Introduction
Colorectal cancer (CRC) is one of the most common
malignancies and a major cause of cancer-related death worldwide.
It is the second most common type of cancer in both genders (women:
10.1%; men: 12.4%) and the number of newly diagnosed CRC cases
continues to grow. CRC, according to the WHO, is the third most
common cancer, with 1,361,000 cases worldwide in 2012 (1).
The primary treatment of CRC is surgical resection.
However, approximately 25% of CRC cases are detected in stage IV
(with distant metastases) and almost 50% of CRC patients will
develop metastasis during their lifetime (2). The treatment outcomes for these patients
are unfavorable, since conventional therapies affect proliferating
and differentiated cancer cells from the tumor mass and save cancer
stem cells (CSCs). This approach seems to explain the initial
post-therapy tumor shrinkage, which is often followed by relapses
resulting from the activity of CSCs (3).
Chemotherapy of patients with CRC can be performed
either as monotherapy (capecitabine, irinotecan) or with a combined
protocol (4–6): LVFU2: 5-fluorouracil (5-FU) + calcium
folinate racemate (or levofolinic acid in equivalent dose);
FOLFOX4: 5-FU + calcium folinate racemate (or levofolinic acid in
equivalent dose) + oxaliplatin; FOLFOXIRI: 5-FU + calcium folinate
racemate (or levofolinic acid in equivalent dose) + oxaliplatin +
irinotecan; FOLFIRI: 5-FU + calcium folinate racemate (or
levofolinic acid in equivalent dose) + irinotecan; CAPOX (XELOX):
Capecitabine + oxaliplatin.
Presurgical radiotherapy can be included in two
different ways. The first involves five fractions of radiotherapy
(5 Gy) each a week before surgical intervention. The second
therapeutic mode involves a total of 50.0–50.4 Gy divided into 1.8
or 2.0 Gy fractions, combined with chemotherapy using fluorouracil,
either with calcium folinate or with capecitabine. In the second
protocol, surgery is delayed and is performed at least six weeks
after the last course of radiotherapy. Both protocols ensure
similar efficacy (7,8).
Additionally, in recent years, new drugs targeting
growth factors or their surface receptors have been introduced as
additional therapy for the treatment of CRC (Table I).
 | Table I.Conventional chemotherapeutics and
monoclonal antibodies used for colorectal cancer therapy. |
Table I.
Conventional chemotherapeutics and
monoclonal antibodies used for colorectal cancer therapy.
|
|
Chemotherapeutics |
|
|---|
|
|
|
|
|---|
| Author, year | Name | Type | Mechanism of
action | (Refs.) |
|---|
| Taieb et al,
2014; | 5-Fluorouracil | Pyrimidine
antimetabolite | Inhibition of
thymidylate synthase activity leading to decreased DNA | (6,158–161) |
| Alberts et
al, 2012; | (5-FU) |
| replication and
cell proliferation |
| Cao et al,
2015; |
|
|
|
|
| Longley et
al, 2003; |
|
|
|
|
| Yamada et
al, 2013 |
|
|
|
|
|
| Schmoll et
al, 2015 | Capecitabine | Pyrimidine
antimetabolite | Inhibition of
thymidylate synthase after cellular thymidine phosphorylase | (162) |
|
|
| (5-FU prodrug) | transforms prodrug
to fluorouracil |
|
| Taieb et al,
2014; | Leucovorin | Folic acid
antagonist | Increase in
fluorouracil efficacy | (6,158,159,161) |
| Alberts et
al, 2012; |
|
|
|
|
| Cao et al,
2015; |
|
|
|
|
| Yamada et
al, 2013 |
|
|
|
|
| Cau et al,
2015; | Irinotecan | Topoisomerase I
inhibitor | Metabolically
activated in the body to 7-ethyl-10-hydroxycamptothecin | (159,163–165) |
| Élez et al,
2015; |
| (SN-38
prodrug) | (SN-38) by
carboxylesterase; reversible stabilization of the |
|
| Sclafani et
al, 2015; |
|
| topoisomerase I
complex results in single-strand DNA breaks; inhibition |
|
| Fujita et
al, 2015 |
|
| of DNA synthesis;
arrest of the cell cycle at the S/G2 phase |
|
| Taieb et al,
2014; | Oxaliplatin | DNA alkylator | Formation of
crosslinks in DNA; arrest of the cell cycle in the G2/M phase; | (6,158,161,166) |
| Alberts et
al, 2012; |
| (platin
derivative) | apoptosis induction
via activation of caspases |
|
| Yamada et
al, 2013; |
|
|
|
|
| de Gramont et
al, 2012 |
|
|
|
|
| Longley et
al, 2003; | Tegafur-uracil | Combinatory therapy
of | Tegafur is
metabolically activated in the body to 5-FU by
dihydropyrimidine | (160,167) |
| Bayoglu et
al, 2015 | (UFT) | CRC with 5-FU
prodrug | dehydrogenase
(DPD); uracil, a competitive inhibitor of DPD, inhibits 5-FU |
|
|
| and uracil | catabolism and
prolongs its life time; uracil decreases 5-FU doses,
protecting |
|
|
|
|
| patients from its
toxicity |
|
| Ben Sahra et
al, 2010; | Metformine | Biguanide
derivative | Activation of
caspase 3; induction of apoptosis; restoration of p53 activity | (136–138) |
| Ben Sahra et
al, 2010; |
|
| Monoclonal
antibodies used in combination with chemotherapeutics |
|
| Miranda et
al, 2016 |
|
|
|
|
| Cao et al,
2015; | Bevacizumab | Humanized
monoclonal | Blocking of the
binding of all known VEGF-A isoforms to VEGF receptors; | (159,166,168–170) |
| de Gramont et
al, 2012; |
| IgG1 antibody | inhibition of tumor
angiogenesis |
|
| Feng et al,
2014; |
|
|
|
|
| Strickler et
al, 2012; |
|
|
|
|
| Roviello et
al, 2017 |
|
|
|
|
| Élez et al,
2015 | Abituzumab | Humanized
monoclonal | Binding to integrin
αv heterodimer; inhibition of cell binding to extracellular | (163) |
|
|
| IgG2 antibody | matrix; inhibition
of cell migration; Induction of apoptosis |
|
| Sclafani et
al, 2015 | Dalotuzumab | Humanized
monoclonal | Inhibition of
ligand (IGF-1, IGF-2) binding and induction of IGFR-1 | (164) |
|
|
| IgG1 antibody | internalization and
degradation; inhibition of signaling pathways responsible |
|
|
|
|
| for proliferation
and resistance to apoptosis |
|
| Cunningham et
al, 2004; | Cetuximab | Chimeric
monoclonal | Antagonist of EGFR;
prevention the signaling and ligand-induced | (4,6,158,164,171,172) |
| Taieb et al,
2014; |
| IgG1 antibody | dimerization of the
receptor; increases susceptibility of EGFR-positive |
|
| Alberts et
al, 2012; |
|
| cells to immune
cytotoxic cells; reduction in tumor growth |
|
| Sclafani et
al, 2015; |
|
|
|
|
| Huang et al,
2014; |
|
|
|
|
| Terazawa et
al, 2017 |
|
|
|
|
| Tay et al,
2015; | Panitumumab | Human
monoclonal | Antagonist of EGFR;
prevention of EGFR autophosphorylation and | (173,174) |
| Bahrami et
al, 2017 |
| IgG2 antibody | signaling;
induction of apoptosis; inhibition of interleukin 8 and VEGF |
|
|
|
|
| production;
reduction of tumor growth |
|
| Françoso and | Ramucirumab | Humanized
monoclonal | Binding of the
extracellular domain of VEGF and VEGFR-2; inhibition of the | (175,176) |
| Simioni, 2017; |
| IgG1 antibody | activation and
signaling of VEGF/VEGFR-2; inhibition of angiogenesis |
|
| Ursem et al,
2016 |
|
|
|
|
Although about 50% of patients respond to
conventional therapy, most develop drug resistance during the
course of treatment, and recurrence of the disease often follows
(9,10). Our review presents the current state
of knowledge concerning experimental CRC treatment protocols
targeting CSCs through the induction of their proliferation,
differentiation, and sensitization to apoptotic signals. The
combined therapy consisting of two distinct constituents:
Conventional drugs and the novel anti-CSC factor; an improvement of
the anticancer therapy efficacy and a reduction in undesirable side
effects is hoped for.
Identification of cancer stem cells in
colorectal cancer
Under physiological conditions, the pool of normal
cells is maintained in tissues and organs due to the presence of
small subpopulation of stem cells with a great capability to
self-renew, proliferate, and differentiate. A tumor can be seen as
an abnormal type of tissue whose growth and development are depend
on a population of stem cells, termed CSCs (11–13). These
CSCs may gain their specific properties-such as self-renewal,
unlimited proliferation potential, and ability to differentiate
into any mature cancer cell type-as a result of neoplastic
transformation caused by the accumulation of some genetic and
epigenetic aberrations. Additionally, they develop specific
protective mechanisms, such as those directed against immune cells,
or insensitivity to standard chemotherapeutics. The CSC hypothesis
remains controversial, but the occurrence of CSCs has been
identified within both hematological and solid tumors, such as
breast and CRCs (11–13).
The identification of CRC-CSCs is based on a set of
CSC-associated protein markers (Table
II). It is not clear if all of these biomarkers influence CRC
progression with the same effect. Furthermore, the great range of
these proteins may result from the genetic heterogeneity of cancer
cells both within the tumors of a particular patient and between
patients (11,14,15).
Experimental data from the rodent model of CRC suggest that only 1
in 25 cells (16), or 1 in 262 cells
(13,16), possesses the characteristic features
of CSCs in the total population of CRC cells. This diversity may
result from the complexity of experimental settings. The initial
verification of new markers for the isolation of CSCs often follows
discoveries in the field concerning either normal tissue stem cells
or CSCs of different tumors. However, the selection of the most
universal and useful CSCs markers has yet to be performed.
| Table II.Markers of colorectal cancer stem
cells. |
Table II.
Markers of colorectal cancer stem
cells.
| Author, year | Marker | Function | (Refs.) |
|---|
| Ricci-Vitiani et
al, 2007; | CD133 | Prominin-1;
membrane glycoprotein, present on | (13,177–179) |
| Botchkina,
2013; |
| the surface of
actively proliferating stem cells; |
|
| Haraguchi et
al, 2008; |
| function
unknown |
|
| Zhu et al,
2009 |
|
|
|
| Manhas et
al, 2016; | CD44 | P Glycoprotein 1;
membrane hyaluronic acid receptor | (2,23,26,178,180) |
| Vermeulen et
al, 2008; |
|
|
|
| Du et al,
2008; |
|
|
|
| Haraguchi et
al, 2008; |
|
|
|
| Botchkina et
al, 2009 |
|
|
|
| Manhas et
al, 2016; | CD166 | ALCAM; membrane
glycoprotein, adhesion molecule | (2,23,180) |
| Vermeulen et
al, 2008; |
|
|
|
| Botchkina et
al, 2009 |
|
|
|
| Huang et al,
2009; | ALDH1 | Aldehyde
dehydrogenase, detoxification enzyme, transforms | (16,181) |
| Zhou et al,
2015 |
| retinol to retinoic
acid, which regulates proliferation of cells |
|
| Vermeulen et
al, 2008 | CD29 | β1 integrin,
adhesion molecule | (23) |
| Manhas et
al, 2016; | CD24 | Heat-stable
antigen; membrane glycoprotein, | (2,23) |
| Vermeulen et
al, 2008 |
| adhesion
molecule |
|
| Manhas et
al, 2016 | ESA | Epithelial specific
antigen, EpCAM, CD326; membrane | (2) |
|
|
| glycoprotein,
adhesion and signaling molecule; |
|
A minor Bmi-1-positive subpopulation of CSCs is
characterized by low mitotic activity, and thus is supposed to
constitute the pool of cells which are resistant to
chemotherapeutics and responsible for tumor relapse through intense
proliferation following therapy (17,18).
According to the CSC hypothesis, conventional chemotherapeutics
reduce the tumor mass, but are not sufficiently efficient to
eliminate all cancer cells, on account of the presence of
chemoresistant CSCs. Efficient DNA repair mechanisms, telomerase
activity, insensitivity to proapoptotic signals, and high levels of
expression of ATP-binding cassette transporters (ABC transporters)
are postulated as the main causes of chemoresistance (19–21).
CD133 protein
The identification and classification of CSCs is
rather controversial, as none of the known markers are universal
and reliable for the identification of CSCs in all experimental
settings (Table II) (22). The most commonly used marker of
CRC-CSCs is prominin-1, also named CD133 (22). CD133+ cells are able to
reproduce a CRC tumor in a mouse xenotransplantation model, whereas
CD133− cells cannot rebuild cancer bulk (11,13).
However, the research groups of Ricci-Vittani and Shmelkov showed
independently that CD133− cells also possess high
proliferative and differentiating potential, comparable to those of
CD133+ CRC-CSCs (13,14).
CD133+ CRC-CSCs isolated from human tumors may be
cultured in vitro for as long as one year without any change
in their phenotype, gaining the ability to form undifferentiated
tumor spheres which maintain the ability to engraft (13). Moreover, it has been shown that even a
single CD133+ cell is able to reproduce the tumor mass
in vivo (23). Human CRCs
resistant to a conventional 5-FU treatment have been found to be
enriched in CD133+ cells; this is directly correlated
with a worse outcome for patients (24). However, knockout of CD133 has been
found not to affect the clonogenicity of cancer cells, suggesting
that CD133 is a passive marker, rather than a CSC-promoting factor
(25–27).
