Introduction
Breast cancer is the most common type of cancer
diagnosed in females and the second leading cause of cancer
mortality in females worldwide, following lung cancer (1). Breast cancer is characterized by a
distinct metastatic pattern involving the regional lymph nodes,
bone marrow, lungs and liver (2). A
family history of breast cancer and other factors (including female
sex, elderly age and ionizing radiation) can increase the risk of
developing breast cancer (3). In
addition, 5–10% of breast cancer cases are caused by inherited gene
mutations (4). Several gene markers
have been identified to predict the response to therapeutic
regimens, including receptor tyrosine-protein kinase ErbB-2 and
Stearoyl-CoA desaturase-1 (5–7). However, the pathological molecular
mechanisms underlying breast cancer remain unknown. Individual
anticancer therapies and the identification of molecular functions
(MFs), on the basis of genes identified as significant, may enable
this challenge to be solved.
Gene ontology (GO) is intended to enable the
annotation of homologous gene and protein sequences in multiple
organisms using a common vocabulary that results in the ability to
query, and retrieve genes and proteins on the basis of shared
biology (8). The ontology covers
three categories: Cellular component, biological process and MF
(8). Studying MFs enables analysis of
the elemental activities of a gene at the molecular level. MF is
defined as the biochemical activity (including specific binding to
ligands or structures) of a gene product and describes only what
happens, without specifying where or when the event occurs
(9). However, a limited number of
studies have been performed with the aim of identifying the roles
of significant MFs in the progression of a disease.
The objective of the present study was to identify
differentially expressed MFs (DEMFs) for breast cancer using the
Gibbs sampling approach. To achieve the objectives, MFs were
evaluated according to the Bayesian Approach for Geneset Selection
(BAGS) package and DEMFs were identified using Gibbs sampling on
the basis of the Markov chain (MC) Monte Carlo (MCMC) algorithm.
Subsequently, additional analyses were performed for DEMFs,
including construction of a co-expression network and pathway
enrichment analysis.
Materials and methods
Gene expression data recruitment and
pre-processing
The publicly available microarray gene expression
profile of breast cancer was recruited from EMBL-EBI ArrayExpress
(www.ebi.ac.uk/arrayexpress; accessing
no. E-GEOD-10780) (10). It was
presented on the A-AFFY-44-Affymetrix GeneChip Human Genome U133
Plus 2.0 platform (HG-U133_Plus_2; Affymetrix; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and was comprised of 185
samples (143 normal controls and 42 breast cancer samples).
To control the quality of the expression data on the
probe-level, standard procedures were performed, including
background correction (11),
normalization (12), probe correction
(13) and summarization (11). Briefly, background correction and
normalization were carried out using the Robust Multi-array Average
algorithm and quantile based algorithm, to eliminate the influence
of nonspecific hybridization. Furthermore, the Micro Array Suite
algorithm was used to revise perfect match and mismatch values, and
the medianpolish method was used to summarize expression values.
Subsequently, the preprocessed probe-level dataset were converted
into expression measures and screened using the feature filter
method to discard duplicated genes (14). A total of 20,102 genes were
obtained.
MFs identification
To identify MFs for breast cancer, the
‘AnnotationMFGO’ function of the BAGS package was utilized. The
BAGS package provides functions to perform statistical
identification of gene functional classes that behave in a distinct
manner between the phenotypes of interest for datasets under
cross-sectional or time series designs (15,16). The
normalized data set was inputted into the package and used to
identify enriched MFs, but only MFs from GO with ≥5 genes were
considered for additional analysis.
Gibbs sampling
In the present study, Gibbs sampling was used to
investigate the significance and functions of MFs in breast cancer.
Gibbs sampling, typically used as a means of statistical inference,
particularly Bayesian inference, is a MCMC algorithm for obtaining
a sequence of observations that are approximated from a specified
multivariate probability distribution (17–19).
MC
To implement the Gibbs sampler procedure, the MFs
were transformed to a data set with functional class expression
measurements that were MCa. An MC is a sequence of random variables
where the distribution of each random variable depends only on the
value of the previous random variable (20). For example, a sample m was
taken from a distribution depending on a parameter vector o
ε O of length T, with prior distribution
g(o1, …, oT). It may be that
T was large and that numerical integration to identify the
marginal densities of the oi may be
computationally expensive. An alternative method of calculating the
marginal densities was to create an MC on the space O by
repeating these two steps: The first step, selecting a random index
1 ≤ j ≤ T; the second step, endowed a new value for
pi according to g(o1, …,
oj-1,., oj+1, …,
oT).
