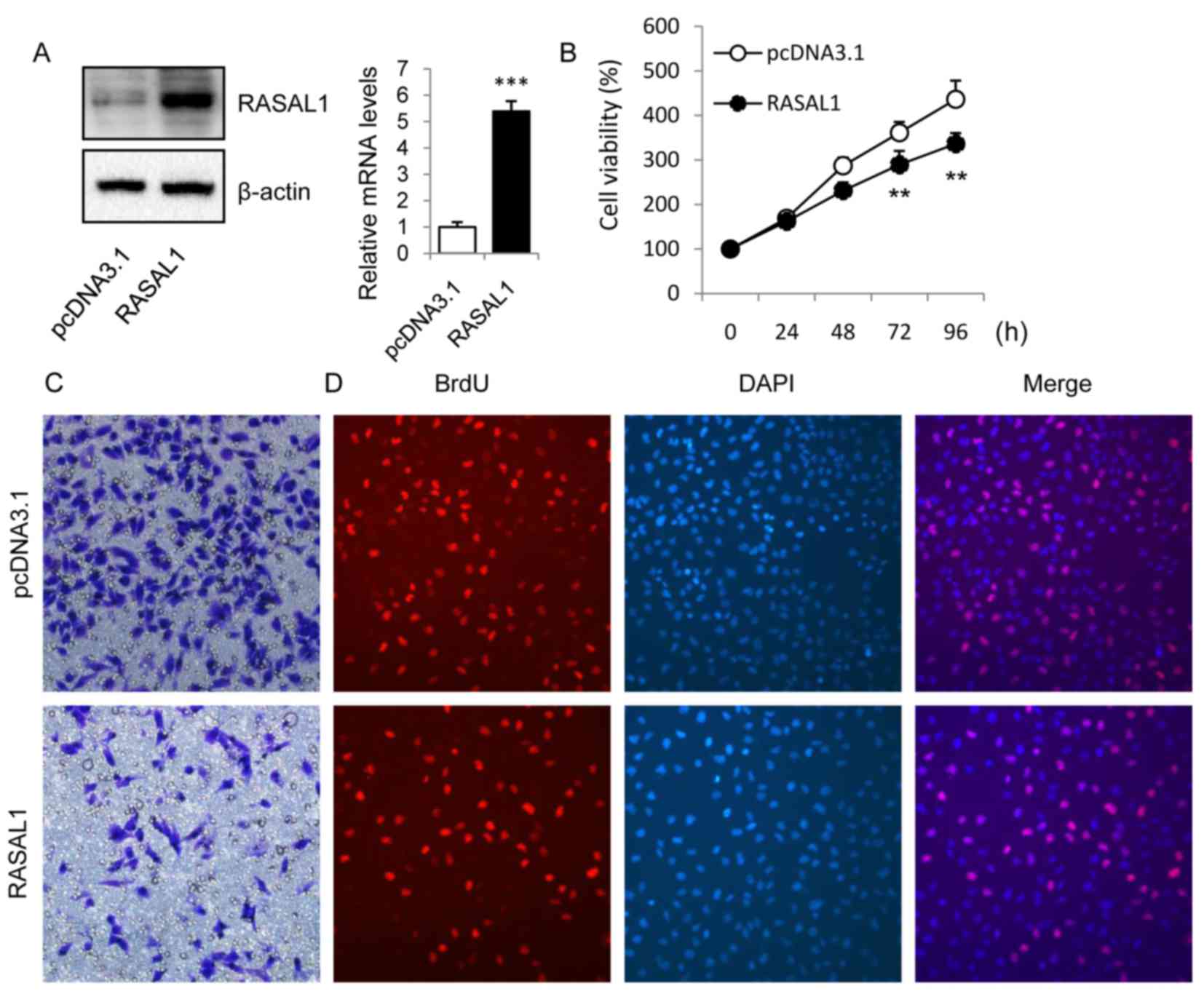
Next, the functional role of RASAL1 in the
proliferation and invasion ability of HepG2 cells was explored
using gain-of-function methods. It was observed that the expression
of RASAL1 was significantly upregulated in HepG2 cells transiently
overexpressing RASAL1 at the protein and mRNA levels, as compared
with in the vector control cell line (Fig. 2A). To examine the effects of RASAL1
overexpression on cell proliferation and invasion, MTT (Fig. 2B), Transwell (Fig. 2C) and BrdU (Fig. 2D) assays were used 48 h following
transfection. As demonstrated, transfection with RASAL1 resulted a
significant inhibition of growth and invasion ability in the HepG2
cell line.