CD44 protein
CD44 is a transmembrane glycoprotein, a receptor of
hyaluronic acid that participates in many cellular processes,
including growth, survival, differentiation and motility.
CD44+ CD133− cells isolated from human CRC
tumors have been shown in vivo to efficiently initiate a
xenograft tumor that possesses similar properties to those of the
primary tumor. Knockdown of CD44 strongly reduced proliferation of
these cells and inhibited tumorigenicity in a mouse xenograft model
(26,27).
Aldehyde dehydrogenase 1
Aldehyde dehydrogenase 1 (ALDH-1) has been
identified in both nonmalignant and malignant stem cells. In many
neoplasms-such as colon, pancreas, breast, and urinary bladder
cancers-this enzyme has been shown to be associated with disease
progression (16,28–31).
Generally, ALDH-1 is responsible for detoxification and defending
against free radicals, although it plays a crucial function in
cancer recurrence due to the downregulation of CSCs' metabolism
during conventional chemotherapy (16,28–31). The
activity of ALDH-1 may be pharmacologically blocked via the
specific inhibitor DAEB (diethylaminobenzaldehyde) (30). A combination of DAEB with conventional
chemotherapeutics, such as doxorubicin and paclitaxel, increases
the level of oxidative stress in cells, enhancing their
susceptibility to free radicals and apoptosis. The first promising
results of such an approach were demonstrated for breast cancer
cell lines (32).
The characteristics of CRC-CSCs being
considered for CSC-targeting therapeutic strategies
The discovery of CSCs in various tumors has provided
new opportunities to overcome chemoresistance and radioresistance
of tumor cells through the targeting of this unique population
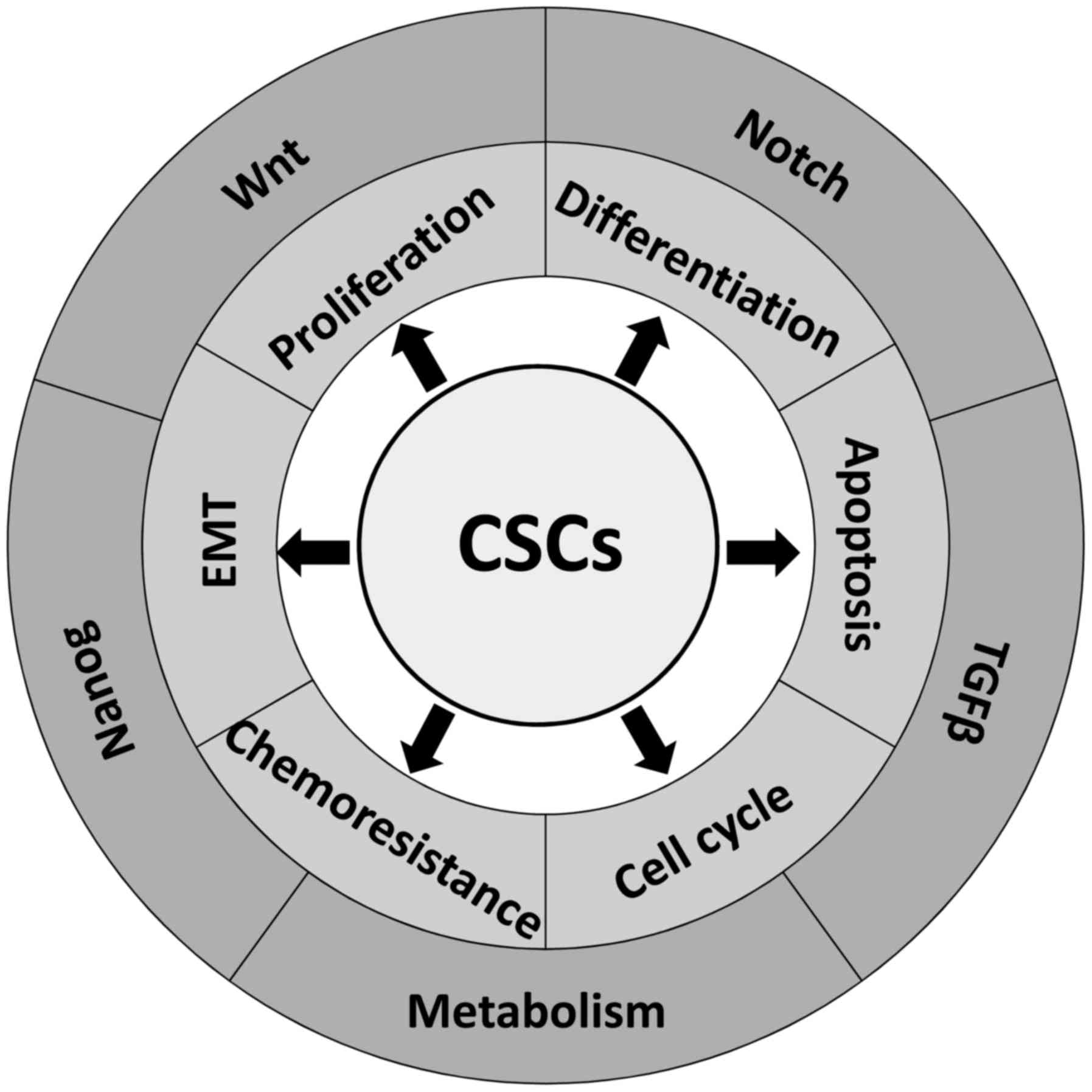
(Fig. 1). To achieve this goal,
diverse strategies have been used: the induction of CSC
differentiation, the inhibition of the epithelial-mesenchymal
transition (EMT), the reduction of angiogenesis, and the
suppression of specific signaling or metabolic pathways.
Significantly, our increasing understanding of the cellular and
molecular mechanisms that regulate CSC quiescence, cell cycle
progression, self-renewal, and resistance to proapoptotic signals
and chemotherapeutics may provide new therapeutic modalities that
will reduce morbidity and increase the overall survival of CRC
patients.
Induction of CRC-CSC
differentiation
The first of the therapeutic approaches is based on
the induction of CSC differentiation into more mature types of
tumor cells, resulting in a reduction of CSC number. In contrast to
CSC, mature cancer cells have no self-renewal ability, cannot
proliferate unlimitedly or induce immunological tolerance, and are
more susceptible to conventional chemotherapy. Such a therapeutic
strategy has been already used in promyelocytic leukemia patients
being treated by retinoic acid (RA). Increased intracellular RA
concentration upregulates the expression of its normal retinoic
acid receptor, RAR, which competitively displaces the
cancer-mutated receptor, PML-RAR. RA functions as an agonist of
steroid hormone receptors and, due to the binding to transcription
factors in the nucleus, may induce the differentiation of abnormal
blasts (33).
Impairment of cell cycle checkpoints
in CRC-CSCs
Blocking of the cell cycle checkpoint proteins
represents a novel approach to treatment aimed at overcoming CSC
resistance to conventional cancer therapy. This approach is based
on the assumption that cells with dysfunctional checkpoints
proliferate in an uncontrolled manner, which could cause genome and
metabolic destabilization and lead to cell death.
The combination of two potential therapeutic
compounds, flavonoid morin and telomerase inhibitor MST-312, has
been demonstrated to lower tumorigenicity of CSCs by targeting
signal transducer and activator of transcription 3 (STAT3) and
telomerase in human CRC cells. A morin/MST-312 combination has been
shown to inhibit the phosphorylation of cellular proteins such as
p53 and check point kinase 2 (Chk2), which are known to play
crucial roles in DNA damage checkpoint control. Inhibition of CRC
HT29 and SW620 cells' proliferation in the morin/MST-312
dose-dependent manner and a decrease in CD44+ CRC-CSC
count were observed (34).
Martino-Echarri et al studied six CRC cell
lines and showed that those expressing a mutated APC gene
exhibited a limited response to 5-FU. The sensitivity of
APC-mutated CRC cells to 5-FU was significantly increased by
deactivating the Chk1 kinase using antisense siRNA-mediated
knockdown (35). These data suggest
that cancer cells (enriched by CSCs) lacking the activity of cell
cycle regulating proteins are much more sensitive to proapoptotic
stimulation (35).
Inhibition of epithelial-mesenchymal
transition
Cancer cells derived from epithelial tissue undergo
differentiation during which they lose the features of their
original tissue and gain some properties of connective tissue
cells, during a process called epithelial-mesenchymal transition
(EMT), which is essential for acquisition of the invasion
phenotype. EMT is regulated by many intracellular signaling
pathways, such as Wnt, Nanog, and transforming growth factor β
(TGFβ), whose functions are impaired during cancer transformation
(36). Moreover, during EMT, non-CSCs
may obtain some characteristics of CSCs through
transdifferentiation, which enables the transition to the more
primitive state and, as it happens, cells have been much better in
acquiring therapy resistance (37,38). EMT
is one of the possible ways to alter the features of cancer cells,
especially of CSCs, which are known to be responsible for the lack
of susceptibility to standard chemotherapy (39). The most frequently diagnosed
metastases in CRC patients occur in the liver, and the mean 5-year
survival rate of such patients is approximately 10% (40).
Crucial signaling pathways associated
with efficient maintenance of CRC-CSCs: Potential targets for
therapy of CRC-CSC
Wnt signaling crucial for CRC-CSC
features and survival
The Wnt/β-catenin pathway has been implicated in the
maintenance of the intestinal crypt stem cell pool, and Wnt
signaling dysregulation (through either loss of APC function or
oncogenic β-catenin mutations) has been identified in 70% CRC
tumors (41,42).
The Wnt pathway is evolutionary conserved and
consists of a family of secreted glycoproteins, known as the 19
distinct Wnt ligands in mammals (1,42–44). The importance of this pathway is
revealed by its role in the establishment of embryonic axis, cell
fate determination, maintenance of adult tissue homeostasis, and
regeneration (45,46). Thus, loss of APC allows
gastrointestinal stem and progenitor cells to continue
proliferating without dying (41,42).
Moreover, in a proof-of-principle assay, β-catenin was demonstrated
to be required for clonal growth of human CRC cell lines, and
targeted deletion of the mutated, constitutively active form of
β-catenin abolished the ability of the CRC cell line SW480 to grow
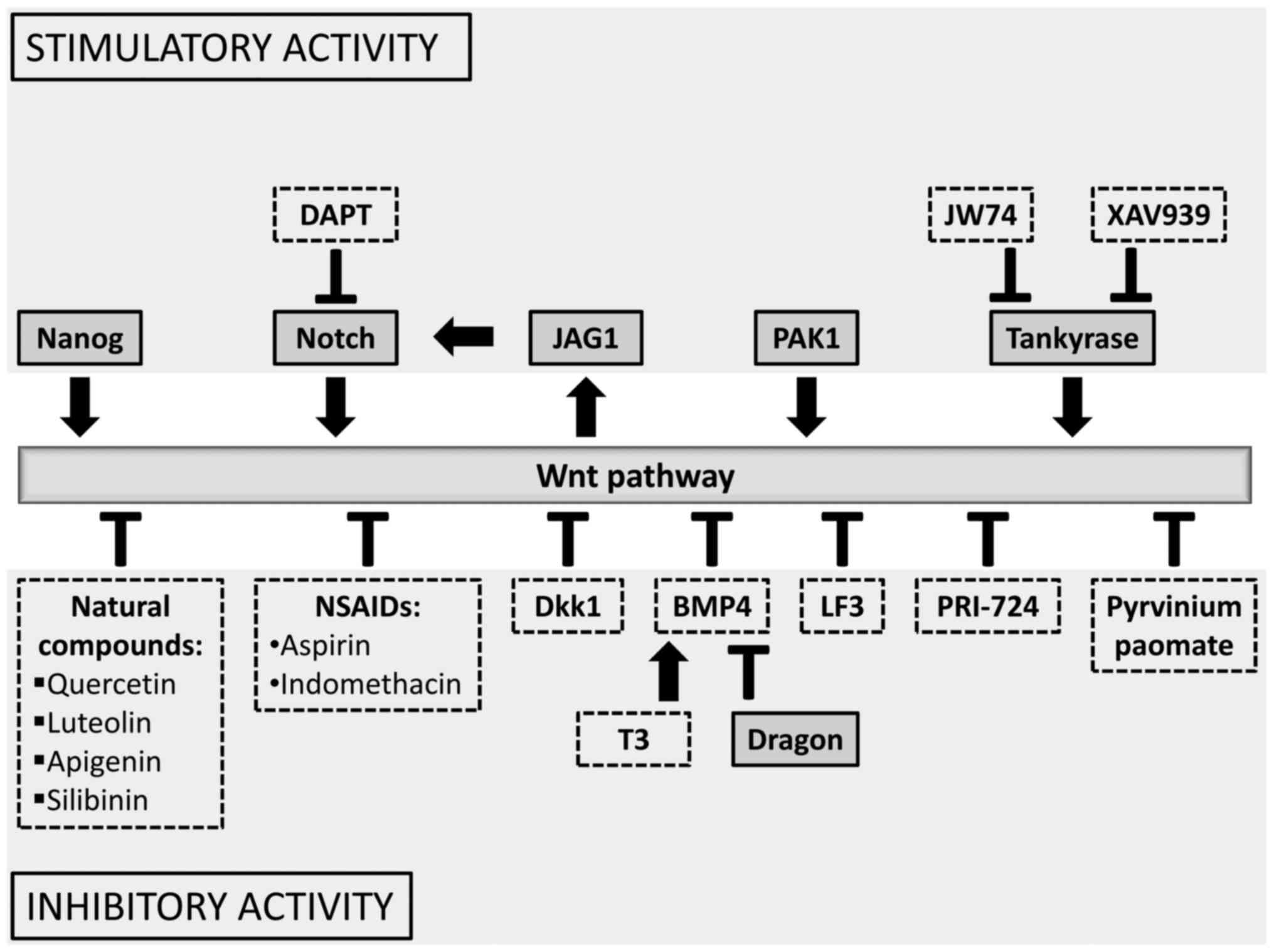
in vitro (47). Our paper is
aimed at presenting a few therapeutic compounds that target the
cytoplasmic β-catenin destruction complex or inhibit expression of
the target genes (1,42) (Fig.