Posterior inference
Following the transformation into MCs, posterior
inferences f were defined to identify the probability distributions
of MFs from breast cancer (21). The
posterior distribution was represented by ρ(x, y), which was
the probability of parameter x provided an observation
y. Provided that prior information is available about the
parameters ρ(x), the posterior distribution may be developed
through the normalized product of the prior and a sampling
distribution (likelihood), ρ(y, x). The algorithm associated
with the objective (target) density f and the conditional
density q produces an MC (X(t)).
Provided x(t) denoted the value of
X(t), y = q(x) it was
defined Yt ~ q
(y|x(t)),
X(t+1)={Ytwith
probabilityρ(x(t),Yt)x(t)with probability1−ρ(x(t),Yt)
Where q(x) was an MF, the probability
was represented by ρ(x, y), which was calculated as
follows:
ρ(x,y)=min{f(y)f(x)q(x|y)q(y|x),1}
The distribution q was termed the
instrumental (or proposal or candidate) distribution and the
probability ρ(x, y). It was to be distinguished from the
acceptance rate, which was the average of the acceptance
probability over iterations.
ρ¯=limT→∞1T∑t=0Tρ(X(t),Yt)=∫ρ(x,y)f(x)q(y|x)dydx
T was the length of the observation vector.
Furthermore, this algorithm satisfied the balance condition
(22),
f(x)K(y | x) =
f(y)K(x | y), from which it was
deduced that f was the stationary distribution of the MC
[X(t)] by integrating each side of the equality
in x; K was transition kernel.
DEMFs
In the present study, probabilities of MFs were
evaluated using the posterior distributions on the basis of Gibbs
sampling. If the posterior probability of an MF was >0.05, this
indicated the reliability of the MF (23). However, there was not a standard for
high frequencies. Based on a previous study (15), if the probability of an MF was ≥0.8,
the MF was differentially expressed. To additionally investigate
the properties of DEMFs, the gene compositions of DEMFs were
studied, and those genes involved in the DEMFs were considered to
be differentially expressed genes (DEGs).
Co-expression network
The empirical Bayes (EB) approach was selected to
construct a co-expression network for the DEGs of breast cancer
(24). An l-by-n matrix
of expression values was created, where l was the number of
genes (or probes) under consideration and n was the total
number of microarrays over all conditions. These values should be
normalized and the E obtained. The conditions array of
length was n. The members of this array should use values in
1-F, where F was the total number of conditions.
Using ‘ebPatterns’ function to define equal
co-expression/differential co-expression (DC) classes.
Using E and the conditions array, the
intra-group associations for all s = l*(l-1)/2
gene pairs were calculated and the resulting D matrix of
associations were s-by-F. Initialization of the
hyper-parameters was performed to identify the component normal
mixture model that fit the associations of D following
transformation. Expectation Maximization calculations were not
conducted and instead the initial estimates of the hyper parameters
were used to generate posterior probabilities of DC (25). Ultimately, the co-expression network
was visualized by Cytoscape 3.5.1 (http://www.cytoscape.org/).
Topological analysis
To understand the functionality of complex systems
of gene signatures in the co-expression network, the biological
importance of genes was characterized using indices of topological
analysis. Degree was counted by summing up the number of the
adjacent genes (26). The degree
D(v) of a node v was defined as follows:
D(v)=∑javj
Pathway enrichment analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analysis for DEGs was performed by using the
online tool Database for Annotation, Visualization and Integrated
Discovery (DAVID) (27). KEGG
pathways with P<0.01 were selected on the basis of Expression
Analysis Systematic Explored (EASE) test implemented in DAVID
(28). The calculated formula of EASE
was as follows:
P=(a+ba)(c+dc)(na+c)
Where n = a' + b + c +
d was the number of background genes, a' was the gene
number of one gene set in the gene lists, a' + b was
the number of genes in the gene list including at least one gene
set, a' + c was the gene number of one gene list in
the background genes; a' was replaced with a =
a'-1.