Overexpression of RASAL1 in HepG2
cells decreases HIF-2α expression and tumor metabolism
To validate whether the inhibition effect of
overexpressed RASAL1 in HepG2 cells was mediated by its decreasing
the expression of HIF proteins, the mRNA levels of HIF-1α, HIF-1β,
HIF-2α and HIF-3α in RASAL1-overexpressing HepG2 cells were
investigated using RT-qPCR. The results indicated that only HIF-2α
was significantly decreased at the mRNA and the protein level, and
that the phosphorylation of ERK1/2 was increased (Fig. 3A and B).
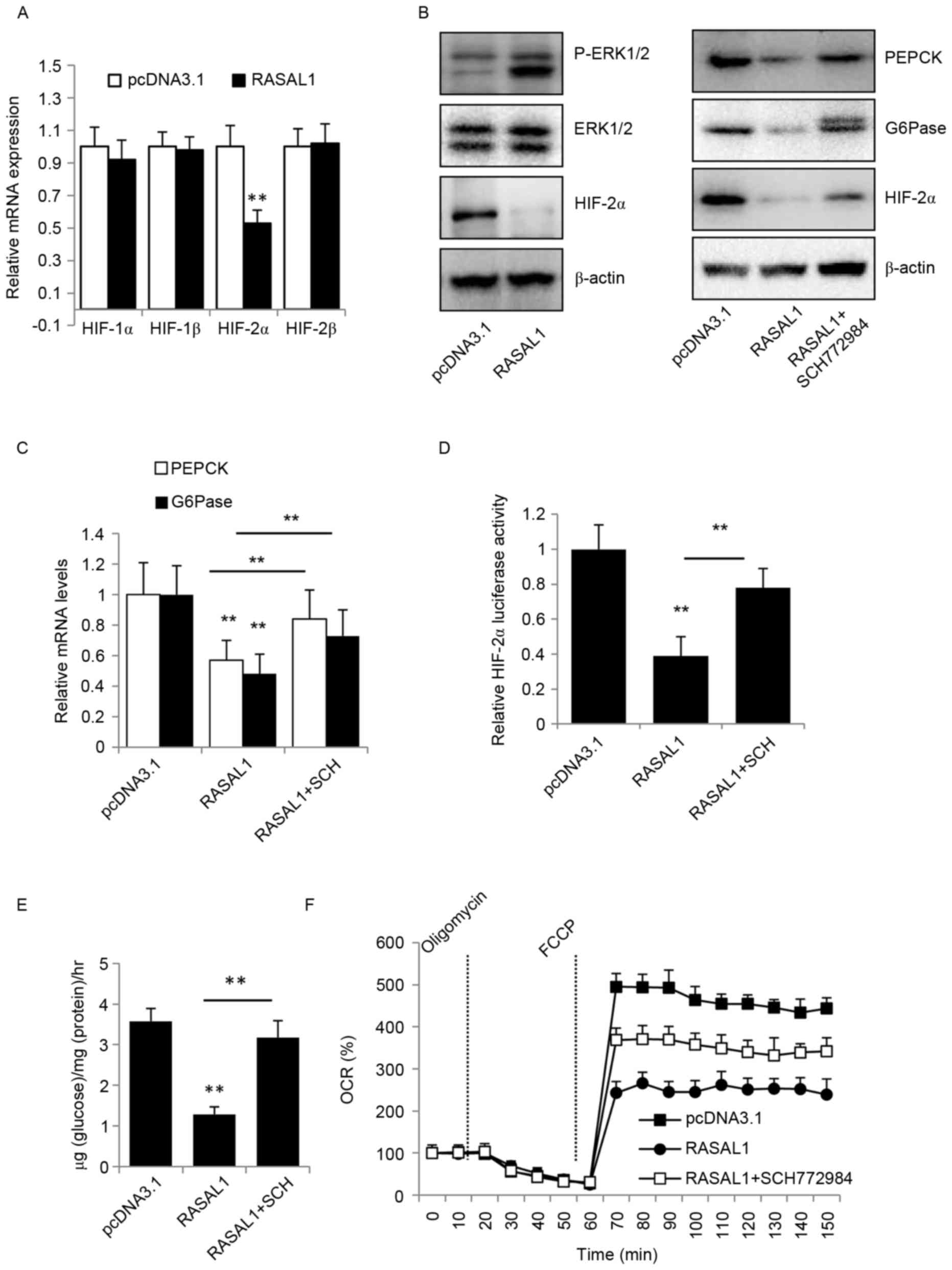 | Figure 3.Overexpression of RASAL1 in HepG2
inhibits HIF-2α expression and tumor metabolism. (A) Reverse
transcription-qPCR was performed to detect the expression of HIF
genes in transfected cells, **P<0.01 vs. pcDNA3.1 group, and (B)
western blot assays were used to detect the levels of HIF-2α,
PEPCK, G6Pase and the phosphorylation of ERK1/2, with or without 1
µM SCH772984 treatment, following the transfection of RASAL1. (C)
The mRNA levels of PEPCK and G6Pase were analyzed with qPCR in
HepG2 cells, with or without 1 µM SCH772984 treatment, following
the transfection of RASAL1, **P<0.01 vs. pcDNA3.1 or RASAL1
group. (D) Luciferase activity of the HIF-2α promoter was detected
via the overexpression of RASAL1 with or without SCH772984,
**P<0.01 vs. pcDNA3.1 or RASAL1 group. (E) Glucose production
from HepG2 cells incubated with 9 mM lactate and 1 mM pyruvate with
overexpression of RASAL1 or inhibition of ERK1/2 phosphorylation,
**P<0.01 vs. pcDNA3.1 or RASAL1 group. (F) The OCR of HepG2
cells was measured 48 h after transfection with RASAL1 or treatment