2).
A recent study has suggested that one protein,
p21-activated kinase 1 (PAK1) is an effective stimulator of the
Wnt/β-catenin pathway and may be a good target for CRC treatment,
since PAK1 inhibition has been found to give a synergistic effect
with 5-FU (48). It has been shown
that PAK1 is associated with the maintenance of stem-cell-like
features of CRC-CSCs, such as the expression of CD44,
tumorigenicity, and spherogenicity in both in vitro and in
xenograft tumor models in vivo (48).
Pyrvinium pamoate, an antiparasitic drug, has been
shown to inhibit LRP6-mediated axin degradation and the potency of
β-catenin stabilization (49).
Pyrvinium treatment of HCT116 and SW480 CRC lines with mutated
APC or β-catenin (CTNNB1) genes inhibited both Wnt
signaling and cell proliferation. Additionally, some other findings
have demonstrated the allosteric activation of CK1α to be an
effective mechanism for inhibiting Wnt signaling (49).
The discovery of tankyrases (ADP-ribosylating
enzymes) and their role in the direction of axin for ubiquitination
and proteasomal degradation (50–53) has
made a significant contribution to this field, as it may provide a
new way of targeting the Wnt pathway (53). Inhibition of tankyrases causes the
stabilization of axin, which enhances the destruction of β-catenin
and reduces Wnt signaling (51).
Tankyrase (Tnk) inhibition with the use of new compounds, such as
JW74 and XAV939, has also been shown to reduce growth, induce
apoptosis and differentiation of cancer cells, and inhibit
stem-cell properties and migration of CSC-like cells in cancer
lines of diverse origins (osteosarcoma, neuroblastoma, colon)
(42). Several small Tnk inhibitors
have been reported to possess anticancer efficacy against cell
lines of diverse origin, both in vitro and in vivo in
xenograft mouse models (50,51,54,55).
However, the clinical utility of Tnk inhibitors is at present
limited by intestinal toxicity and low therapeutic index in a mouse
model (56).
PRI-724, a second generation specific CBP/catenin
(cyclic AMP response element binding protein) antagonist has been
shown to be safe in preclinical studies. PRI-724 disrupts the
complex of β-catenin with CBP, which reduces the expression of a
subset of Wnt target genes that are important in the proliferation
of CRC cells (57). Several phase
I/II trials are ongoing in hematological malignancies, pancreatic
cancer, and colon cancer, testing the effectiveness of PRI-724
compound (58). Moreover, PRI-724
induced differentiation of CRC xenografts, accompanied by tumor
growth suppression (42,59). Recently, the antineoplastic activity
of the LF3 compound, which directly inhibits β-catenin/TCF4
interaction, was reported (59). LF3
treatment of colon, head, and neck cancer cells resulted in the
suppression of Wnt activity and reduced self-renewal properties of
CSCs (54).
Dickkopf-1 (Dkk1) as a potential
target in CRC therapy
Dkk1-a potent, soluble Wnt pathway inhibitor-is
reported to be a promising molecule in potential therapy of CRC
(60). It has an affinity to one of
the coreceptors LRP5/6 affecting the formation of the active
receptor complex, Frizzl/LRP5/6, which induces endocytosis of those
receptors, inhibiting Wnt signaling.
Additionally, Dkk1 has a role in embryogenesis and
its level is regulated by negative feedback with Wnt pathway
effectors, such as β-catenin (61).
However, in CRC, this mechanism is disturbed by mutations and
epigenetic changes in genes encoding β-catenin (62). It has been shown in 217 CRC patients
that Dkk1 overexpression is inversely related to tumor grade,
presence of metastasis, and the recurrence rate of colon cancer. In
samples obtained from patients with high Dkk1 levels, increased
expression of E-cadherin and cytoplasmic β-catenin, and a reduced
level of vimentin (an EMT marker) were observed in comparison to
Dkk1-negative samples (62).
Additionally, the overexpression of Dkk1 in CRC HCT-116 cells
allowed the maintenance of epithelial phenotype and led to
diminished expression of transcription factors characteristic of
EMT (such as Snail and Twist), but also decreased the expression of
CSC markers (such as CD133 and Lgr-5) (63). An immunocytochemical analysis has
shown a correlation direct between Dkk1 expression and decreased
microvessel density, as well as VEGF expression in CRC tumors. CRC
cells overexpressing Dkk1 formed smaller tumors following
xenotransplantation, with a significantly lower number of small
blood vessels (64). Hence, the Dkk1
protein can suppress the progression of the colon cancer, possibly
through EMT inhibition, and could serve as a potent target of
antitumor therapy (64,65).
Natural compounds targeting the Wnt
signaling pathway
Flavonoids, polyphenolic compounds, constitute a
very large group of natural products and are one of the most
characteristic classes of compounds in plant metabolism (42). Their therapeutic anticancer properties
have been studied for decades and are related to the ability of
these molecules to modulate the Wnt/β-catenin signaling pathway
(66–68). Flavonoids have been shown to affect
different elements of this signaling pathway, varying from ligand
receptor recognition and binding (Wnt/Frizzled/LRP5/6), to the
methylation of genes encoding Wnt components (1).
Quercetin, one of the most studied flavonoids in
clinical trials, has been suggested as a potential anticancer drug
in CRC (1,67,68) due to
its modulation of Wnt activity (1,56,57). Quercetin interacts with β-catenin and
inhibits the binding between β-catenin and TCF (69). Moreover, quercetin, as well as the
flavonoids luteolin and apigenin, inhibits GSK-3β, which is a
multifunctional serine-threonine kinase involved in the formation
of β-catenin destructive complex in cytoplasm (67,68).
Silibinin, a flavonolignan from milk thistles, has
been shown to exert chemoprevention of intestinal cancer in
vitro and in vivo in a mouse model (70,71). The
pilot study on CRC patients who were administered silipide, an oral
formulation of silibinin and phosphatidylcholine, demonstrated
increased levels of silipide in blood, liver, and tumor tissue
(70–72). In an experimental follow-up to that
study, silibinin has been shown to suppress the growth of CRC SW480
cells in culture and the growth of xenografts through
downregulation of β-catenin-dependent signaling (71). The effect of silibinin on CRC-CSCs
from the HT29, SW480, and LoVo lines has been shown to be mediated
by blocking IL-4/-6 protumorigenic signaling and is associated with
decreased mRNA and protein levels of various CSC-associated
transcription factors, signaling molecules, and surface markers
(such as CD44, NANOG, TERT, SOX-2, SOX-9, and WT1). Furthermore,
differentiation assays have indicated the formation of more
differentiated clones by silibinin due to the shifting of CSC cell
division to asymmetric. These findings support the clinical
usefulness of silibinin in CRC intervention and therapy (73).
Recent clinical trials thus suggest that targeting
downstream components of the neoplastic Wnt pathway may be a novel
therapeutic approach for CRC treatment.
Nanog is crucial in CRC-CSC
activity
Nanog is a crucial transcription factor involved in
the maintenance of pluripotency and self-renewal ability in
undifferentiated embryonic stem cells (74). This protein is thought to be
responsible for many aspects of cancer development typical of CSCs,
such as proliferation, self-renewal, migration,
epithelial-mesenchymal transition, and resistance to conventional
chemotherapy. Its increased expression has been found to correlate
with worse prognosis in many types of cancer, including liver,
kidney, colon, prostate, brain, and endometrial cancers (74–82).
NANOG activation in cancer cell cultures promotes their
transformation into CSCs, as has been shown following ectotopic
overexpression of NANOG/NANOG8 in the colon and prostate
cancer cell lines (75,80,81).
Meng et al (83) provided evidence that Nanog can be used
as a prognostic factor of CRC after they examined 75 human CRC
samples, which showed that overexpression of NANOG strongly
correlated with poor prognosis and lymph node metastasis (83). Another study conducted
immunohistochemical analysis of the expression patterns of
CSC-specific markers (such as CD44, CD133, Nanog, and Oct3/4) and
of immunosuppressive molecules HLA-G and HLA-E in advanced CRC
tumor tissues and noncancerous colon biopsies. Statistically
significant increased expression of these genes in CRC tumor
tissues has been found in comparison to colon biopsies of healthy
subjects. These findings suggest that CRC-CSCs may have increased
expression of HLA-G and HLA-E, which may be considered as an
immune-evasive mechanism and may thus become new potential targets
in the elimination of CRC-CSCs (84).
Lentivirus-mediated Nanog overexpression has been
revealed to significantly improve the proliferation and migratory
abilities of CRC cells; Nanog was thus supposed to induce EMT
through upregulation of the Slug and Snail transcription factors.
Moreover, Nanog silencing mediated by interfering RNA in breast
cancer MCF-7 and MDA-MB-231 cells resulted in a reduced size of the
tumor in a xenotransplantation model and decreased proliferation of
these cells (85). Silencing of the
NANOG gene was associated with diminishing activation of
cyclin D1 and cyclin-dependent kinases (85,86).
Downregulation of Nanog in embryonic stem P19 cells resulted in the
reduction of pluripotency markers such as Fgf4, Klf2, Mtf2, Oct-4,
Rex1, Sox1, Yes, and Zfp143, whereas overexpression of NANOG
restored their primary expression levels (86). Interestingly, the expression of cyclin
D1 and c-myc were markedly downregulated, and the cell cycle was
blocked at the G0/G1 phase following the knockdown of NANOG,
while the expression of cyclin E and signal transducers and
activators of transcription 3 (STAT3) remained unaffected in breast
cancer cells (85).
Embryonic NANOG is considered an important
regulator of pluripotency, whereas NANOGP8
(NANOG-pseudogene) plays a crucial role in tumorigenesis
(75). NANOGP8 can substitute
for NANOG in directly promoting stemness in CRC; this
conclusion was drawn from the observation that 80% of human CRC
liver metastases expressed NANOG and 75% of the metastases
contained NANOGP8 transcripts (76). The effects of NANOG
inhibition-such as reduced spherogenicity, growth, and expression
of embryonic-like transcription factors (Oct4, Sox2)-were partially
reversed by the overexpression of NANOGP8 (76,81).
Recent studies have suggested that the knockdown of
NANOG/NANOG8 genes impairs the ability to migrate and
metastasize in xenograft mouse models, as well as the progression
of the cell cycle and resistance to apoptosis in CRC cells and
embryonic carcinoma cells (75,76,86). Nanog
inhibitors administered with cisplatin and other chemotherapeutics
had a synergistic effect, and led to apoptosis of esophageal cancer
cells (25). The lentivirus
vector-mediated inhibition of NANOG/NANOG8 in CRC cells
(Clone A, CX-1, LS 174T) decreased the level of Bcl-2 antiapoptotic
protein and increased sensitivity to proapoptotic factors ABT-737
and ABT-199 (87). Such combined cell
treatment, including inhibitors of Nanog and the modulation of
proapoptotic Bcl-2 family proteins, may provide a new potential
therapeutic approach for CRC-CSCs.
Immunocytochemistry and microarray examination
showed that NANOG1 expression was limited only to very small
population of CSCs, which made up 0.5–2% of all CRC cells (75). Furthermore, NANOG1 expression
showed a positive correlation with c-JUN and Wnt/β-catenin/TCF4
expression (75), which are known to
be disrupted in CRC oncogenic transformation (41,42). The
ectopic expressions of OCT4 and NANOG in lung
adenocarcinoma cells led to an increased percentage of
CD133+ cells and sphere formation rate, and promoted
drug resistance and epithelial-mesenchymal transition (EMT)
(88,89). Since Nanog directly inhibited EMT, it
has been suggested that it should be considered as a potential
therapeutic approach (89).
TGFβ inhibitors target the
epithelial-mesenchymal transition
TGFβ belongs to a superfamily of approximately 30
different pleiotropic proteins that control cell proliferation,
migration, adhesion and apoptosis, maintaining tissue homeostasis.
Of the three isoforms of TGFβ (TGFβ-1, TGFβ-2 and TGFβ-3), TGFβ-1
has been most widely studied. Signaling is mediated by binding to
cell membrane receptors (TGFβR1 and TGFβR2), which results in the
phosphorylation of cytoplasmic SMAD proteins being translocated to
the nucleus and binding with activators or repressors of genes
associated with proliferation, survival, and migration. However, in
specific situations, such as the advanced stages of cancer, TGFβ
promotes the progression of the disease. During neoplastic
transformation, cells lose their susceptibility to TGFβ signaling,
which then acts as an autocrine promoter of invasion and metastasis
(90,91).