Results
Identification of MFs
Under the condition of GOs with ≥5 genes, a total
of 396 MFs were obtained based on the BAGS package. Of all the MFs,
32 possessed between 100 and 200 genes, 34 possessed between 200
and 300 genes, and 24 terms possessed >300 genes. DNA binding
contained 561 genes, receptor activity contained 542 genes, and
transferase activity transferring phosphorus containing groups
included 401 genes. Transmembrane receptor activity with 390 genes
and substrate specific transporter activity of 378 genes were
markedly MF terms for breast cancer. The MFs with ≥200 genes are
listed in Table I.
 | Table I.MFs with ≥200 genes for breast
cancer. |
Table I.
MFs with ≥200 genes for breast
cancer.
| MFs | n |
|---|
| DNA binding | 561 |
| Receptor
activity | 542 |
| Transferase
activity transferring phosphorus containing groups | 401 |
| Transmembrane
receptor activity | 390 |
| Substrate specific
transporter activity | 378 |
| Transmembrane
transporter activity | 362 |
| Receptor
binding | 357 |
| Kinase
activity | 347 |
| Substrate specific
transmembrane transporter activity | 332 |
| Transcription
factor activity | 330 |
| Phosphotransferase
activity alcohol group as acceptor | 316 |
| Enzyme regulator
activity | 311 |
| Transcription
factor binding | 289 |
| Identical protein
binding | 288 |
| Oxidoreductase
activity | 272 |
| Protein kinase
activity | 269 |
| Ion transmembrane
transporter activity | 268 |
| Ion binding | 260 |
| Hydrolase activity
acting on ester bonds | 253 |
| Structural molecule
activity | 222 |
| Hydrolase activity
acting on acid anhydrides | 216 |
| RNA binding | 214 |
| Pyrophosphatase
activity | 214 |
| Transcription
cofactor activity | 214 |
| Nucleotide
binding | 212 |
| Cation binding | 206 |
| Cation
transmembrane transporter activity | 205 |
| Purine nucleotide
binding | 201 |
| Nucleotide
triphosphatase activity | 200 |
Identification of DEMFs
Prior to evaluating the probabilities of MFs, using
Gibbs sampling on the basis of the MCMC algorithm, the 396 MFs in
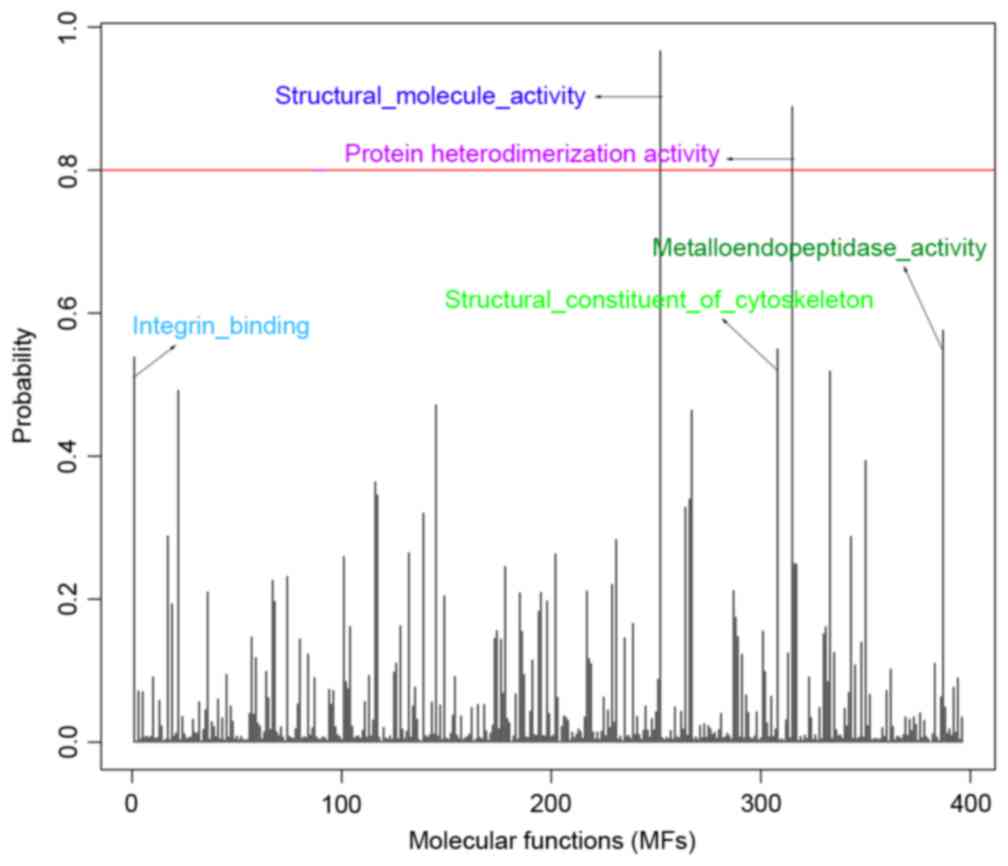
expression data structure were converted to MCs. Fig. 1 illustrates the association between
probability distribution and each MF, and lists the top five MFs
with high probabilities, including: Structural molecule activity,
0.967; protein heterodimerization activity, 0.889;
metalloendopeptidase activity, 0.576; structural constituent of the
cytoskeleton, 0.550; integrin binding, 0.539. In addition, an MF
with probability ≥0.8 was defined as DEMF, thus two DEMFs were
