| hsa04510 | Focal adhesion | 57 |
1.02×10−20 | FLT4, CAV2, MET,
COMP, LAMA3, RELN, ITGA6, TLN2, ERBB2, MYLK, IGF1, LAMA2, IBSP,
ROCK2, PDGFD, CCND2, PIK3R2, COL6A6, PDGFRA, KDR, FLNC, PDGFA,
AKT3, PAK4, MAPK10, PRKCA, TNXB, LAMC1, VWF, COL5A2, CAV1, ARHGAP5,
LAMB3, FIGF, LAMA4, EGFR, COL3A1, COL1A2, COL1A1, PIK3R1, LAMA1,
JUN, ITGA9, THBS2, LAMC3, PPP1CA, LAMC2, PAK3, COL5A1, PAK7, ITGB8,
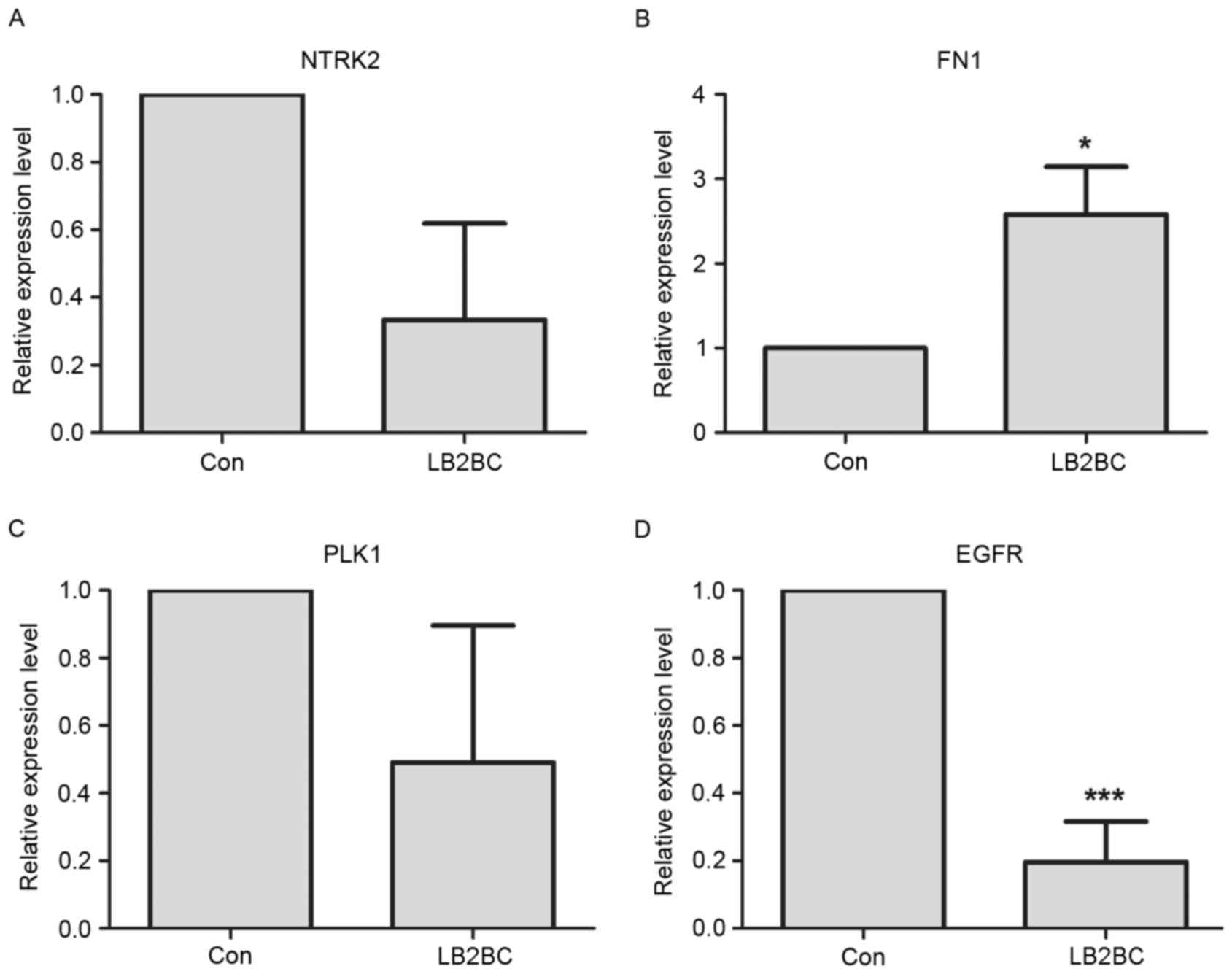
MYL9, COL11A1, ITGA11, FN1, VAV3, ITGA7 |
| hsa05200 | Pathways in
cancer | 74 |
1.50×10−20 | CDK6, CCNE2, PTCH1,
FGF9, TCF7L1, MET, LEF1, STAT1, LAMA3, FOS, RARB, FGF1, WNT7B,
FGF2, ITGA6, KIT, BIRC5, TGFA, FOXO1, EPAS1, ERBB2, IGF1, RUNX1T1,
FZD7, LAMA2, BMP2, PLCG2, CCNE1, TGFBR2, FGFR3, PIK3R2, E2F1,
RAD51, TPM3, E2F2, WNT2, PDGFRA, PDGFA, AKT3, TCF7, FZD5, MAPK10,
TCEB1, PRKCA, FADD, LAMC1, LAMB3, FGFR1, PTCH2, FIGF, MECOM, LAMA4,
EGFR, PIK3R1, FZD4, LAMA1, MYC, JUN, LAMC3, LAMC2, TCF7L2, FGF7,
ZBTB16, RET, CEBPA, PLD1, PPARG, DAPK2, STAT5B, TCEB2, STAT5A, FN1,
PTGS2, MMP1 |
| hsa04512 | ECM-receptor
interaction | 31 |
1.15×10−14 | SDC1, COMP, LAMA3,
RELN, ITGA6, LAMA2, IBSP, HMMR, COL6A6, SV2B, TNXB, LAMC1, VWF,
COL5A2, LAMB3, LAMA4, COL3A1, COL1A2, COL1A1, LAMA1, ITGA9, THBS2,
LAMC3, LAMC2, COL5A1, CD36, ITGB8, COL11A1, ITGA11, FN1, ITGA7 |
| hsa04060 | Cytokine-cytokine
receptor interaction | 51 |
1.50×10–11 | INHBB, FLT4, MET,
TNFRSF10D, KIT, CXCL9, LEPR, BMP2, CXCL2, CNTFR, TGFBR2, CXCL10,
IL28RA, CXCR7, TNFRSF21, CXCL3, CX3CR1, CCR8, LIFR, EDA, IL22RA2,