TGFβ is a positive regulator of processes associated
with EMT. Among other effects, it stimulates the modification of
morphology and the loss of cell polarity, decreases E-cadherin
expression, and increases the expression of key transcription
factors, such as Snail1/2, Twist, and Zeb1/2 (90). The synthetic proteins P17 and P144,
designed to inhibit TGFβ1-mediated pathways, have recently been
considered as a useful tool in a clinical approach aimed at
reducing liver metastases from CRCs, lymphomas, and thymomas
(92,93). Additionally, the administration of
peptide P17 blocked the adhesion of cancer cells to cancer
fibroblasts and significantly reduced metastasis to the liver,
proliferation, and angiogenesis in xenotransplantation model
(94). In a CRC CT26 cell line, P17
peptide was involved in the blockage of the T regulatory (Treg)
lymphocytes, which synergistically increased the total effect of
this compound (92).
The complex analysis of the role of Kindlin-1 in the
TGFβ pathway strongly suggests that this regulatory molecule may be
a new anticancer target. Kindlin-1 is known to be essential for the
maintenance of the structure of cell-matrix adhesion (95). Recently, Kindling-1 has been
identified as directly interacting with the key TGFβ/SMAD3
signaling components in numerous CRC cell lines (SW1116, SW480,
SW620, Caco2, HCT116, RKO, LST and HT29). Kindlin-1 expression has
been found to correlate with the progression of CRC and poor
prognosis (96).
BMP regulates differentiation and
maturation
Bone morphogenetic protein 4 (BMP4), belonging to
the TGFβ superfamily, has been suggested to be a key regulatory
factor in the differentiation of CSCs in CRC (50,97). BMP4,
which is secreted by the connective tissue cells of the intestinal
wall, has been shown to regulate the maturation and differentiation
of normal epithelial cells via paracrine signaling (30,98,99). The
distribution of BMP4 increases along the colon crypt axis from
bottom to top, and thus its signaling increases toward the top of
the crypt. The loss of BMP4 activity in the intestinal epithelium
may lead to altered maturation of epithelial cells and, in
consequence, to the development of CRC (100). Recently, BMP pathway suppression has
been suggested as an essential factor leading to
inflammation-induced tumorigenesis of CRC in a mouse model of
colonic polypoidogenesis where adenomatous polyps arise several
months after induction (97).
Additionally, silencing the BMP4 gene by transplacental RNAi
administration appeared to be sufficient to induce the formation of
colorectal polyps in mice (101).
It has been shown that BMP4 stimulates the
maturation and apoptosis of CSCs by reducing β-catenin levels in
the nucleus (98,102). Recombinant BMP4 was able to
stimulate maturation, differentiation, and apoptosis, leading
eventually to higher susceptibility to chemotherapy in human
CRC-CSCs. Administration of this protein to nude mice bearing a
tumor originating from CRC-CSCs improved the antitumor effect of
oxaliplatin and 5-FU. The observed effects did not depend on either
SMAD4 expression or microsatellite stability (103).
Additionally, a meta-analysis has demonstrated that
the locus rs4444235 of the BMP4 gene may be considered as a risk
factor for CRC in some ethnic populations (East Asians and
Caucasians) (104). Moreover, Dragon
(RGMb, a member of the repulsive guidance molecule family) has been
found to be upregulated in CRC. Both mRNA and protein levels were
increased in tumor tissue proportionally to CRC progression. The
knockdown of the Dragon gene with the use of shRNA (small hairpin
RNA) led to a lowered proportion of CD133+ CRC-CSCs in
CT26.WT and CMT93 cell lines (105).
Dragon, as a co-receptor for BMP signaling (106), has been suggested as a new target
for anti-CRC therapy (105).
Recently, triiodothyronine (T3) has been described
as playing a role in the regulation of BMP4 signaling by
sensitizing CRC-CSCs to chemotherapeutics via significant
attenuation of Wnt pathway signaling, and, by extension, via
reduction of their tumorigenicity. The influence of T3 on BMP4/Wnt
pathway was demonstrated when sphere-forming CSCs from patient
samples treated with 5-FU and oxaliplatin presented increased cell
death (up to 75%) (107).
Blocking Notch pathway increases the
efficiency of anticancer therapy
Under normal circumstances, Notch signaling clearly
plays an important role in the maintenance of colon crypt
homeostasis. However, the inappropriate activation of the Notch
signaling pathway has been reported to be associated with CRC-CSCs.
An upregulated Notch pathway has been found to play a role in CSC
viability, tumorigenicity, and self-renewal (108,109).
In humans, Notch signaling shows high activity in
adenomas and early stage CRCs (65,110), but
low activity in advanced, later stage, and metastatic CRCs
(111). The molecular mechanisms
that cause Notch signaling to be important for early stage CRC
initiation are not understood, and only a few mechanistic studies
of Notch signaling in human CRC cell lines have been performed
(109). Moreover, Hoey et al
demonstrated that, by inhibiting DLL4 (Delta-Like 4 Ligand), an
important component of the Notch pathway, with human monoclonal
antibody in colon carcinoma xenografts, tumor growth and the
frequency of CSCs were reduced in comparison to the control
(112). Combination treatment with
irinotecan and anti-hDLL4 reduced tumor growth and CRC stem cell
frequency at higher levels than the anti-DLL4 treatment alone
(112,113). This indicates that inhibiting Notch
signaling reduces CSC frequencies and sensitizes tumor cells for
irinotecan treatment.
However, treatment with anti-DLL4 antibody leads to
serious toxic effects in the liver, including sinusoidal dilation
and centrilobular hepatocyte atrophy, as observed in mice, monkeys,
and rats (114). Using athymic nude
mice as a model system, prominent thymic atrophy in
immune-competent animals treated with anti-DLL4 antibody was
observed. Chronic DLL4 blockade has been shown to activate
endothelial cells, disrupt the homeostasis of organs (including the
heart, lung, liver, and skin) and induce vascular tumors (114). These reservations notwithstanding,
further studies were conducted on both CRC patient-derived
specimens (in colon tumor xenografts in NOD/SCID mice) and CRC
lines (HCT116 and SW480), and these confirmed the efficacy of such
potential therapeutic strategy (115,116).
Van Es and colleagues (117) demonstrated that the blocking of the
Notch cascade with a γ-secretase inhibitor dibenzazepine (DBZ)
induced goblet cell differentiation in adenomas, even in mice
carrying a mutation of the Apc gene, and subsequent tumor growth
arrest (117). Additionally, another
group induced expression of the Notch intracellular domain in the
intestinal epithelium of transgenic mice, impairing both
differentiation of the goblet and enteroendocrine cells and
resulting in intensive proliferation of immature intestinal
progenitor cells (118).
Notch signaling plays an important role in the
determination of cell fate. In recent years, this signaling pathway
has been shown to play a critical role in regulating the balance
between proliferation, differentiation, and apoptosis of cells in
various tissues (108,109). The interaction between Notch
receptors and their ligands (Jagged 1 and 2, and Delta-like 1, 3,
and 4) results in the proteolytic cleavage of Notch receptors by
γ-secretase and other proteases, which releases the Notch
intracellular domain (NICD) from the plasma membrane and initiates
its subsequent translocation into the nucleus. After nuclear
translocation, NICD binds to and forms a complex with one of three
transcriptional regulators (119–121).
Moreover, the Jagged1 gene (JAG1), which
encodes a Notch ligand, has been reported to be transcriptionally
activated by the β-catenin/TCF4 complex (122). The expression of JAG1 was
limited to enteroendocrine cells of the human small intestine
epithelium and was undetectable in the mucosa of human large
intestine. In contrast, increased expression was found in half of
human colon tumors, although not all tumors with elevated Wnt
signaling displayed elevated Jagged1 (122). Experiments on mice have demonstrated
that elevated levels of Notch signaling in most intestinal tumors
co-occurred with increased JAG1 expression. Targeting of
Jagged1 could thus be effective in downregulating Notch signaling
in a subset of tumors, as shown in the human HT29Cl16E CRC line
(122).
Endothelial cells have been reported to promote the
CSC phenotype of human CRC cells through the secretion of the
soluble form of Jagged1. In human CRC specimens, CD133−
(a basic CRC-CSC markers) and NICD-positive CRC cells have been
found to colocalize in perivascular regions (119,123).
Microarray analysis has identified a group of
Wnt/β-catenin downstream genes that are directly regulated by Notch
(65). These genes were repressed by
γ-secretase inhibitors and upregulated by active Notch1, even in
the absence of β-catenin signaling, through β-catenin-mediated
transcriptional activation of the Notch-ligand Jagged1 in Ls174T
CRC cells. Consistently, the expression of activated Notch1
partially reversed the effects of blocking Wnt/β-catenin pathway in
tumors implanted into nude mice. These results suggest that Notch
activation, accomplished by β-catenin-mediated upregulation of
Jagged1, is required for tumorigenesis in the intestine (65).
Moreover, a recent study in nude mice indicated
that a subpopulation of CRC HCT116 cells chemoresistant to 5-FU and
oxaliplatin, enriched in CD133+CD44+ CSCs,
was more sensitive to γ-secretase inhibitor (DAPT), which depleted
the cells in vitro and reduced the growth of tumors derived
from these cells (124). Another
study reported that upregulation of Notch1 in colonic cancer cells
may provide a specific protective mechanism in response to
conventional chemotherapeutics (125). These findings suggested that
inhibiting the Notch pathway may be an effective strategy for
targeting CRC-CSCs and overcoming the resistance of CRC cells to
conventional chemotherapeutics.
Metabolic target strategy
Although it has been commonly accepted that
neoplastic transformation is caused by many genetic and epigenetic
factors, little is known of how it affects the metabolism of cancer
cells. There are only few reports concerning selected aspects of
cancer cell metabolic adaptations which impede cancer
progression.
Recent studies have demonstrated overexpression of
SIRT1 (silent mating type information regulation 2 homolog 1) in
cancer cells resistant to 5-FU and described its implication for
the promotion of tumorigenesis and the development of drug
resistance (126). SIRT1 is a
NAD+-dependent histone deacetylase that can deacetylate
histones and a number of nonhistone proteins. SIRT1 has been shown
to regulate various cellular processes, including senescence and
cell survival under genotoxic and oxidative stress (127,128). A
recent meta-analysis showed that, in CRC patients, SIRT1 expression
correlates with the development of invasion, lymph node metastasis,
and TNM stage, thus suggesting that SIRT1 may be regarded as a
negative prognostic marker of the overall survival rate of CRC
patients (128). SIRT1 has also been
shown to be one of the target genes of miR-34a, a small noncoding
RNAs that may control gene expression (126,129,130).
It has been found that miR-34a inhibits SIRT1 expression directly
through binding to the 3′-UTR of its mRNA in HCT116 CRC cells
(129). The introduction of miR-34a
into 5-FU-resistant DLD-1 cells significantly limited their
resistance to 5-FU, which was accompanied by the reduced expression
of SIRT1 and E2F family proteins (126,129).
These findings suggest that targeting the SIRT1 gene could decrease
resistance to 5-FU in human CRC by increasing p53
apoptosis-promoting activity (129).
SIRT1 has been suggested as a key protein in
maintaining stem-like features of CRC-CSCs, since SIRT1 was
coexpressed with the CD133 marker, and overexpressed in colorectal
CSC-like cells (131). Moreover,
SIRT1 deficiency decreased percentage of CD133+ cells
and their tumorigenicity and the abilities to form colonies and
spheres (131). Additionally, the
knockdown of SIRT1 gene in CRC SW620 cells reduced expression of
several stemness-associated genes (such as Oct4, Nanog, and Tert)
(131). These findings suggest that
SIRT1 can be considered as a novel prognostic marker or a new
target for anti-CRC therapy.
Other studies have focused on a different aspect of
cancer cell biology-the Warburg effect, the strong tendency of
cancer cells to switch their metabolism into anaerobic respiration
(glycolysis), to secrete lactate, and take up high levels of
glucose, even in the presence of oxygen in their niche; it
particularly affects CSCs (132).
This unusual phenomenon has been found to be associated with
carcinogenesis due to the inactivation in cancer cells of some
metabolic checkpoints, such as dysregulation of AMPK (energy
rheostat AMP-activated protein kinase) (10,133,134).
The Warburg effect is postulated to create an environment favorable
to CSC survival and the reprogramming of non-CSCs into CSCs
(135). These observations imply
that the elimination of CSCs alone may not be an effective
therapeutic approach, because they can be regenerated from
non-CSCs. Thus, an optimally effective cancer therapy should rely
on the administration of drugs targeting different types of cells
within the tumor mass.
Metformin improves anticancer therapy
effectiveness
Recently, some inhibitors of AMPK have been
considered as potential anticancer therapeutic agents (136–138).
Metformin (MET) is the best-established compound in this group of
anticancer molecules. MET is an extensively prescribed and
well-tolerated first-line therapeutic drug for type-2 diabetes
mellitus, which has demonstrated more effective anticancer effects
in cancers characterized by hyperinsulinemia, such as breast and
colon cancers (139,140). This evidence supports the
qualification of MET to preclinical and clinical trials of cancer
therapy (136–138).
Metformin has been described as agent capable of
directly and indirectly influencing cancer cells through the
reduction of glucose and insulin levels in the cancer niche, which
decreases cancer progression (139,141).
The very first observations of the effects of MET on cancer
development were demonstrated in diabetes complicated with CRC; in
such patients, CSCs showed lower proliferation and higher rates of
apoptosis than patients not pretreated with MET (141). In the same study, it was reported
that MET enhanced the antiproliferative effects of 5-FU on
CD133+ CSCs in SW620, SW480, and HCT116 CRC cell lines
(141–143). Recent analyses of the role of MET
treatment in the occurrence of CRC among type-2 diabetes mellitus
patients have shown that MET may reduce CRC incidence (144–146).