obtained (structural molecule activity and protein
heterodimerization activity) for breast cancer.
Gene composition of DEMFs
To investigate the properties and functions of
DEMFs, the gene composition was analyzed for two DEMFs. The results
of the present study demonstrated that structural molecule activity
was comprised of 222 genes; whereas, protein heterodimerization
activity consisted of 75 genes. Genes in the DEMFs were denoted as
DEGs in the present study; therefore, a total of 297 DEGs were
identified, all of which may serve functions in the progression of
breast cancer.
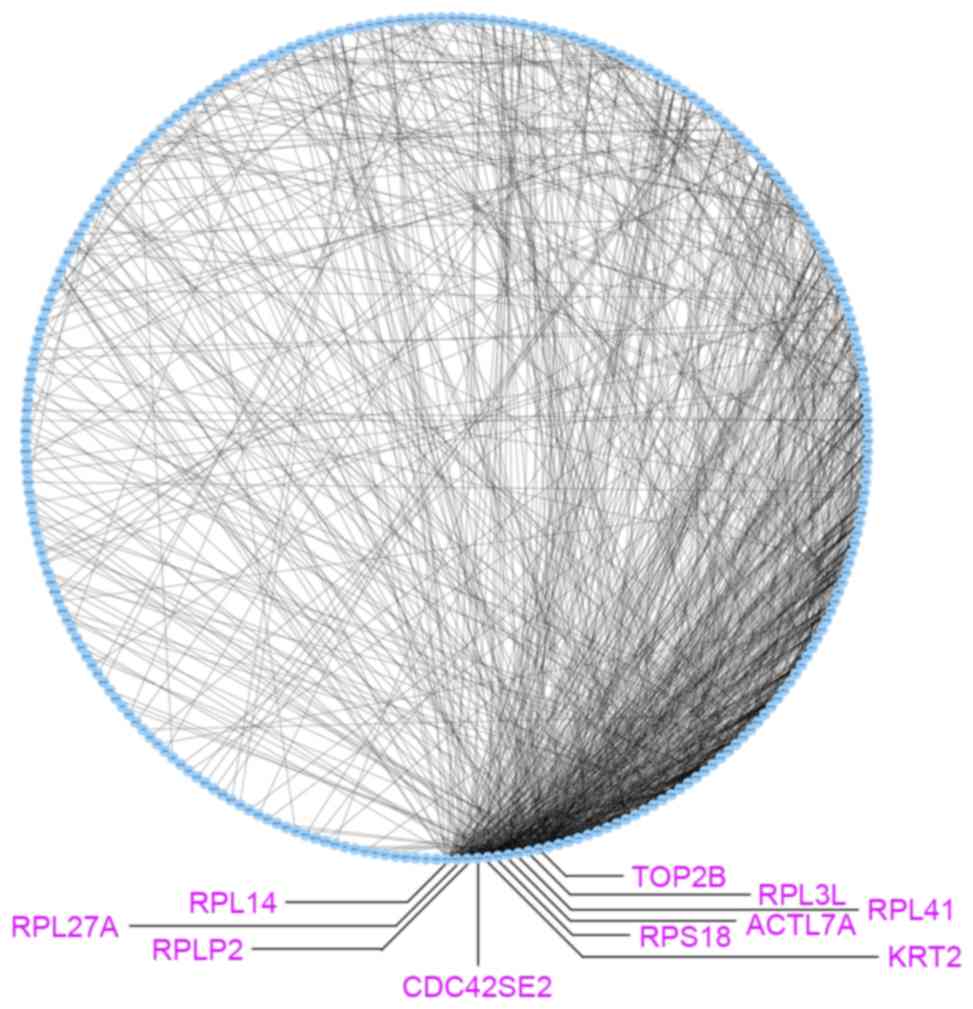
The co-expression network for 297 DEGs was
constructed using the EB approach and subsequently visualized using
Cytoscape 3.5.1 (Fig. 2). There were
259 nodes and 985 edges in the co-expression network. Subsequently,
topological analysis was implemented for the co-expression network
to identify significant genes. As presented in Fig. 2, the top 10 nodes with higher degrees
were ribosomal protein (RP)L14 (degree, 49), RPL27A
(degree, 45), RPLP2 (degree, 45), CDC42 small effector 2
(degree, 40), RPS2 (degree, 40), actin like 7A (degree, 35),
RPL3 L (degree, 35), RPL41 (degree, 35), RPS18
(degree, 35) and topoisomerase (DNA) II β (degree, 34).
KEGG enrichment analysis for DEGs was conducted to
gain additional insight into DEMFs and to identify the associated
biological pathways. A total of 10 significant pathways with
P<0.01 were identified (Table
II). Ribosome (P=1.27×10−64), hypertrophic
cardiomyopathy (P=1.09×10−7) and dilated cardiomyopathy
(P=3.02×10−7) were identified as the three most
significant pathways in breast cancer. When calculating the counts
for each pathway, the ribosome pathway was enriched by the maximum
numbers of 55 DEGs, a number of which exhibited a high degree
(including RPL14, RPL27A and RPLP2), thus it may be a
potential biomarker for the detection and treatment of breast
cancer.
| Table II.Pathways with P<0.01, based on
differentially expressed genes from differentially expressed
molecular functions. |
Table II.
Pathways with P<0.01, based on
differentially expressed genes from differentially expressed
molecular functions.
| Pathway | Count | P-value | Gene |
|---|
| Ribosome | 55 |
1.27×10−64 | RPL18, RPL19,
RPL14, RPL13, RPL15, RPLP2, RPS2, RPS3, RPLP0, RPLP1, FAU, RPL11,
RPS4X, RPS18, RPS19, RPL41, RPS16, RPS15, RPS12, RPL3L, RPS4Y1,
RPS10, RPS11, RPL27A, RPL35, RPS15A, RPL37, RPL38, RPL39, RPS25,
