Introduction
5′ Adenosine monophosphate-activated protein kinase
(AMPK) is a master regulator of cellular energy metabolism in
normal and transformed cells (1,2).
Activation of AMPK suppresses adenosine triphosphate
(ATP)-dependent metabolic functions, and induces ATP generation via
glycolysis and fatty acid oxidation (3). AMPK is activated by cellular stress,
including glucose deprivation, hypoxia, and ischemia. During
carcinogenesis and tumor progression, cancer cells generate and
promote metabolically strained microenvironments (4,5). The
phosphorylation of AMPK by the tumor suppressor serine/threonine
kinase 11 (liver kinase B1) regulates cell growth and proliferation
in response to stress (6). AMPK also
supports the growth of aggressive orthotopic tumors prepared from
MDA-MB-231 and DU4475 breast cancer cells (7); however, paradoxically, activation of
AMPK promotes cellular apoptosis through the downstream signal
mediators c-Jun N-terminal protein kinase (JNK) and p53 (8,9). Treatment
of HCT-116 colon cancer cells with quercetin induces apoptosis
under hypoxic conditions by suppressing AMPK activity (10), whereas activation of AMPK by plumbagin
inhibits HT-29 colon cancer cell growth via the JNK-p53 signal axis
(11). Based on the aforementioned
results, the role of AMPK in colon cancer remains
controversial.
The endoplasmic reticulum (ER) is the principal
organelle responsible for protein synthesis and folding. It also
serves as a Ca2+ reservoir. Conditions of cellular
stress, such as hypoxia or exposure to chemotherapeutic drugs,
change the redox state of proteins as well as the Ca2+
concentration in the ER, resulting in the induction of the unfolded
protein response (UPR) and the release of Ca2+ from the
ER (12,13). Ca2+ leakage from the ER
into other cellular compartments stimulates autophagy through
Ca2+/calmodulin-dependent kinase kinase β (CaMKKβ) and
subsequent activation of AMPK (14,15). In
addition, the activation of AMPK by metformin triggers ER stress
via an AMPK-dependent mechanism in acute lymphoblastic leukemia
(16). The induction of AMPK activity
by metformin also mediates the anti-cancer effect of dasatinib in
head and neck squamous cell carcinoma by activating AMPK-dependent
ER stress (17). Despite supporting
evidence that patients with metabolic syndrome exhibit decreased
AMPK activity and increased cancer-associated mortality (18), the association between AMPK activity
and ER stress in chemotherapy-treated colon cancer cells remains
unclear.
Numerous types of cancer aberrantly express
inhibitors of apoptosis (IAPs) (19).
X-linked IAP (XIAP)-associated factor 1 (XAF1), a novel antagonist
of XIAP (20), interacts with
endogenous XIAP to suppress its anti-caspase activity (21). XAF1 expression is not only associated
with colon cancer cell survival, but is also upregulated by various
anti-cancer therapies (22–24). However, the connection between XAF1,
AMPK, and ER stress in colon cancer remains to be elucidated.
Ampelopsin (Amp), which is extracted from
Ampelopsis grossedentata, is reported to have various
biological functions, including anti-oxidative (25) and hepatoprotective effects (26). This flavonoid, also called
dihydromyricetin, has potent anti-cancer activities in several
types of malignancy (27,28). For example, treatment of breast cancer
cells with Amp decreases cell viability and induces apoptosis in a
dose-dependent manner through the generation of reactive oxygen
species (ROS) and induction of ER stress (29). In contradiction, another previous
study reported that Amp-induced ER stress leads to the activation
of autophagy, which protects breast cancer cells from apoptosis
(30). In addition, treatment with
Amp protects human umbilical vein endothelial cells from
hyperglycemia-induced cell damage by upregulating autophagy via the
enhancement of AMPK activity (31).
Based on these reports, the effects of Amp on cancer cells are
controversial.
In the present study, the chemotherapeutic effect of
Amp on colon cancer cells was evaluated, and the relationship
between ER stress, AMPK activation and downstream signaling was
determined. In addition, the effects of AMPK activation or ER
stress responses on the induction of XAF1 in Amp-treated colon
cancer cells were examined.