Moreover, MET has recently been identified as a
potential and attractive anticancer adjuvant drug, combined with
conventional chemotherapeutics to improve treatment efficacy and
decrease chemotherapeutic doses. The molecular mechanisms
underlying the anticancer effects of MET include insulin-dependent
and AMPK-dependent effects, selective targeting of CSCs, reversion
of multidrug resistance and inhibition of tumor metastasis
(147–149). Positive effects of such synergistic
combinatory therapy have been described for a broad spectrum of
cancers, including CRC, gastric, hepatic, pancreatic, breast, lung,
and prostate cancers (139,148).
The combination of MET and 2-deoxyglucose induces
p53-dependent apoptosis via the AMPK pathway and expression of a
functional p53 in p53-deficient prostate cancer cells. In addition,
such combined therapy arrests prostate cancer cells in the G2/M
phase and switched the cell death pattern from autophagic to
apoptotic, independently of p53 (136). In CRC SW480 cells, MET inhibited
cell growth mainly by blocking the cell cycle at the G0/G1 phase,
downregulating the expression of cyclin D1, and decreasing
telomerase activity (143).
Another study demonstrated that MET effectively
sensitizes human DLD-1, HT29, Colo205, and HCT116 cell lines to the
proapoptotic activity of tumor-necrosis-factor-related
apoptosis-inducing ligand (TRAIL) (134). At the same time, MET has been shown
to upregulate Bax and downregulate antiapoptotic myeloid cell
leukemia 1 (Mcl-1) levels in CRC cells, responsible for increased
TRAIL-mediated cell death in those human CRC cell lines (134).
MET has been shown to inhibit cancer transformation
and selectively kill CSCs in four genetically different types of
breast cancer (MCF-7, MCF10A ER-Src, SKBR3, and MDA-MB-486). The
administration of MET and doxorubicin collectively reduced the
number of both CD44highCD24low CSCs and
non-CSCs during in vitro culture. Furthermore, this
combinatorial therapy reduced tumor mass and prevented relapse
significantly more effectively than doxorubicin alone in a
xenograft mouse model (150).
Moreover, MET-treated breast cancer cell lines
showed downregulation of the CD44+CD24−/low
cell proportion via repression of EMT, including through decreasing
the level of ZEB, Twist, and Snail2 transcription factors (151). Surprisingly, this combination was
effective with a fourfold lower dose of doxorubicin than used in
treatment with the chemotherapeutic alone, which enables the
reduction of toxicity and an increase in the effectiveness of this
therapeutic approach.
However, the therapeutic anticancer activity of MET
seems to be controversial, as some groups have not shown its
antiproliferative and proapoptotic effect in CRC lines (141,143).
Sui et al suggested that MET cannot induce these therapeutic
effects as a single agent (152). A
possible explanation of these diverse results may be the dependency
of MET effectiveness on the experimental settings and cell lines
used, as Sui et al (152)
used HT29, HCT116, and RKO cells, while the other authors used
SW620, and SW480 CRC cell lines (141,143).
Chemoprevention: Nonsteroid
anti-inflammatory drugs in CRC therapy
After the discovery of increased prostaglandin
levels within cancer tissue, including CRC (153,154),
the regular use of nonsteroidal anti-inflammatory drugs (NSAIDs)
was hoped to provide new therapeutic anticancer effects that would
slow the progression of the disease. The issue of NSAID use in
cancer prevention has been supported by growing evidence from a
number of observational studies and post-trial follow-up data
(153). Of all cancers, aspirin and
indomethacin have been shown to be most effective at reducing the
risk of CRC, and even at lower doses demonstrate a 30–40%
effectiveness in preventing CRC (153). A case-control study conducted
between 1976 and 2011 and including 8634 CRC patients (and 8553
control patients) from the United States, Canada, Australia, and
Germany has demonstrated that regular use of aspirin or NSAIDs
reduces the risk of CRC (153,155).
In a genome-wide investigation of interactions between genes and
environment, the use of aspirin or NSAIDs was associated with a
lower risk of CRC, and this association differed depending on
genetic variation at two SNPs (single-nucleotide polymorphisms) on
chromosomes 12 and 15 (154).
The common mechanism through which NSAIDs and their
derivatives act is the inhibition of β-catenin/TCF transcriptional
activity and, consequently, downregulation of target genes such as
cyclin D1. Indomethacin is a cyclooxygenase 1 (COX-1) and COX-2
inhibitor and exhibits anti-inflammatory and analgesic properties.
In addition to the more general inhibition of the β-catenin/TCF
pathway mentioned above, indomethacin impairs β-catenin gene
expression, as shown by the significant reduction of the
corresponding mRNA in CRC cell lines (SW480, SW948, LoVo, and
HCT-116) (43,153,156,157).
Furthermore, indomethacin stimulates β-catenin degradation in a
manner independent of APC/GSK3β and proteasome (the Wnt
‘noncanonical’ pathway), even in cells bearing a mutated APC
or β-catenin gene CTNNB1.
Aspirin downregulates the Wnt/β-catenin pathway in
CRC cells, leading to reduced transcription of the target genes.
Unlike other NSAIDs, this effect seems to be mediated by
stabilization of β-catenin in its transcriptionally inactive form
(i.e., its phosphorylated form), hampering its activity as a
transcription factor (155). All
NSAIDs, in addition to their effects on β-catenin and related
pathways, act as ligands of PPARγ (Peroxisome
Proliferator-Activated Receptors) by stimulating PPARγ-dependent
effects, such as cell cycle block, differentiation, and apoptosis.
PPARγ costimulates the expression of cell cycle inhibitors, such as
p18, p21 and p27 (155).
Although aspirin and NSAIDs have an undisputable
preventive role in CRC development, their wider use in cancer
prevention needs to be carefully considered, on account of the
increased risk of bleeding from the gastrointestinal tract
(153,154,156).
Conclusions
In this review, we have summarized the state-of-art
in experimental CRC treatment targeting CSCs to prevent or reverse
their chemoresistance and reduce their metastatic potential. It is
hypothesized that creating combined therapy regimens, in which
conventional drugs are supplemented with novel CSC-targeting drugs,
might offer improved overall and cancer-free survival rates. A
potential dose reduction of conventional chemotherapeutics would
help limit their toxicity and improve patients' quality of
life.
Acknowledgements
The study was supported by the Polish National
Science Center (grant no. NN402684040).
Glossary
Abbreviations
Abbreviations:
|
5-FU
|
5-fluorouracil
|
|
AKT
|
protein kinase B
|
|
ALDH-1
|
aldehyde dehydrogenase 1
|
|
AMPK
|
energy rheostat AMP-activated protein
kinase
|
|
APC
|
adenomatous polyposis coli
|
|
BMP4
|
bone morphogenetic protein 4
|
|
CBP
|
cyclic AMP response element binding
protein
|
|
CK1
|
casein kinase 1
|
|
Chk2
|
check point kinase 2
|
|
COX-1/2
|
cyclooxygenase 1/2
|
|
CRC
|
colorectal cancer
|
|
CSC
|
cancer stem cell
|
|
Dkk1
|
Dickkopf-1
|
|
DLL4
|
delta-Like 4 ligand
|
|
EGF
|
epidermal growth factor
|
|
EGFR
|
epidermal growth factor receptor
|
|
EMT
|
epithelial-mesenchymal transition
|
|
GSK-3β
|
glycogen synthase kinase 3β
|
|
IGF-1/2
|
insulin-like growth factor 1/2
|
|
IGFR
|
insulin-like growth factor-1
receptor
|
|
Mcl-1
|
myeloid cell leukemia 1
|
|
MET
|
metformin
|
|
NICD
|
notch intracellular domain
|
|
NSAIDs
|
non-steroidal anti-inflammatory
drugs
|
|
PAK1
|
p21-activated kinase 1
|
|
PI3K
|
phosphatidylinositol-4,5-bisphosphate
3-kinase
|
|
PPARγ
|
peroxisome proliferator-activated
receptors
|
|
RTKs
|
receptors tyrosine kinases
|
|
RYK
|
receptor-like tyrosine kinase
|
|
shRNA
|
small hairpin RNA
|
|
siRNA
|
small interfering RNA
|
|
SIRT1
|
silent mating type information
regulation 2 homolog 1
|
|
SMAD3
|
SMAD family member 3
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
TCF/LEF
|
T-cell factor/lymphoid enhancer
factor
|
|
TERT
|
telomerase reverse transcriptase
|
|
TGFβ
|
transforming growth factor β
|
|
Tnk
|
tankyrase
|
|
TRAIL
|
tumor necrosis factor related
apoptosis-inducing ligand
|
|
VEGF
|
vascular endothelial growth
factor
|
References
|
1
|
Amado NG, Predes D, Moreno MM, Carvalho
IO, Mendes FA and Abreu JG: Flavonoids and Wnt/β-catenin signaling:
Potential role in colorectal cancer therapies. Int J Mol Sci.
15:12094–12106. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Manhas J, Bhattacharya A, Agrawal SK,
Gupta B, Das P, Deo SV, Pal S and Sen S: Characterization of cancer
stem cells from different grades of human colorectal cancer. Tumour
Biol. 37:14069–14081. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Franco S, Todaro M, Dieli F and Stassi
G: Colorectal cancer defeating? Mol Aspects Med. 39:61–81. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cunningham D, Humblet Y, Siena S, Khayat
D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype
C, et al: Cetuximab monotherapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van Cutsem E, Cervantes A, Nordlinger B
and Arnold D; ESMO Guidelines Working Group, : Metastatic
colorectal cancer: ESMO clinical practice guidelines for diagnosis,
treatment and follow-up. Ann Oncol. 25 Suppl 3:iii1–9. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taieb J, Tabernero J, Mini E, Subtil F,
Folprecht G, Van Laethem JL, Thaler J, Bridgewater J, Petersen LN,
Blons H, et al: Oxaliplatin, fluorouracil, and leucovorin with or
without cetuximab in patients with resected stage III colon cancer
(PETACC-8): An open-label, randomised phase 3 trial. Lancet Oncol.
15:862–873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Binefa G, Rodríguez-Moranta F, Teule A and
Medina-Hayas M: Colorectal cancer: From prevention to personalized
medicine. World J Gastroenterol. 20:6786–6808. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sauer R, Liersch T, Merkel S, Fietkau R,
Hohenberger W, Hess C, Becker H, Raab HR, Villanueva MT, Witzigmann
H, et al: Preoperative versus postoperative chemoradiotherapy for
locally advanced rectal cancer: Results of the German
CAO/ARO/AIO-94 randomized phase III trial after a median follow-up
of 11 years. J Clin Oncol. 30:1926–1933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khan K, Wale A, Brown G and Chau I:
Colorectal cancer with liver metastases: Neoadjuvant chemotherapy,
surgical resection first or palliation alone? World J
Gastroenterol. 20:12391–12406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paldino E, Tesori V, Casalbore P,
Gasbarrini A and Puglisi MA: Tumor initiating cells and
chemoresistance: Which is the best strategy to target colon cancer
stem cells? Biomed Res Int 2014. 8598712014.
|
|
11
|
O'Brien CA, Pollett A, Gallinger S and
Dick JE: A human colon cancer cell capable of initiating tumour
growth in immunodeficient mice. Nature. 445:106–110. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Puglisi MA, Barba M, Corbi M, Errico MF,
Giorda E, Saulnier N, Boninsegna A, Piscaglia AC, Carsetti R,
Cittadini A, et al: Identification of Endothelin-1 and NR4A2 as
CD133-regulated genes in colon cancer cells. J Pathol. 225:305–314.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shmelkov SV, Butler JM, Hooper AT, Hormigo
A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, et
al: CD133 expression is not restricted to stem cells, and both
CD133+ and CD133- metastatic colon cancer cells initiate tumors. J
Clin Invest. 118:2111–2120. 2008.PubMed/NCBI
|
|
15
|
Todaro M, Perez Alea M, Scopelliti A,
Medema JP and Stassi G: IL-4-mediated drug resistance in colon
cancer stem cells. Cell Cycle. 7:309–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang EH, Hynes MJ, Zhang T, Ginestier C,
Dontu G, Appelman H, Fields JZ, Wicha MS and Boman BM: Aldehyde
dehydrogenase 1 is a marker for normal and malignant human colonic
stem cells (SC) and tracks SC overpopulation during colon
tumorigenesis. Cancer Res. 69:3382–3389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin SP, Lee YT, Yang SH, Miller SA, Chiou
SH, Hung MC and Hung SC: Colon cancer stem cells resist
antiangiogenesis therapy-induced apoptosis. Cancer Lett.
328:226–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Z and Huang J: Intestinal stem
cells-types and markers. Cell Biol Int. 37:406–414. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P,
van Buren G II, Samuel S, Kim MP, Lim SJ and Ellis LM:
Chemoresistant colorectal cancer cells, the cancer stem cell
phenotype, and increased sensitivity to insulin-like growth
factor-I receptor inhibition. Cancer Res. 69:1951–1957. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang EH and Wicha MS: Colon cancer stem
cells: Implications for prevention and therapy. Trends Mol Med.
14:503–509. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu ZQ, Zhang C, Wang H, Lao XY, Chai R,
Gao XH, Cao GW and Fu CG: Downregulation of ATP-binding cassette
subfamily C member 4 increases sensitivity to neoadjuvant
radiotherapy for locally advanced rectal carcinoma. Dis Colon
Rectum. 56:600–608. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kozovska Z, Gabrisova V and Kucerova L:
Colon cancer: Cancer stem cells markers, drug resistance and
treatment. Biomed Pharmacother. 68:911–916. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vermeulen L, Todaro M, de Sousa Mello F,
Sprick MR, Kemper K, Perez Alea M, Richel DJ, Stassi G and Medema
JP: Single-cell cloning of colon cancer stem cells reveals a
multi-lineage differentiation capacity. Proc Natl Acad Sci USA.