RPL30, RPS27, RPS28, RPS29, RPL7, RPL6, RPL31, RPL9, RPL34, RPL8,
RPL3, RPS21, RPS23, RPS24, RPS9, RPL27, RPL23A, RPL24, RPS6, RPS5,
RPS8, RPL28, RPL29, RPS7, RPL22 |
| Hypertrophic
cardiomyopathy | 15 |
1.09×10−7 | ACTB, LAMA2,
ACTC1, DES, MYL2, MYL3, DMD, MYBPC3, MYH7, MYH6, TTN, TPM2, TPM1,
TPM4, TGFB2 |
| Dilated
cardiomyopathy | 15 |
3.02×10−7 | ACTB, LAMA2,
ACTC1, DES, MYL2, MYL3, DMD, MYBPC3, MYH7, MYH6, TTN, TPM2, TPM1,
TPM4, TGFB2 |
| Tight junction | 17 |
1.25×10−6 | ACTB, PPP2R1B,
PPP2R1A, MYL5, MYL2, EPB41, VAPA, MYLPF, MYH7, ACTN2, MYH6, ACTN3,
LLGL1, MYL9, PPP2CA, MYH11, SPTAN1 |
| Focal adhesion | 20 |
4.41×10−6 | EGFR, COL4A4,
ACTB, CAV1, COL4A2, MYL5, MYL2, ERBB2, MYLPF, ACTN2, ACTN3, MYL9,
LAMA2, LAMA1, LAMA4, LAMC3, BCL2, COMP, LAMC1, LAMB1 |
| Small cell lung
cancer | 10 |
6.72×10−4 | COL4A4, LAMA2,
LAMA1, LAMA4, COL4A2, LAMC3, BCL2, TP53, LAMC1, LAMB1 |
| ECM-receptor
interaction | 10 |
6.72×10−4 | COL4A4, LAMA2,
LAMA1, LAMA4, COL4A2, LAMC3, COMP, AGRN, LAMC1, LAMB1 |
| Cardiac muscle
contraction | 8 |
7.21×10−3 | ACTC1, MYL2,
MYL3, MYH7, MYH6, TPM2, TPM1, TPM4 |
| ABC
transporters | 6 |
8.50×10−3 | ABCG8, ABCG5,
TAP2, TAP1, ABCG4, ABCG1 |
| Regulation of actin
cytoskeleton | 14 |
9.35×10−3 | EGFR, ACTB,
MYL5, MYL2, MYLPF, ACTN2, ARPC4, ARPC5, ACTN3, MYL9, ARPC1B, ARPC3,
ARPC2, MSN |
Discussion
In the present paper, two DEMFs were identified for
breast cancer, including structural molecule activity and protein
heterodimerization activity. Structural molecule activity is
defined as the action of a molecule that contributes to the
structural integrity of a complex or assembly within or outside a
cell (29). A previous study
demonstrated that structural molecule activity was among the top
ten gain/loss spatial functional hotspots of hepatocellular
carcinoma with enriched functions (29). In breast cancer, Hong et al
(30) reported that the dominant
function of the identified protein was involved in the structural
molecule activity. It has been suggested that extracellular matrix
protein expression and nuclear gene expression are associated with
a negative feedback regulatory mechanism of breast cancer, and
differential genes actively participated in structural molecule
activity (31). In addition, ~9.3% of
upregulated genes in breast tumors were identified to belong to
categories of structural molecule activity (32). In the present study, the DEMF,
structural molecule activity, was associated with breast cancer,
which validated the feasibility of Gibbs sampling to identify DEMFs
for breast cancer.