CXCR2, PDGFRA, KDR, IL6R, PDGFA, IL21R, EDAR, EDA2R, CSF1, CX3CL1,
FIGF, EGFR, CCL13, RELT, IFNGR1, LEP, CSF3, INHBA, NGFR, GHR,
CXCL11, IL11RA, CXCL12, CCL21, TNFRSF18, CCL14, TNFSF4, TPO, CCL28,
TSLP |
| hsa04360 | Axon guidance | 32 |
4.70×10−10 | SLIT3, GNAI1, MET,
ROBO3, EFNA4, SEMA5A, DPYSL2, EPHB6, SEMA7A, SEMA6D, ROCK2, CFL2,
EFNB3, SEMA3G, ABLIM3, EPHB1, EFNB1, SLIT2, PAK4, ABLIM1, UNC5B,
SEMA3D, NFATC2, SEMA3F, NFAT5, PAK3, CFL1, PAK7, SEMA3A, EPHA2,
CXCL12, RND1 |
| hsa04110 | Cell cycle | 30 |
3.32×10−9 | CDK6, CCNE2, ESPL1,
MCM4, CCNB2, CDC20, CCNA2, PTTG1, CCNE1, CCND2, E2F1, CDC25C, E2F2,
CCNB1, MAD2L1, CDKN2C, MYC, PLK1, BUB1B, ANAPC11, CDC45, CDC25A,
PKMYT1, TTK, BUB1, CDC14B, CDKN1C, PCNA, MCM2, CDK1 |
| hsa04810 | Regulation of actin
cytoskeleton | 39 |
2.69×10−8 | MRAS, DIAPH3,
ARHGEF6, FGF9, FGF1, FGF2, ITGA6, ARPC1B, MYLK, EZR, ROCK2, IQGAP3,
PDGFD, CFL2, FGFR3, PIK3R2, PDGFRA, PDGFA, PAK4, GSN, DIAPH2,
FGFR1, EGFR, PIK3R1, ITGA9, RRAS2, PPP1CA, WASF2, FGF7, PAK3, CFL1,
ARHGEF4, PAK7, ITGB8, MYL9, ITGA11, FN1, VAV3, ITGA7 |
| hsa04080 | Neuroactive ligand-
receptor interaction | 45 |
4.29×10−8 | GRIA3, DRD2,
AVPR1A, ADRB2, S1PR1, GABRE, HTR2A, CNR1, EDNRB, LEPR, HTR1D,
GLP2R, PTGFR, NMUR1, SSTR1, VIPR1, ADRA2A, GABRD, ADCYAP1R1, CTSG,
LHCGR, OXTR, GRIN2D, PTGER4, GABRP, NR3C1, PTGER3, FPR3, P2RX6,
LEP, GRIN2A, GRIA4, GHR, NPY2R, AVPR2, THRB, CRHR1, GRM4, GRIK1,
PTH1R, P2RY13, TACR1, ADRA1A, LPAR6, P2RY14 |
| hsa05215 | Prostate
cancer | 23 |
7.92×10−8 | CCNE2, TCF7L1,