105:pp. 13427–13432. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ong CW, Kim LG, Kong HH, Low LY, Iacopetta
B, Soong R and Salto-Tellez M: CD133 expression predicts for
non-response to chemotherapy in colorectal cancer. Mod Pathol.
23:450–457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Du Y, Shi L, Wang T, Liu Z and Wang Z:
Nanog siRNA plus cisplatin may enhance the sensitivity of
chemotherapy in esophageal cancer. J Cancer Res Clin Oncol.
138:1759–1767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Du L, Wang H, He L, Zhang J, Ni B, Wang X,
Jin H, Cahuzac N, Mehrpour M, Lu Y and Chen Q: CD44 is of
functional importance for colorectal cancer stem cells. Clin Cancer
Res. 14:6751–6760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Berretta M, Alessandrini L, De Divitiis C,
Nasti G, Lleshi A, Di Francia R, Facchini G, Cavaliere C, Buonerba
C and Canzonieri V: Serum and tissue markers in colorectal cancer:
State of art. Crit Rev Oncol Hematol. 111:103–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Honoki K, Fujii H, Kubo A, Kido A, Mori T,
Tanaka Y and Tsujiuchi T: Possible involvement of stem-like
populations with elevated ALDH1 in sarcomas for chemotherapeutic
drug resistance. Oncol Rep. 24:501–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim MP, Fleming JB, Wang H, Abbruzzese JL,
Choi W, Kopetz S, McConkey DJ, Evans DB and Gallick GE: ALDH
activity selectively defines an enhanced tumor-initiating cell
population relative to CD133 expression in human pancreatic
adenocarcinoma. PLoS One. 6:e206362011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koppaka V, Thompson DC, Chen Y, Ellermann
M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD and
Vasiliou V: Aldehyde dehydrogenase inhibitors: A comprehensive
review of the pharmacology, mechanism of action, substrate
specificity, and clinical application. Pharmacol Rev. 64:520–539.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su Y, Qiu Q, Zhang X, Jiang Z, Leng Q, Liu
Z, Stass SA and Jiang F: Aldehyde dehydrogenase 1 A1-positive cell
population is enriched in tumor-initiating cells and associated
with progression of bladder cancer. Cancer Epidemiol Biomarkers
Prev. 19:327–337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Croker AK and Allan AL: Inhibition of
aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and
radiation resistance of stem-like ALDHhiCD44+ human breast cancer
cells. Breast Cancer Res Treat. 133:75–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nowak D, Stewart D and Koeffler HP:
Differentiation therapy of leukemia: 3 decades of development.
Blood. 113:3655–3665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chung SS, Oliva B, Dwabe S and Vadgama JV:
Combination treatment with flavonoid morin and telomerase inhibitor
MST-312 reduces cancer stem cell traits by targeting STAT3 and
telomerase. Int J Oncol. 49:487–498. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martino-Echarri E, Henderson BR and
Brocardo MG: Targeting the DNA replication checkpoint by
pharmacologic inhibition of Chk1 kinase: A strategy to sensitize
APC mutant colon cancer cells to 5-fluorouracil chemotherapy.
Oncotarget. 5:9889–9900. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pulito C, Donzelli S, Muti P, Puzzo L,
Strano S and Blandino G: microRNAs and cancer metabolism
reprogramming: The paradigm of metformin. Ann Transl Med.
2:582014.PubMed/NCBI
|
|
38
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zubeldia IG, Bleau AM, Redrado M, Serrano
D, Agliano A, Gil-Puig C, Vidal-Vanaclocha F, Lecanda J and Calvo
A: Epithelial to mesenchymal transition and cancer stem cell
phenotypes leading to liver metastasis are abrogated by the novel
TGFβ1-targeting peptides P17 and P144. Exp Cell Res. 319:12–22.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brenner H, Kloor M and Pox CP: Colorectal
cancer. Lancet. 383:1490–1502. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
de Sousa E, Melo F and Vermeulen L: Wnt
signaling in cancer stem cell biology. Cancers (Basel). 8:pii:
E602016. View Article : Google Scholar
|
|
43
|
Anastas JN and Moon RT: WNT signalling
pathways as therapeutic targets in cancer. Nat Rev Cancer.
13:11–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wierzbicki PM and Rybarczyk A: The Hippo
pathway in colorectal cancer. Folia Histochem Cytobiol. 53:105–119.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tian H, Biehs B, Warming S, Leong KG,
Rangell L, Klein OD and de Sauvage FJ: A reserve stem cell
population in small intestine renders Lgr5-positive cells
dispensable. Nature. 478:255–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yan KS, Chia LA, Li X, Ootani A, Su J, Lee
JY, Su N, Luo Y, Heilshorn SC, Amieva MR, et al: The intestinal
stem cell markers Bmi1 and Lgr5 identify two functionally distinct
populations. Proc Natl Acad Sci USA. 109:pp. 466–471. 2012;
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kim JS, Crooks H, Foxworth A and Waldman
T: Proof-of-principle: Oncogenic beta-catenin is a valid molecular
target for the development of pharmacological inhibitors. Mol
Cancer Ther. 1:1355–1359. 2002.PubMed/NCBI
|
|
48
|
Huynh N, Shulkes A, Baldwin G and He H:
Up-regulation of stem cell markers by P21-activated kinase 1
contributes to 5-fluorouracil resistance of colorectal cancer.
Cancer Biol Ther. 17:813–823. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Thorne CA, Hanson AJ, Schneider J, Tahinci
E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson
AG, et al: Small-molecule inhibition of Wnt signaling through
activation of casein kinase 1α. Nat Chem Biol. 6:829–836. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan
CW, Wei S, Hao W, Kilgore J, Williams NS, et al: Small
molecule-mediated disruption of Wnt-dependent signaling in tissue
regeneration and cancer. Nat Chem Biol. 5:100–107. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huang SM, Mishina YM, Liu S, Cheung A,
Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner
S, et al: Tankyrase inhibition stabilizes axin and antagonizes Wnt
signalling. Nature. 461:614–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Riffell JL, Lord CJ and Ashworth A:
Tankyrase-targeted therapeutics: Expanding opportunities in the
PARP family. Nat Rev Drug Discov. 11:923–936. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Polakis P: Drugging Wnt signalling in
cancer. EMBO J. 31:2737–2746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Waaler J, Machon O, Tumova L, Dinh H,
Korinek V, Wilson SR, Paulsen JE, Pedersen NM, Eide TJ, Machonova
O, et al: A novel tankyrase inhibitor decreases canonical Wnt
signaling in colon carcinoma cells and reduces tumor growth in
conditional APC mutant mice. Cancer Res. 72:2822–2832. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lau T, Chan E, Callow M, Waaler J, Boggs
J, Blake RA, Magnuson S, Sambrone A, Schutten M, Firestein R, et
al: A novel tankyrase small-molecule inhibitor suppresses APC
mutation-driven colorectal tumor growth. Cancer Res. 73:3132–3144.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhong Y, Katavolos P, Nguyen T, Lau T,
Boggs J, Sambrone A, Kan D, Merchant M, Harstad E, Diaz D, et al:
Tankyrase inhibition causes reversible intestinal toxicity in mice
with a therapeutic index <1. Toxicol Pathol. 44:267–278. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lenz HJ and Kahn M: Safely targeting
cancer stem cells via selective catenin coactivator antagonism.
Cancer Sci. 105:1087–1092. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Duchartre Y, Kim YM and Kahn M: The Wnt
signaling pathway in cancer. Crit Rev Oncol Hematol. 99:141–149.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Fang L, Zhu Q, Neuenschwander M, Specker
E, Wulf-Goldenberg A, Weis WI, von Kries JP and Birchmeier W: A
small-molecule antagonist of the β-catenin/TCF4 interaction blocks
the self-renewal of cancer stem cells and suppresses tumorigenesis.
Cancer Res. 76:891–901. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Aguilera Ó, González-Sancho JM, Zazo S,
Rincón R, Fernández AF, Tapia O, Canals F, Morte B, Calvanese V,
Orgaz JL, et al: Nuclear DICKKOPF-1 as a biomarker of
chemoresistance and poor clinical outcome in colorectal cancer.
Oncotarget. 6:5903–5917. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
González-Sancho JM, Aguilera O, García JM,
Pendás-Franco N, Peña C, Cal S, García de Herreros A, Bonilla F and
Muñoz A: The Wnt antagonist DICKKOPF-1 gene is a downstream target
of beta-catenin/TCF and is downregulated in human colon cancer.
Oncogene. 24:1098–1103. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Qi L, Sun B, Liu Z, Li H, Gao J and Leng
X: Dickkopf-1 inhibits epithelial-mesenchymal transition of colon
cancer cells and contributes to colon cancer suppression. Cancer
Sci. 103:828–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu Z, Sun B, Qi L, Li Y, Zhao X, Zhang D
and Zhang Y: Dickkopf-1 expression is down-regulated during the
colorectal adenoma-carcinoma sequence and correlates with reduced
microvessel density and VEGF expression. Histopathology.
67:158–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rodilla V, Villanueva A, Obrador-Hevia A,
Robert-Moreno A, Fernández-Majada V, Grilli A, López-Bigas N,
Bellora N, Albà MM, Torres F, et al: Jagged1 is the pathological
link between Wnt and Notch pathways in colorectal cancer. Proc Natl
Acad Sci USA. 106:pp. 6315–6320. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Atashpour S, Fouladdel S, Movahhed TK,
Barzegar E, Ghahremani MH, Ostad SN and Azizi E: Quercetin induces
cell cycle arrest and apoptosis in CD133(+) cancer stem cells of
human colorectal HT29 cancer cell line and enhances anticancer
effects of doxorubicin. Iran J Basic Med Sci. 18:635–643.
2015.PubMed/NCBI
|
|
67
|
Temraz S, Mukherji D and Shamseddine A:
Potential targets for colorectal cancer prevention. Int J Mol Sci.
14:17279–17303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Johnson JL, Rupasinghe SG, Stefani F,
Schuler MA and Gonzalez de Mejia E: Citrus flavonoids luteolin,
apigenin, and quercetin inhibit glycogen synthase kinase-3β
enzymatic activity by lowering the interaction energy within the
binding cavity. J Med Food. 14:325–333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ning Y, Zhang W, Hanna DL, Yang D, Okazaki
S, Berger MD, Miyamoto Y, Suenaga M, Schirripa M, El-Khoueiry A and
Lenz HJ: Clinical relevance of EMT and stem-like gene expression in
circulating tumor cells of metastatic colorectal cancer patients.
Pharmacogenomics J. Aug 9–2016.(Epub ahead of print). View Article : Google Scholar
|
|
70
|
Rajamanickam S, Velmurugan B, Kaur M,
Singh RP and Agarwal R: Chemoprevention of intestinal tumorigenesis
in APCmin/+ mice by silibinin. Cancer Res. 70:2368–2378. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kaur M, Velmurugan B, Tyagi A, Agarwal C,
Singh RP and Agarwal R: Silibinin suppresses growth of human
colorectal carcinoma SW480 cells in culture and xenograft through
down-regulation of beta-catenin-dependent signaling. Neoplasia.
12:415–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hoh C, Boocock D, Marczylo T, Singh R,
Berry DP, Dennison AR, Hemingway D, Miller A, West K, Euden S, et
al: Pilot study of oral silibinin, a putative chemopreventive
agent, in colorectal cancer patients: Silibinin levels in plasma,
colorectum, and liver and their pharmacodynamic consequences. Clin
Cancer Res. 12:2944–2950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kumar S, Raina K, Agarwal C and Agarwal R:
Silibinin strongly inhibits the growth kinetics of colon cancer
stem cell-enriched spheroids by modulating interleukin 4/6-mediated
survival signals. Oncotarget. 5:4972–4989. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang ML, Chiou SH and Wu CW: Targeting
cancer stem cells: Emerging role of Nanog transcription factor.
Onco Targets Ther. 6:1207–1220. 2013.PubMed/NCBI
|
|
75
|
Ibrahim EE, Babaei-Jadidi R, Saadeddin A,
Spencer-Dene B, Hossaini S, Abuzinadah M, Li N, Fadhil W, Ilyas M,
Bonnet D and Nateri AS: Embryonic NANOG activity defines colorectal
cancer stem cells and modulates through AP1- and TCF-dependent
mechanisms. Stem Cells. 30:2076–2087. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang J, Espinoza LA, Kinders RJ, Lawrence
SM, Pfister TD, Zhou M, Veenstra TD, Thorgeirsson SS and Jessup JM:
NANOG modulates stemness in human colorectal cancer. Oncogene.
32:4397–4405. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lee TK, Castilho A, Cheung VC, Tang KH, Ma
S and Ng IO: CD24(+) liver tumor-initiating cells drive
self-renewal and tumor initiation through STAT3-mediated NANOG
regulation. Cell Stem Cell. 9:50–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Niu CS, Li DX, Liu YH, Fu XM, Tang SF and
Li J: Expression of NANOG in human gliomas and its relationship
with undifferentiated glioma cells. Oncol Rep. 26:593–601.
2011.PubMed/NCBI
|
|
79
|
Zhou X, Zhou YP, Huang GR, Gong BL, Yang
B, Zhang DX, Hu P and Xu SR: Expression of the stem cell marker,
Nanog, in human endometrial adenocarcinoma. Int J Gynecol Pathol.