with SCH772984 using the Seahorse Bio analyzer. Data are normalized
to basal OCR and are representative of three independent
experiments. Each bar represents the mean ± standard error of the
mean of three independent experiments. **P<0.01 vs. pcDNA3.1 or
RASAL1 group. HIF, hypoxic-inducible factor; qPCR, quantitative
polymerase chain reaction; OCR, oxygen consumption rate; p,
phosphorylated; ERK, extracellular signal-regulated kinase; RASAL1,
RAS protein activator like 1; SCH, SCH772984; FCCP, Carbonyl
cyanide 4-(trifluoromethoxy) phenylhydrazone; PEPCK,
phosphoenolpyruvate carboxykinase; G6Pase, glucose
6-phosphatase. |
As previously demonstrated, the upregulation of
HIF-1 and/or HIF-2 may decrease hepatic expression of the glucose
transporter Glut2 and the gluconeogenic gene G6Pase, and also
decrease the levels of the PEPCK rate-limiting enzyme of
gluconeogenesis (21). Therefore, the
protein and mRNA levels of PEPCK and G6Pase were detected by
western blot analysis to determine whether RASAL1 inhibited tumor
growth through the metabolism pathway. As presented in Fig. 3B and C, the two key gluconeogenic
enzymes were downregulated at the protein and mRNA levels, while
the inhibitor of ERK1/2, SCH772984, upregulated PEPCK and G6Pase
protein and mRNA levels. These data also suggest that HIF-2α
transcription activity and glucose production was suppressed in
RASAL1-overexpressing HepG2 cells, but that SCH772984 weakened the
inhibitory effect (Fig. 3D and E).
OCRs were additionally measured in vitro in the presence and
absence of RASAL1 in HepG2 cells, and it was identified that RASAL1
caused a significant decrease, while SCH772984 induced an increase
(Fig. 3F).