Protein heterodimerization activity interacts
selectively and non-covalently with a non-identical protein to form
a heterodimer (33). Previous studies
had revealed that Protein heterodimerization activity was involved
in a number of cancer types, including nasopharyngeal carcinoma
(33), esophageal cancer (34) and hepatocellular carcinoma (35). Topoisomerase IIα is a prognostic
biomarker and a novel therapeutic target of nasopharyngeal
carcinoma, which was identified to be associated with tumor
aggressiveness and significantly enriched in protein
heterodimerization activity (33).
However, there are a limited number of studies that have focused on
the association between breast cancer and protein
heterodimerization activity directly. The Erb-b2 receptor tyrosine
kinase (ErbB) 2/Erb-b3 heterodimer functioned as an
oncogenic unit to drive breast tumor cell proliferation (36). The receptor tyrosine kinase Erb-b2
receptor tyrosine kinase 2 was demonstrated to be overexpressed in
~25% breast cancer cases and to serve a signaling function, via
self-constitutive phosphorylation and the transactivation of
heterodimerization partners (37).
The results of the present study revealed that the
heterodimerization activity of proteins or genes were identified
and associated with the formation of breast cancer.
In the present study, the gene compositions of
DEMFs were subsequently studied and the results identified that the
ribosome was the most significant pathway. In the ribosome pathway,
RPL14, RPL27A and RPLP2 were the three DEGs that
exhibited the highest degrees. Ribosomes, the organelles that
catalyze protein synthesis, consist of a small 40S subunit and a
large 60S subunit. RPL14, RPL27A and RPLP2 belong to
members of ribosomal protein L (RPL) family. A number of
human pathological conditions, not associated with genetic
alterations of oncogenes or tumor suppressors, are associated with
an increased risk of developing cancer and may be characterized by
quantitative and/or qualitative changes in ribosomes (38). Therefore, in the present study it was
considered that DEG-associated analysis demonstrated the two DEMFs
were associated with breast cancer.
The results of the present study identified two
DEMFs (structural molecule activity and protein heterodimerization
activity) utilizing Gibbs sampling, and validated the associations
between these DMEFs and breast cancer. Thus, the results of the
present study may provide a novel insight into the underlying
pathological mechanisms of breast cancer.