30:262–270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bussolati B, Bruno S, Grange C, Ferrando U
and Camussi G: Identification of a tumor-initiating stem cell
population in human renal carcinomas. FASEB J. 22:3696–3705. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jeter CR, Liu B, Liu X, Chen X, Liu C,
Calhoun-Davis T, Repass J, Zaehres H, Shen JJ and Tang DG: NANOG
promotes cancer stem cell characteristics and prostate cancer
resistance to androgen deprivation. Oncogene. 30:3833–3845. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zbinden M, Duquet A, Lorente-Trigos A,
Ngwabyt SN, Borges I and Ruiz i Altaba A: NANOG regulates glioma
stem cells and is essential in vivo acting in a cross-functional
network with GLI1 and p53. EMBO J. 29:2659–2674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Meng HM, Zheng P, Wang XY, Liu C, Sui HM,
Wu SJ, Zhou J, Ding YQ and Li J: Over-expression of Nanog predicts
tumor progression and poor prognosis in colorectal cancer. Cancer
Biol Ther. 9:295–302. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Özgül Özdemir RB, Özdemir AT, Oltulu F,
Kurt K, Yiğittürk G and Kirmaz C: A comparison of cancer stem cell
markers and nonclassical major histocompatibility complex antigens
in colorectal tumor and noncancerous tissues. Ann Diagn Pathol.
25:60–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Han J, Zhang F, Yu M, Zhao P, Ji W, Zhang
H, Wu B, Wang Y and Niu R: RNA interference-mediated silencing of
NANOG reduces cell proliferation and induces G0/G1 cell cycle
arrest in breast cancer cells. Cancer Lett. 321:80–88. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Choi SC, Choi JH, Park CY, Ahn CM, Hong SJ
and Lim DS: Nanog regulates molecules involved in stemness and cell
cycle-signaling pathway for maintenance of pluripotency of P19
embryonal carcinoma stem cells. J Cell Physiol. 227:3678–3692.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Mattoo AR, Zhang J, Espinoza LA and Jessup
JM: Inhibition of NANOG/NANOGP8 downregulates MCL-1 in colorectal
cancer cells and enhances the therapeutic efficacy of BH3 mimetics.
Clin Cancer Res. 20:5446–5455. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong
CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS and Wu CW:
Coexpression of Oct4 and Nanog enhances malignancy in lung
adenocarcinoma by inducing cancer stem cell-like properties and
epithelial-mesenchymal transdifferentiation. Cancer Res.
70:10433–10444. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pan Q, Meng L, Ye J, Wei X, Shang Y, Tian
Y, He Y, Peng Z, Chen L, Chen W, et al: Transcriptional repression
of miR-200 family members by Nanog in colon cancer cells induces
epithelial-mesenchymal transition (EMT). Cancer Lett. 392:26–38.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Abetov D, Mustapova Z, Saliev T and
Bulanin D: Biomarkers and signaling pathways of colorectal cancer
stem cells. Tumour Biol. 36:1339–1353. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yokobori T and Nishiyama M: TGF-β
signaling in gastrointestinal cancers: Progress in basic and
clinical research. J Clin Med. 6:pii: E112017. View Article : Google Scholar
|
|
92
|
Gil-Guerrero L, Dotor J, Huibregtse IL,
Casares N, López-Vázquez AB, Rudilla F, Riezu-Boj JI,
López-Sagaseta J, Hermida J, Van Deventer S, et al: In vitro and in
vivo down-regulation of regulatory T cell activity with a peptide
inhibitor of TGF-beta1. J Immunol. 181:126–135. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Llopiz D, Dotor J, Casares N, Bezunartea
J, Díaz-Valdés N, Ruiz M, Aranda F, Berraondo P, Prieto J, Lasarte
JJ, et al: Peptide inhibitors of transforming growth factor-beta
enhance the efficacy of antitumor immunotherapy. Int J Cancer.
125:2614–2623. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Gonzalez-Zubeldia I, Dotor J, Redrado M,
Bleau AM, Manrique I, de Aberasturi AL, Villalba M and Calvo A:
Co-migration of colon cancer cells and CAFs induced by TGFβ1
enhances liver metastasis. Cell Tissue Res. 359:829–839. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhan J, Yang M, Zhang J, Guo Y, Liu W and
Zhang H: Kindler syndrome protein Kindlin-1 is mainly expressed in
adult tissues originating from ectoderm/endoderm. Sci China Life
Sci. 58:432–441. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kong J, Du J, Wang Y, Yang M, Gao J, Wei
X, Fang W, Zhan J and Zhang H: Focal adhesion molecule Kindlin-1
mediates activation of TGF-β signaling by interacting with TGF-βRI,
SARA and Smad3 in colorectal cancer cells. Oncotarget.
7:76224–76237. 2016.PubMed/NCBI
|
|
97
|
Karagiannis GS, Afaloniati H, Karamanavi
E, Poutahidis T and Angelopoulou K: BMP pathway suppression is an
early event in inflammation-driven colon neoplasmatogenesis of
uPA-deficient mice. Tumour Biol. 37:2243–2255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Kim JS, Crooks H, Dracheva T, Nishanian
TG, Singh B, Jen J and Waldman T: Oncogenic beta-catenin is
required for bone morphogenetic protein 4 expression in human
cancer cells. Cancer Res. 62:2744–2748. 2002.PubMed/NCBI
|
|
99
|
Lubbe SJ, Pittman AM, Matijssen C, Twiss
P, Olver B, Lloyd A, Qureshi M, Brown N, Nye E, Stamp G, et al:
Evaluation of germline BMP4 mutation as a cause of colorectal
cancer. Hum Mutat. 32:E1928–E1938. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Kosinski C, Li VS, Chan AS, Zhang J, Ho C,
Tsui WY, Chan TL, Mifflin RC, Powell DW, Yuen ST, et al: Gene
expression patterns of human colon tops and basal crypts and BMP
antagonists as intestinal stem cell niche factors. Proc Natl Acad
Sci USA. 104:pp. 15418–15423. 2007; View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Jin X, Chen Z, Xiang L, Luo Q, Guo Z, Ding
X and Jin X: Colorectal polyp model established by transplacental
BMP4 RNAi. Mol Med Rep. 10:33–38. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Dou J and Gu N: Emerging strategies for
the identification and targeting of cancer stem cells. Tumour Biol.
31:243–253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lombardo Y, Scopelliti A, Cammareri P,
Todaro M, Iovino F, Ricci-Vitiani L, Gulotta G, Dieli F, de Maria R
and Stassi G: Bone morphogenetic protein 4 induces differentiation
of colorectal cancer stem cells and increases their response to
chemotherapy in mice. Gastroenterology. 140:297–309. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhou L, Xie J, Gu EL, Huang Y, Qu Y, Xu
AP, Zhu Y and Wang H: Common genetic variant on BMP4 contributes to
colorectal adenoma and cancer: A meta-analysis based on 15 studies.
Cytokine. 72:154–159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Shi Y, Chen GB, Huang XX, Xiao CX, Wang
HH, Li YS, Zhang JF, Li S, Xia Y, Ren JL and Guleng B: Dragon
(repulsive guidance molecule b, RGMb) is a novel gene that promotes
colorectal cancer growth. Oncotarget. 6:20540–20554. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Halbrooks PJ, Ding R, Wozney JM and Bain
G: Role of RGM coreceptors in bone morphogenetic protein signaling.
J Mol Signal. 2:42007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Catalano V, Dentice M, Ambrosio R, Luongo
C, Carollo R, Benfante A, Todaro M, Stassi G and Salvatore D:
Activated thyroid hormone promotes differentiation and
chemotherapeutic sensitization of colorectal cancer stem cells by
regulating Wnt and BMP4 signaling. Cancer Res. 76:1237–1244. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Miyamoto S and Rosenberg DW: Role of Notch
signaling in colon homeostasis and carcinogenesis. Cancer Sci.
102:1938–1942. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Sikandar SS, Pate KT, Anderson S, Dizon D,
Edwards RA, Waterman ML and Lipkin SM: NOTCH signaling is required
for formation and self-renewal of tumor-initiating cells and for
repression of secretory cell differentiation in colon cancer.
Cancer Res. 70:1469–1478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Reedijk M, Odorcic S, Zhang H, Chetty R,
Tennert C, Dickson BC, Lockwood G, Gallinger S and Egan SE:
Activation of Notch signaling in human colon adenocarcinoma. Int J
Oncol. 33:1223–1229. 2008.PubMed/NCBI
|
|
111
|
Fre S, Pallavi SK, Huyghe M, Laé M,
Janssen KP, Robine S, Artavanis-Tsakonas S and Louvard D: Notch and
Wnt signals cooperatively control cell proliferation and
tumorigenesis in the intestine. Proc Natl Acad Sci USA. 106:pp.
6309–6314. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hoey T, Yen WC, Axelrod F, Basi J,
Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, et
al: DLL4 blockade inhibits tumor growth and reduces
tumor-initiating cell frequency. Cell Stem Cell. 5:168–177. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu Z, Fan F, Wang A, Zheng S and Lu Y:
Dll4-Notch signaling in regulation of tumor angiogenesis. J Cancer
Res Clin Oncol. 140:525–536. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Yan M, Callahan CA, Beyer JC, Allamneni
KP, Zhang G, Ridgway JB, Niessen K and Plowman GD: Chronic DLL4
blockade induces vascular neoplasms. Nature. 463:E6–E7. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fischer M, Yen WC, Kapoun AM, Wang M,
O'Young G, Lewicki J, Gurney A and Hoey T: Anti-DLL4 inhibits
growth and reduces tumor-initiating cell frequency in colorectal
tumors with oncogenic KRAS mutations. Cancer Res. 71:1520–1525.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
He P, Liang J, Shao T, Guo Y, Hou Y and Li
Y: HDAC5 promotes colorectal cancer cell proliferation by
up-regulating DLL4 expression. Int J Clin Exp Med. 8:6510–6516.
2015.PubMed/NCBI
|
|
117
|
van Es JH, Sato T, van de Wetering M,
Lyubimova A, Gregorieff A, Zeinstra L, van den Born M, Korving J,
Martens ACM, van den Oudenaarden A and Clevers H: Dll1+ secretory
progenitor cells revert to stem cells upon crypt damage. Nat Cell
Biol. 14:1099–1104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Fre S, Huyghe M, Mourikis P, Robine S,
Louvard D and Artavanis-Tsakonas S: Notch signals control the fate
of immature progenitor cells in the intestine. Nature. 435:964–968.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Grochowski CM, Loomes KM and Spinner NB:
Jagged1 (JAG1): Structure, expression, and disease associations.
Gene. 576:381–384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Bray SJ: Notch signalling: A simple
pathway becomes complex. Nat Rev Mol Cell Biol. 7:678–689. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
121
|
van Es JH, van Gijn ME, Riccio O, van den
Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ,
Radtke F and Clevers H: Notch/gamma-secretase inhibition turns
proliferative cells in intestinal crypts and adenomas into goblet
cells. Nature. 435:959–963. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Guilmeau S, Flandez M, Mariadason JM and
Augenlicht LH: Heterogeneity of Jagged1 expression in human and
mouse intestinal tumors: Implications for targeting Notch
signaling. Oncogene. 29:992–1002. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Lu J, Ye X, Fan F, Xia L, Bhattacharya R,
Bellister S, Tozzi F, Sceusi E, Zhou Y, Tachibana I, et al:
Endothelial cells promote the colorectal cancer stem cell phenotype
through a soluble form of Jagged-1. Cancer Cell. 23:171–185. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Huang R, Wang G, Song Y, Tang Q, You Q,
Liu Z, Chen Y, Zhang Q, Li J, Muhammand S and Wang X: Colorectal
cancer stem cell and chemoresistant colorectal cancer cell
phenotypes and increased sensitivity to Notch pathway inhibitor.
Mol Med Rep. 12:2417–2424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Meng RD, Shelton CC, Li YM, Qin LX,
Notterman D, Paty PB and Schwartz GK: Gamma-secretase inhibitors
abrogate oxaliplatin-induced activation of the Notch-1 signaling
pathway in colon cancer cells resulting in enhanced
chemosensitivity. Cancer Res. 69:573–582. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Akao Y, Noguchi S, Iio A, Kojima K, Takagi
T and Naoe T: Dysregulation of microRNA-34a expression causes
drug-resistance to 5-FU in human colon cancer DLD-1 cells. Cancer
Lett. 300:197–204. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Horio Y, Hayashi T, Kuno A and Kunimoto R:
Cellular and molecular effects of sirtuins in health and disease.
Clin Sci (Lond). 121:191–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zu G, Ji A, Zhou T and Che N:
Clinicopathological significance of SIRT1 expression in colorectal
cancer: A systematic review and meta analysis. Int J Surg.