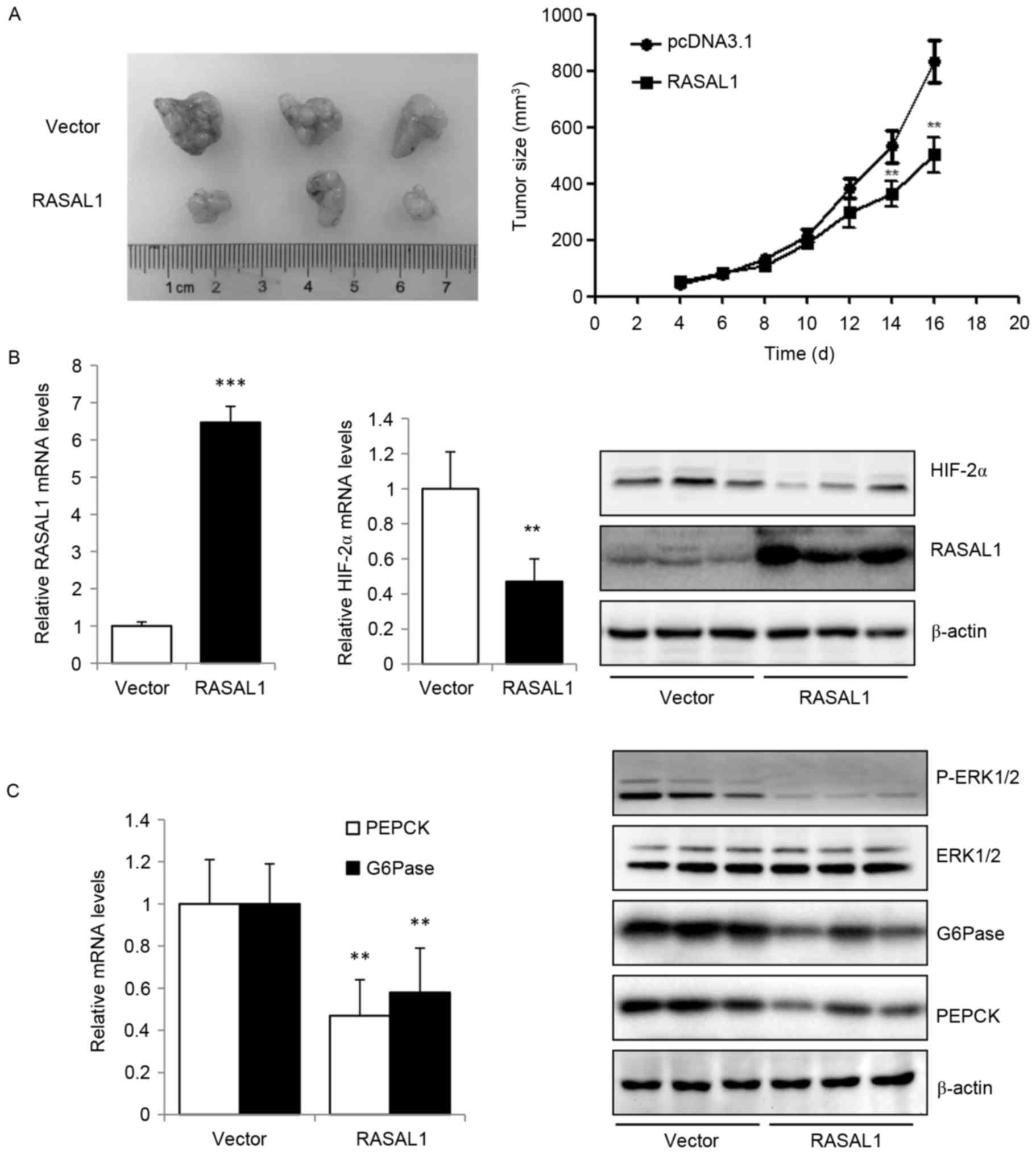
Upregulation of RASAL1 in HepG2
inhibits tumor growth via HIF-2α-mediated glucose metabolism in
vivo
Based on the data from the present study that the
exogenous expression of RASAL1 in liver cancer cells inhibited
cancer cell proliferation and invasion in vitro, which maybe
mediated by reducing the expression of HIF-2α, the effect of RASAL1
overexpression in tumor xenografts in vivo was additionally
explored. The results indicated that RASAL1 exhibited an inhibitory
effect on tumor size compared with the vector control group
(Fig. 4A). The higher expression
levels of RASAL1 in tumor xenografts transfected with
RASAL1-overexpressing HepG2 stable cells, as compared with those
transfected with vector control cells, were validated by RT-qPCR
and western blotting, are concomitant with the decreased expression
of HIF-2α at the mRNA and protein levels (Fig. 4B). The key gluconeogenic enzymes PEPCK
and G6Pase were downregulated at the mRNA and protein levels, and
the phosphorylation of ERK1/2 was significantly increased (Fig. 4C). These in vivo data were
concordant with the in vitro observations, and suggest that
RASAL1 may elicit a tumor suppressive effect through the inhibition
of HIF-2α expression, gluconeogenesis and oxygen consumption rate.