References
|
1
|
DeSantis C, Ma J, Bryan L and Jemal A:
Breast cancer statistics, 2013. CA Cancer J Clin. 64:52–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Müller A, Homey B, Soto H, Ge N, Catron D,
Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al:
Involvement of chemokine receptors in breast cancer metastasis.
Nature. 410:50–56. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DeSantis C, Siegel R, Bandi P and Jemal A:
Breast cancer statistics, 2011. CA Cancer J Clin. 61:409–418. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DiSipio T, Rye S, Newman B and Hayes S:
Incidence of unilateral arm lymphoedema after breast cancer: A
systematic review and meta-analysis. Lancet Oncol. 14:500–515.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Charif M, Lower EE, Kennedy D, Kumar H,
Khan S, Radhakrishnan N and Zhang X: Abstract P3-05-16: The effect
of HER-2/neu inhibition on prolonging clinical benefit with
fulvestrant treatment for metastatic estrogen receptor positive
breast cancer patients treated with trastuzumab. Cancer Res.
75:3–5. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mittendorf EA, Clifton GT, Holmes JP,
Clive KS, Patil R, Benavides LC, Gates JD, Sears AK, Stojadinovic
A, Ponniah S and Peoples GE: Clinical trial results of the
HER-2/neu (E75) vaccine to prevent breast cancer recurrence in
high-risk patients: From US Military Cancer Institute Clinical
Trials Group Study I-01 and I-02. Cancer. 118:2594–2602. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Holder AM, Gonzalez-Angulo AM, Chen H,
Akcakanat A, Do KA, Symmans Fraser W, Pusztai L, Hortobagyi GN,
Mills GB and Meric-Bernstam F: Increased stearoyl-CoA desaturase 1
expression is associated with shorter survival in breast cancer
patients. Cancer Res. 72:6822012. View Article : Google Scholar
|
|
8
|
Gene Ontology Consortium, . Blake JA,
Dolan M, Drabkin H, Hill DP, Li N, Sitnikov D, Bridges S, Burgess
S, Buza T, et al: Gene Ontology annotations and resources. Nucleic
Acids Res. 41:(Database issue). D530–D535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen DT, Nasir A, Culhane A, Venkataramu
C, Fulp W, Rubio R, Wang T, Agrawal D, McCarthy SM, Gruidl M, et
al: Proliferative genes dominate malignancy-risk gene signature in
histologically-normal breast tissue. Breast Cancer Res Treat.
119:335–346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Irizarry RA, Bolstad BM, Collin F, Cope
LM, Hobbs B and Speed TP: Summaries of Affymetrix GeneChip probe
level data. Nucleic Acids Res. 31:e152003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bolstad BM, Irizarry RA, Astrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bolstad B: Affy: Built-in Processing
Methods. 2013.
|
|
14
|
Lee J and Kim DW: Efficient multivariate
feature filter using conditional mutual information. Electronics
Lett. 48:161–162. 2012. View Article : Google Scholar
|
|
15
|
Quiroz-Zárate A, Haibe-Kains B and
Quackenbush J: BAGS: A Bayesian Approach for Geneset Selection.
2013.
|
|
16
|
Rasnake N, Haddad L, Rasnake M and Heidel
E: 487: Bacterial colonization of manual resuscitation bags. Crit
Care Med. 40:1–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kozumi H and Kobayashi G: Gibbs sampling
methods for Bayesian quantile regression. J Statistical Computa
Simulation. 81:1565–1578. 2011. View Article : Google Scholar
|
|
18
|
El-Hay T, Friedman N and Kupferman R:
Gibbs sampling in factorized continuous-time Markov processes.
arXiv preprint arXiv. 1206:32512012.
|
|
19
|
Chib S and Winkelmann R: Markov chain
Monte Carlo analysis of correlated count data. J Bus Eco
Statistics. 19:428–435. 2012. View Article : Google Scholar
|
|
20
|
Sinclair A: Algorithms for random
generation and counting: A Markov chain approach. Springer Sci Bus
Media. 2012.