26:32–37. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Yamakuchi M, Ferlito M and Lowenstein CJ:
miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci
USA. 105:pp. 13421–13426. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Stiegelbauer V, Perakis S, Deutsch A, Ling
H, Gerger A and Pichler M: MicroRNAs as novel predictive biomarkers
and therapeutic targets in colorectal cancer. World J
Gastroenterol. 20:11727–11735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Chen X, Sun K, Jiao S, Cai N, Zhao X, Zou
H, Xie Y, Wang Z, Zhong M and Wei L: High levels of SIRT1
expression enhance tumorigenesis and associate with a poor
prognosis of colorectal carcinoma patients. Sci Rep. 4:74812014.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Gentric G, Mieulet V and Mechta-Grigoriou
F: Heterogeneity in cancer metabolism: New concepts in an old
field. Antioxid Redox Signal. 26:462–485. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Stubbs M and Griffiths JR: The altered
metabolism of tumors: HIF-1 and its role in the Warburg effect. Adv
Enzyme Regul. 50:44–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Park SH, Lee DH, Kim JL, Kim BR, Na YJ, Jo
MJ, Jeong YA, Lee SY, Lee SI, Lee YY and Oh SC: Metformin enhances
TRAIL-induced apoptosis by Mcl-1 degradation via Mule in colorectal
cancer cells. Oncotarget. 7:59503–59518. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Menendez JA, Joven J, Cufi S,
Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B,
López-Bonet E, Alarcón T and Vazquez-Martin A: The Warburg effect
version 2.0: Metabolic reprogramming of cancer stem cells. Cell
Cycle. 12:1166–1179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Ben Sahra I, Laurent K, Giuliano S,
Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y,
Giorgetti-Peraldi S, Cormont M, Bertolotto C, et al: Targeting
cancer cell metabolism: The combination of metformin and
2-deoxyglucose induces p53-dependent apoptosis in prostate cancer
cells. Cancer Res. 70:2465–2475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Ben Sahra I, Le Marchand-Brustel Y, Tanti
JF and Bost F: Metformin in cancer therapy: A new perspective for
an old antidiabetic drug? Mol Cancer Ther. 9:1092–1099. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Miranda VC, Braghiroli MI, Faria LD,
Bariani G, Alex A, Bezerra Neto JE, Capareli FC, Sabbaga J, Lobo
Dos Santos JF, Hoff PM and Riechelmann RP: Phase 2 trial of
metformin combined with 5-fluorouracil in patients with refractory
metastatic colorectal cancer. Clin Colorectal Cancer.
15:321–328.e1. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zhang HH and Guo XL: Combinational
strategies of metformin and chemotherapy in cancers. Cancer
Chemother Pharmacol. 78:13–26. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Hosono K, Endo H, Takahashi H, Sugiyama M,
Sakai E, Uchiyama T, Suzuki K, Iida H, Sakamoto Y, Yoneda K, et al:
Metformin suppresses colorectal aberrant crypt foci in a short-term
clinical trial. Cancer Prev Res (Phila). 3:1077–1083. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zhang Y, Guan M, Zheng Z, Zhang Q, Gao F
and Xue Y: Effects of metformin on CD133+ colorectal cancer cells
in diabetic patients. PLoS One. 8:e812642013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Montales MT, Simmen RC, Ferreira ES, Neves
VA and Simmen FA: Metformin and soybean-derived bioactive molecules
attenuate the expansion of stem cell-like epithelial subpopulation
and confer apoptotic sensitivity in human colon cancer cells. Genes
Nutr. 10:492015. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Zhou XZ, Xue YM, Zhu B and Sha JP: Effects
of metformin on proliferation of human colon carcinoma cell line
SW-480. Nan Fang Yi Ke Da Xue Xue Bao. 30:1935–1938. 1942.2010 (In
Chinese).
|
|
144
|
Nie Z, Zhu H and Gu M: Reduced colorectal
cancer incidence in type 2 diabetic patients treated with
metformin: A meta-analysis. Pharm Biol. 54:2636–2642. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Sehdev A, Shih YC, Vekhter B, Bissonnette
MB, Olopade OI and Polite BN: Metformin for primary colorectal
cancer prevention in patients with diabetes: A case-control study
in a US population. Cancer. 121:1071–1078. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Cho YH, Ko BM, Kim SH, Myung YS, Choi JH,
Han JP, Hong SJ, Jeon SR, Kim HG, Kim JO and Lee MS: Does metformin
affect the incidence of colonic polyps and adenomas in patients
with type 2 diabetes mellitus? Intest Res. 12:139–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lonardo E, Cioffi M, Sancho P, Crusz S and
Heeschen C: Studying pancreatic cancer stem cell characteristics
for developing new treatment strategies. J Vis Exp.
e528012015.PubMed/NCBI
|
|
148
|
Iliopoulos D, Hirsch HA and Struhl K:
Metformin decreases the dose of chemotherapy for prolonging tumor
remission in mouse xenografts involving multiple cancer cell types.
Cancer Res. 71:3196–3201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Hirsch HA, Iliopoulos D and Struhl K:
Metformin inhibits the inflammatory response associated with
cellular transformation and cancer stem cell growth. Proc Natl Acad
Sci USA. 110:pp. 972–977. 2013; View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells, and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Vazquez-Martin A, Oliveras-Ferraros C,
Cufi S, Del Barco S, Martin-Castillo B and Menendez JA: Metformin
regulates breast cancer stem cell ontogeny by transcriptional
regulation of the epithelial-mesenchymal transition (EMT) status.
Cell Cycle. 9:3807–3814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Sui X, Xu Y, Yang J, Fang Y, Lou H, Han W,
Zhang M, Chen W, Wang K, Li D, et al: Use of metformin alone is not
associated with survival outcomes of colorectal cancer cell but
AMPK activator AICAR sensitizes anticancer effect of 5-fluorouracil
through AMPK activation. PLoS One. 9:e977812014. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Burn J and Sheth H: The role of aspirin in
preventing colorectal cancer. Br Med Bull. 119:17–24. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Nan H, Hutter CM, Lin Y, Jacobs EJ, Ulrich
CM, White E, Baron JA, Berndt SI, Brenner H, Butterbach K, et al:
Association of aspirin and NSAID use with risk of colorectal cancer
according to genetic variants. JAMA. 313:1133–1142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Sabatino L, Pancione M, Votino C,
Colangelo T, Lupo A, Novellino E, Lavecchia A and Colantuoni V:
Emerging role of the β-catenin-PPARγ axis in the pathogenesis of
colorectal cancer. World J Gastroenterol. 20:7137–7151. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Dihlmann S, Siermann A and von Knebel
Doeberitz M: The nonsteroidal anti-inflammatory drugs aspirin and
indomethacin attenuate beta-catenin/TCF-4 signaling. Oncogene.
20:645–653. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Hawcroft G, D'Amico M, Albanese C, Markham
AF, Pestell RG and Hull MA: Indomethacin induces differential
expression of beta-catenin, gamma-catenin and T-cell factor target
genes in human colorectal cancer cells. Carcinogenesis. 23:107–114.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Alberts SR, Sargent DJ, Nair S, Mahoney
MR, Mooney M, Thibodeau SN, Smyrk TC, Sinicrope FA, Chan E, Gill S,
et al: Effect of oxaliplatin, fluorouracil, and leucovorin with or
without cetuximab on survival among patients with resected stage
III colon cancer: A randomized trial. JAMA. 307:1383–1393. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Cao R, Zhang S, Ma D and Hu L: A
multi-center randomized phase II clinical study of bevacizumab plus
irinotecan, 5-fluorouracil, and leucovorin (FOLFIRI) compared with
FOLFIRI alone as second-line treatment for Chinese patients with
metastatic colorectal cancer. Med Oncol. 32:3252015. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Longley DB, Harkin DP and Johnston PG:
5-fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Yamada Y, Takahari D, Matsumoto H, Baba H,
Nakamura M, Yoshida K, Yoshida M, Iwamoto S, Shimada K, Komatsu Y,
et al: Leucovorin, fluorouracil, and oxaliplatin plus bevacizumab
versus S-1 and oxaliplatin plus bevacizumab in patients with
metastatic colorectal cancer (SOFT): An open-label,
non-inferiority, randomised phase 3 trial. Lancet Oncol.
14:1278–1286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Schmoll HJ, Tabernero J, Maroun J, de
Braud F, Price T, Van Cutsem E, Hill M, Hoersch S, Rittweger K and
Haller DG: Capecitabine plus oxaliplatin compared with
fluorouracil/folinic acid as adjuvant therapy for stage iii colon
cancer: Final results of the NO16968 randomized controlled phase
III trial. J Clin Oncol. 33:3733–3740. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Élez E, Kocáková I, Höhler T, Martens UM,
Bokemeyer C, Van Cutsem E, Melichar B, Smakal M, Csőszi T, Topuzov
E, et al: Abituzumab combined with cetuximab plus irinotecan versus
cetuximab plus irinotecan alone for patients with KRAS wild-type
metastatic colorectal cancer: The randomised phase I/II POSEIDON
trial. Ann Oncol. 26:132–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Sclafani F, Kim TY, Cunningham D, Kim TW,
Tabernero J, Schmoll HJ, Roh JK, Kim SY, Park YS, Guren TK, et al:
A randomized phase II/III study of dalotuzumab in combination with
cetuximab and irinotecan in chemorefractory, KRAS wild-type,
metastatic colorectal cancer. J Natl Cancer Inst. 107:djv2582015.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Fujita K, Kubota Y, Ishida H and Sasaki Y:
Irinotecan, a key chemotherapeutic drug for metastatic colorectal
cancer. World J Gastroenterol. 21:12234–12248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
de Gramont A, Van Cutsem E, Schmoll HJ,
Tabernero J, Clarke S, Moore MJ, Cunningham D, Cartwright TH, Hecht
JR, Rivera F, et al: Bevacizumab plus oxaliplatin-based
chemotherapy as adjuvant treatment for colon cancer (AVANT): A
phase 3 randomised controlled trial. Lancet Oncol. 13:1225–1233.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Bayoglu IV, Yildiz I, Varol U, Cokmert S,
Alacacıoğlu A, Kucukzeybek Y, Akyol M, Demir L, Dirican A and
Tarhan O: Uracil/tegafur as a possible salvage therapy in
chemo-refractory colorectal cancer patients: A single institutional
retrospective study. Contemp Oncol (Pozn). 19:385–390.
2015.PubMed/NCBI
|
|
168
|
Feng QY, Wei Y, Chen JW, Chang WJ, Ye LC,
Zhu DX and Xu JM: Anti-EGFR and anti-VEGF agents: Important
targeted therapies of colorectal liver metastases. World J
Gastroenterol. 20:4263–4275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Strickler JH and Hurwitz HI:
Bevacizumab-based therapies in the first-line treatment of
metastatic colorectal cancer. Oncologist. 17:513–524. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Roviello G, Bachelot T, Hudis CA,
Curigliano G, Reynolds AR, Petrioli R and Generali D: The role of
bevacizumab in solid tumours: A literature based meta-analysis of
randomised trials. Eur J Cancer. 75:245–258. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Huang J, Nair SG, Mahoney MR, Nelson GD,
Shields AF, Chan E, Goldberg RM, Gill S, Kahlenberg MS, Quesenberry
JT, et al: Comparison of FOLFIRI with or without cetuximab in
patients with resected stage III colon cancer; NCCTG (Alliance)
intergroup trial N0147. Clin Colorectal Cancer. 13:100–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Terazawa T, Nishitani H, Kato K, Hashimoto
H, Akiyoshi K, Ito Y, Nakamoto A, Iwasa S, Nakajima TE, Hamaguchi
T, et al: Phase II study of cetuximab with irinotecan for KRAS
wild-type colorectal cancer in Japanese patients. Asia Pac J Clin
Oncol. 13:e132–e137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Tay RY, Wong R and Hawkes EA: Treatment of
metastatic colorectal cancer: Focus on panitumumab. Cancer Manag
Res. 7:189–198. 2015.PubMed/NCBI
|
|
174
|
Bahrami A, Hassanian SM, ShahidSales S,
Farjami Z, Hasanzadeh M, Anvari K, Aledavood A, Maftouh M, Ferns
GA, Khazaei M and Avan A: Targeting RAS signaling pathway as a
potential therapeutic target in the treatment of colorectal cancer.
J Cell Physiol. Mar 6–2017.(Epub ahead of print). View Article : Google Scholar
|
|
175
|
Françoso A and Simioni PU: Immunotherapy
for the treatment of colorectal tumors: Focus on approved and
in-clinical-trial monoclonal antibodies. Drug Des Devel Ther.
11:177–184. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Ursem C, Van Loon K and Venook A: Adjuvant
therapy trials. Cancer J. 22:196–198. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Botchkina G: Colon cancer stem cells-from
basic to clinical application. Cancer Lett. 338:127–140. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Haraguchi N, Ohkuma M, Sakashita H,
Matsuzaki S, Tanaka F, Mimori K, Kamohara Y, Inoue H and Mori M:
CD133+CD44+ population efficiently enriches colon cancer initiating
cells. Ann Surg Oncol. 15:2927–2933. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Zhu L, Gibson P, Currle DS, Tong Y,
Richardson RJ, Bayazitov IT, Poppleton H, Zakharenko S, Ellison DW
and Gilbertson RJ: Prominin 1 marks intestinal stem cells that are
susceptible to neoplastic transformation. Nature. 457:603–607.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Botchkina IL, Rowehl RA, Rivadeneira DE,
Karpeh MS Jr, Crawford H, Dufour A, Ju J, Wang Y, Leyfman Y and
Botchkina GI: Phenotypic subpopulations of metastatic colon cancer
stem cells: Genomic analysis. Cancer Genomics Proteomics. 6:19–29.
2009.PubMed/NCBI
|
|
181
|
Zhou F, Mu YD, Liang J, Liu ZX, Zhou D,
Ning WL, Li YZ, Ding D and Zhang JF: Aldehyde dehydrogenase 1: A
specific cancer stem cell marker for human colorectal carcinoma.
Mol Med Rep. 11:3894–3899. 2015. View Article : Google Scholar : PubMed/NCBI
|