Thus, the RASAL1/HIF-2α axis maybe a novel therapeutic target for
liver cancer treatment.
Discussion
RASAL1 has been suggested to be a tumor suppressor
gene in colorectal, thyroid and gastric cancer (4,22,23), through its negative modulation of the
RAS signaling pathway, and also to function as an RasGAP that
catalyzes RAS inactivation (24,25).
Although efforts have been made, the specific molecular mechanisms
of its tumor suppressor function remain unknown. Therefore, the
present study focused on the tumor suppressive effects of RASAL1 in
liver cancer.
In the present study, it was identified that the
average levels of RASAL1 in liver cancer tissues were significantly
low when compared with those in corresponding non-tumor tissues.
The low RASAL1 expression levels of patients with liver cancer are
associated with advanced pathological stage and larger tumor size.
Consistent with previous data, RASAL1 gene expression was decreased
in gastric carcinoma tissues and cell lines (26). HepG2 is a hepatoblastoma cell line
that has been previously misidentified as hepatocellular carcinoma.
However, it may be used to investigate the functional role of
RASAL1 in the development and treatment of liver cancer (18,27). The
in vitro experiments conducted withRASAL1-overexpressing
HepG2 cells in the present study demonstrated that RASAL1 can
inhibit the proliferation and invasion ability of HepG2 cells.
These results indicate that RASAL1 may have a crucial role in liver
cancer development and progression.
As a metabolic regulator, HIF-2α has been identified
to be involved in cancer progression via a regulatory role in
cancer cell metabolism (28,29). It has been demonstrated previously
that the overexpression of HIF-2α in rat glioma tumors may reduce
growth by increasing caspase-3-mediated tumor cell apoptosis
(30). However, HIF-2α has been
revealed to promote tumor growth in a renal carcinoma xenograft
model, suggesting a unique role for HIF-2α in tumor growth
(31). In the present study,
increased expression of RASAL1 in HepG2 cells was observed at the
mRNA and protein levels, as compared with the vector control cell
line, 48 h after transfection, and this upregulation was
concomitant with reduced expression of HIF-2α and increased
phosphorylation of ERK1/2. The phosphorylation of ERK1/2 has been
indicated to modulate HIF-1 or HIF-2 activity in several cell types
(32). HIF-2α restored the expression
of the gluconeogenic genes Pepck and G6Pase, and
rescued the hypoglycemic phenotype of Vhlh mutants, supporting a
role as a regulator of hepatic lipid metabolism (33). Furthermore, the present study also
identified that the overexpression of RASAL1 in HepG2 cells
decreases PEPCK and G6Pase mRNA and protein levels, the luciferase
assay conducted in the present study indicated that RASAL1
decreased HIF-2α transcription activity, and measurement of the OCR
in vitro in the presence and absence of RASAL1 in HepG2
cells demonstrated a significant decrease in oxygen consumption in
liver cancer cell lines. Notably, inhibition of the activation of
ERK1/2 with SCH772984 rescued the effect of RASAL1 downregulation
on gluconeogenesis induced by HIF-2α. These data provide evidence
that RASAL1 may be a critical inhibitor in liver cancer, via
HIF-2α-mediated glucose metabolism.
Based on the in vitro data obtained in the
present study, which indicated that RASAL1 may be involved in
HIF-2α-mediated metabolism in liver cancer cells, the in
vivo efficacy of the RASAL1-inhibition effect was explored. The
results demonstrated that the enhanced tumor growth inhibition
efficacy induced by RASAL1-overexpressing HepG2 stable cells with
decreased HIF-2α, PEPCK and G6Pase mRNA and protein expression in
tumor xenografts, indicates that the RASAL1/HIF-2α axis maybe a
potential therapeutic target for current liver cancer therapy.