|
|
21
|
Moradkhani H, DeChant CM and Sorooshian S:
Evolution of ensemble data assimilation for uncertainty
quantification using the particle filter-Markov chain Monte Carlo
method. Water Resources Res. 48:2012. View Article : Google Scholar
|
|
22
|
Gonzalez J, Low Y, Gretton A and Guestrin
C: Parallel gibbs sampling: From colored fields to thin junction
trees. International Conference on Artificial Intelligence and
Statistics. pp. 324–332. 2011
|
|
23
|
Yan Y and Zhang S: An improved estimation
method and empirical properties of the probability of informed
trading. J Banking Finance. 36:454–467. 2012. View Article : Google Scholar
|
|
24
|
Dawson JA, Ye S and Kendziorski C:
R/EBcoexpress: An empirical Bayesian framework for discovering
differential co-expression. Bioinformatics. 28:1939–1940. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moon TK: The expectation-maximization
algorithm. Signal Pro Magazine IEEE. 13:47–60. 1996. View Article : Google Scholar
|
|
26
|
Haythornthwaite C: Social network
analysis: An approach and technique for the study of information
exchange. Library Inf Sci Res. 18:323–342. 1996. View Article : Google Scholar
|
|
27
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Grossglauser M and Vetterli M: Locating
nodes with EASE: Last encounter routing in ad hoc networks through
mobility diffusion. In: INFOCOM 2003. Twenty-Second Annual Joint
Conference of the IEEE Computer and Communications. IEEE Societies
IEEE. 1954–1964. 2003.
|
|
29
|
Hsiao TH, Chen HI, Roessler S, Wang XW and
Chen Y: Identification of genomic functional hotspots with copy
number alteration in liver cancer. EURASIP J Bioinform Syst Biol.
2013:142013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hong WX, Huang A, Lin S, Yang X, Yang L,
Zhou L, Huang H, Wu D, Huang X, Xu H and Liu J: Differential
expression profile of membrane proteins in L-02 cells exposed to
trichloroethylene. Toxicol Ind Health. 32:1774–1783. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hao X, Sun B, Hu L, Lähdesmäki H, Dunmire
V, Feng Y, Zhang SW, Wang H, Wu C, Wang H, et al: Differential gene
and protein expression in primary breast malignancies and their
lymph node metastases as revealed by combined cDNA microarray and
tissue microarray analysis. Cancer. 100:1110–1122. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cizkova M, Cizeron-Clairac G, Vacher S,
Susini A, Andrieu C, Lidereau R and Bièche I: Gene expression
profiling reveals new aspects of PIK3CA mutation in
ERalpha-positive breast cancer: Major implication of the Wnt
signaling pathway. PLoS One. 5:e156472010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lan J, Huang HY, Lee SW, Chen TJ, Tai HC,
Hsu HP, Chang KY and Li CF: TOP2A overexpression as a poor
prognostic factor in patients with nasopharyngeal carcinoma. Tumor
Biol. 35:179–187. 2014. View Article : Google Scholar
|
|
34
|
Ogawa R, Ishiguro H, Kuwabara Y, Kimura M,
Mitsui A, Mori Y, Mori R, Tomoda K, Katada T, Harada K and Fujii Y:
Identification of candidate genes involved in the radiosensitivity
of esophageal cancer cells by microarray analysis. Dis Esophagus.
21:288–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang L, Huang J, Jiang M and Sun L:
Survivin (BIRC5) cell cycle computational network in human no-tumor
hepatitis/cirrhosis and hepatocellular carcinoma transformation. J
Cell Biochem. 112:1286–1294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Holbro T, Beerli RR, Maurer F, Koziczak M,
Barbas CF III and Hynes NE: The ErbB2/ErbB3 heterodimer functions
as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor
cell proliferation. Proc Natl Acad Sci USA. 100:pp. 8933–8938.
2003, View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Emde A, Köstler WJ and Yarden Y:
Association of Radiotherapy and Oncology of the Mediterranean arEa
(AROME): Therapeutic strategies and mechanisms of tumorigenesis of
HER2-overexpressing breast cancer. Crit Rev Oncol Hematol. 84 Suppl
1:e49–e57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Montanaro L, Treré D and Derenzini M:
Changes in ribosome biogenesis may induce cancer by down-regulating
the cell tumor suppressor potential. Biochim Biophys Acta.
1825:101–110. 2012.PubMed/NCBI
|