LEF1, TGFA, FOXO1, ERBB2, CREB3L1, IGF1, CCNE1, PDGFD, PIK3R2,
E2F1, E2F2, PDGFRA, PDGFA, CREB3L4, AKT3, TCF7, CREB5, FGFR1, EGFR,
PIK3R1, TCF7L2 |
| hsa04914 |
Progesterone-mediated oocyte
maturation | 22 |
2.40×10−7 | GNAI1, CCNB2,
CCNA2, IGF1, CPEB1, PIK3R2, CDC25C, CCNB1, MAD2L1, AKT3, MAPK10,
ADCY4, PDE3B, RPS6KA2, PIK3R1, PLK1, ANAPC11, CDC25A, PKMYT1, BUB1,
RPS6KA3, CDK1 |
| hsa03320 | PPAR signaling
pathway | 19 | 6.97×10
−7 | ACSL1, AQP7, ACADL,
SLC27A4, FABP4, PPARA, PLTP, ADIPOQ, SLC27A6, ACSL5, ANGPTL4,
PLIN1, ACSL4, CD36, SORBS1, LPL, PPARG, MMP1, OLR1 |
| hsa05222 | Small cell lung
cancer | 21 |
7.22×10−7 | CDK6, CCNE2, LAMA3,
RARB, ITGA6, LAMA2, CCNE1, PIK3R2, E2F1, E2F2, AKT3, LAMC1, LAMB3,
LAMA4, PIK3R1, LAMA1, MYC, LAMC3, LAMC2, FN1, PTGS2 |
| hsa04610 | Complement and
coagulation cascades | 18 |
1.04×10−6 | F10, PLAUR, F3, C3,
PROS1, SERPING1, CFI, C6, F12, MASP2, VWF, C4BPA, MASP1, C7, TFPI,
PROC, PLAU, F7 |
| hsa04114 | Oocyte meiosis | 24 |
1.06×10−6 | CCNE2, ESPL1,
CCNB2, CDC20, PTTG1, IGF1, CPEB1, SGOL1, CCNE1, CDC25C, CCNB1,
MAD2L1, PPP2R1B, ADCY4, ITPR2, RPS6KA2, PLK1, PPP1CA, ANAPC11,
PKMYT1, BUB1, RPS6KA3, AURKA, CDK1 |
| hsa04010 | MAPK signaling
pathway | 40 |
2.61×10−6 | NTRK2, MRAS,
CACNA2D1, FGF9, MEF2C, FOS, FGF1, FGF2, MAPK8IP2, CACNG1, DUSP1,
TGFBR2, FGFR3, CACNG4, PDGFRA, FLNC, RAPGEF2, PDGFA, AKT3, DUSP6,
MAPK10, PRKCA, FGFR1, MAP3K5, RPS6KA2, MECOM, EGFR, MYC, JUN,
PLA2G4A, NR4A1, RRAS2, NFATC2, FGF7, HSPB1, RPS6KA3, STMN1, CACNB3,
NTF4, MAP3K8 |