The present study demonstrated that RASAL1 may
partially abrogate HIF-2α-mediated gluconeogenesis through the
activation of ERK1/2. Using in vitro and in vivo
bioassays, it was demonstrated, that RASAL1 is an important
inhibitory factor for patients with liver cancer, and that it
modulates HepG2 cell proliferation. Regulation of HIF-2α, as a
component of RASAL1-mediated metabolism, participates in the
occurrence and development of liver cancer. Thus, the present study
may present a novel strategy for targeting with the RASAL1/HIF-2α
interaction as a novel therapeutic application for patients with
liver cancer.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rebollo A and Martı́nez-A C: Ras proteins:
Recent advances and new functions. Blood. 94:2971–2980.
1999.PubMed/NCBI
|
|
3
|
Qiao F, Su X, Qiu X, Qian D, Peng X, Chen
H, Zhao Z and Fan H: Enforced expression of RASAL1 suppresses cell
proliferation and the transformation ability of gastric cancer
cells. Oncol Rep. 28:1475–1481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu D, Yang C, Bojdani E, Murugan AK and
Xing M: Identification of RASAL1 as a major tumor suppressor gene
in thyroid cancer. J Natl Cancer Inst. 105:1617–1627. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tao H, Huang C, Yang JJ, Ma TT, Bian EB,
Zhang L, Lv XW, Jin Y and Li J: MeCP2 controls the expression of
RASAL1 in the hepatic fibrosis in rats. Toxicology. 290:327–333.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ko KS, Tomasi ML and Iglesiasas AL: MeCP2
controls the expression of RASAL1 in the hepatic fibrosis in rats.
Toxicology 290: Rosis, and hepatocellular carcinoma in mice.
Hepatology. 52:2096–2108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Semenza GL: Hypoxia-inducible factors:
Mediators of cancer progression and targets for cancer therapy.
Trends Pharmacol Sci. 33:207–214. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ryan HE, Poloni M, McNulty W, Elson D,
Gassmann M, Arbeit JM and Johnson RS: Hypoxia-inducible
factor-1alpha is a positive factor in solid tumor growth. Cancer
Res. 60:4010–4015. 2000.PubMed/NCBI
|
|
9
|
Weidemann A and Johnson R: Biology of
HIF-1alpha. Cell Death Differ. 15:621–627. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Löfstedt T, Fredlund E,
Holmquist-Mengelbier L, Pietras A, Ovenberger M, Poellinger L and
Påhlman S: Hypoxia inducible factor-2alpha in cancer. Cell Cycle.
6:919–926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peng J, Zhang L, Drysdale L and Fong GH:
The transcription factor EPAS-1/hypoxia-inducible factor 2alpha
plays an important role in vascular remodeling. Proc Natl Acad Sci.
97:pp. 8386–8391. 2000, View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian H, Hammer RE, Matsumoto AM, Russell
DW and McKnight SL: The hypoxia-responsive transcription factor
EPAS1 is essential for catecholamine homeostasis and protection
against heart failure during embryonic development. Genes Dev.
12:3320–3324. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei K, Piecewicz SM, McGinnis LM,
Taniguchi CM, Wiegand SJ, Anderson K, Chan CW, Mulligan KX, Kuo D,
Yuan J, et al: A liver Hif-2 α-Irs2 pathway sensitizes hepatic
insulin signaling and is modulated by Vegf inhibition. Nat Med.
19:1331–1337. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Taniguchi CM, Finger EC, Krieg AJ, Wu C,
Diep AN, LaGory EL, Wei K, McGinnis LM, Yuan J, Kuo CJ and Giaccia
AJ: Cross-talk between hypoxia and insulin signaling through Phd3
regulates hepatic glucose and lipid metabolism and ameliorates
diabetes. Nat Med. 19:1325–1330. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ramakrishnan SK, Zhang H, Takahashi S,
Centofanti B, Periyasamy S, Weisz K, Chen Z, Uhler MD, Rui L,
Gonzalez FJ and Shah YM: HIF2α is an essential molecular brake for
postprandial hepatic glucagon response independent of insulin
signaling. Cell Metab. 23:505–516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
DeBerardinis RJ, Sayed N, Ditsworth D and
Thompson CB: Brick by brick: Metabolism and tumor cell growth. Curr
Opin Genet Dev. 18:54–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sikes RS: The Animal Care and Use
Committee of the American Society of Mammalogists: 2016 Guidelines
of the American Society of Mammalogists for the use of wild mammals
in research and education. J Mammalogy. 97:663–688. 2016.
View Article : Google Scholar
|
|
18
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S
and Muramatsu T: A small interfering RNA targeting vascular
endothelial growth factor as cancer therapeutics. Cancer Res.
64:3365–3370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
McClain DA, Abuelgasim KA, Nouraie M,
Salomon-Andonie J, Niu X, Miasnikova G, Polyakova LA, Sergueeva A,
Okhotin DJ, Cherqaoui R, et al: Decreased serum glucose and
glycosylated hemoglobin levels in patients with Chuvash
polycythemia: A role for HIF in glucose metabolism. J Mol Med
(Berl). 91:59–67. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ohta M, Seto M, Ijichi H, Miyabayashi K,
Kudo Y, Mohri D, Asaoka Y, Tada M, Tanaka Y, Ikenoue T, et al:
Decreased expression of the RAS-GTPase activating protein RASAL1 is
associated with colorectal tumor progression. Gastroenterology.
136:206–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiao F, Su X, Qiu X, Qian D, Peng X, Chen
H, Zhao Z and Fan H: Enforced expression of RASAL1 suppresses cell
proliferation and the transformation ability of gastric cancer
cells. Oncol Rep. 28:1475–1481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin H, Wang X, Ying J, Wong AH, Cui Y,
Srivastava G, Shen ZY, Li EM, Zhang Q, Jin J, et al: Epigenetic
silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL
defines a new mechanism of Ras activation in human cancers. Proc
Natl Acad Sci USA. 104:pp. 12353–12358. 2007, View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kolfschoten IG, van Leeuwen B, Berns K,
Mullenders J, Beijersbergen RL, Bernards R, Voorhoeve PM and Agami
R: A genetic screen identifies PITX1 as a suppressor of RAS
activity and tumorigenicity. Cell. 121:849–858. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen H, Yang XW, Zhang H, Yang Q, Wang Z,
Liu Y, Lu FL, Zhou BY, Qiu-Xi CH and Lu SL: In vivo and in vitro
expression of the RASAL1 gene in human gastric adenocarcinoma and
its clinicopathological significance. Oncol Lett. 3:535–540.
2012.PubMed/NCBI
|
|
27
|
Jin W, Chen L, Cai X, Zhang Y, Zhang J, Ma
D, Cai X, Fu T, Yu Z, Yu F and Chen G: Long non-coding RNA TUC338
is functionally involved in sorafenib-sensitized hepatocarcinoma
cells by targeting RASAL1. Oncol Rep. 37:273–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blancher C, Moore JW, Talks KL, Houlbrook
S and Harris AL: Relationship of hypoxia-inducible factor
(HIF)-1alpha and HIF-2alpha expression to vascular endothelial
growth factor induction and hypoxia survival in human breast cancer
cell lines. Cancer Res. 60:7106–7113. 2000.PubMed/NCBI
|
|
29
|
Beasley NJ, Leek R, Alam M, Turley H, Cox
GJ, Gatter K, Millard P, Fuggle S and Harris AL: Hypoxia-inducible
factors HIF-1alpha and HIF-2 alpha in head and neck cancer:
Relationship to tumor biology and treatment outcome in surgically
resected patients. Cancer Res. 62:2493–2497. 2002.PubMed/NCBI
|
|
30
|
Acker T, Diez-Juan A, Aragones J, Tjwa M,
Brusselmans K, Moons L, Fukumura D, Moreno-Murciano MP, Herbert JM,
et al: Genetic evidence for a tumor suppressor role of HIF-2alpha.
Cancer Cell. 8:131–141. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kondo K, Klco J, Nakamura E, Lechpammer M
and Kaelin WG Jr: Inhibition of HIF is necessary for tumor
suppression by the von Hippel-Lindau protein. Cancer Cell.
1:237–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hur E, Chang KY, Lee E, Lee S-K and Park
H: Mitogen-activated protein kinase kinase inhibitor PD98059 blocks
the trans-activation but not the stabilization or DNA binding
ability of hypoxia-inducible factor-1alpha. Mol Pharmacol.
59:1216–1224. 2001.PubMed/NCBI
|
|
33
|
Rankin EB, Rha J, Selak MA, Unger TL,
Keith B, Liu Q and Haase VH: Hypoxia-inducible factor 2 regulates
hepatic lipid metabolism. Mol Cell Biol. 29:4527–4538. 2009.
View Article : Google Scholar : PubMed/NCBI
|