Materials and methods
Cell culture
The human colon cancer cell lines HCT-116, HCT-8 and
HT-29 were purchased from ATCC (Manassas, VA, USA). These cells
were maintained in RPMI-1640 medium (Corning Inc., Corning, NY,
USA), supplemented with 10% FBS (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA), antibiotics and glutamine (HyClone; GE
Healthcare Life Sciences) at 37°C in a humidified atmosphere with
5% CO2.
Drugs and chemicals
Amp and N-acetyl-L-cysteine (NAC; a ROS scavenger)
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Amp was dissolved in sterile dimethyl sulfoxide (DMSO) as a 200-mM
stock solution, and diluted in medium to the indicated
concentrations prior to use. Salubrinal (an ER stress inhibitor),
SB203580 [a p38 mitogen-activated protein kinase (MAPK) inhibitor]
and SP600125 (a JNK inhibitor) were purchased from Calbiochem (EMD
Millipore, Billerica, MA, USA).
Analysis of apoptotic cells by flow
cytometry
The percentages of human colon cancer HCT-116, HCT-8
or HT-29 cells undergoing apoptosis were determined by flow
cytometry using fluorescein isothiocyanate (FITC)-labeled Annexin V
and 7-aminoactinomycin D (7-AAD). To determine optimal conditions,
experiments were performed using different concentrations of Amp
(0, 10, 20, 50, 100, and 200 µM) and different periods of
incubation (2, 4, 8, 16 and 24 h). For comparison, DMSO (0.05%) was
used as a vehicle control and the negative group represented
untreated cells. These cells were maintained in RPMI-1640 medium
(Corning Inc.), supplemented with 10% FBS (HyClone; GE Healthcare
Life Sciences), antibiotics and glutamine (HyClone; GE Healthcare
Life Sciences) at 37°C in a humidified atmosphere with 5%
CO2. For staining, cells were harvested, rinsed with
PBS, and resuspended in 100 µl 1X Annexin V binding buffer [10 mM
HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2].
Following resuspension, 3 µl Annexin V-FITC and 3 µl of 7-AAD (both
purchased from BD Biosciences, San Diego, CA, USA) were added, and
the cells were incubated at room temperature for 15 min in the dark
with gentle vortexing. The stained cells were analyzed using a
FACSCalibur flow cytometer (BD Biosciences) equipped with
CellQuestpro software version 5.1 (BD Biosciences).
Measurement of mitochondrial membrane
potential (Δψm) and intracellular ROS production
The changes in Δψm were assessed using
3,3′-dihexyloxacarbocyanine iodide (DiOC6; Molecular
Probes, Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cells
were treated with Amp or DMSO for 24 h, harvested, washed twice in
PBS, resuspended in PBS supplemented with DiOC6 (20 nM),
incubated at 37°C for 15 min in the dark, and immediately analyzed
with a flow cytometer using the FL-1 filter. The intracellular
accumulation of ROS was monitored by flow cytometry after staining
with the fluorescent probe 2′,7′-dichlorodihydro-fluorescein
diacetate (DCFH-DA; 10 µM; Molecular Probes; Thermo Fisher
Scientific, Inc.) as previously described (32), with slight modification. DCFH-DA is
deacetylated in cells by esterases to a nonfluorescent compound,
DCFH, which remains trapped within the cell and is cleaved and
oxidized by ROS in the presence of endogenous peroxidase to yield a
highly fluorescent compound, 2′,7′-dichlorofluorescein (DCF). Cells
were pre-incubated with 10 µM DCFH-DA for 30 min at 37°C, seeded in
6-well plates (5×105 cells/ml), and treated with or
without Amp for 24 h. Cells were then washed, resuspended in PBS,
and ROS levels were determined using a FACSCalibur flow
cytometer.
Western blot analysis
Cells were washed in PBS and lysed in NP-40 buffer
(Elpis Biotech, Daejeon, Korea) supplemented with a protease
inhibitor cocktail (Sigma-Aldrich; Merck Millipore). To evaluate
phosphorylation events, phosphatase inhibitors (Cocktail II;
Sigma-Aldrich; Merck Millipore) were added to the NP-40 buffer. The
protein concentration was determined using a Pierce BCA Assay kit
(Thermo Fisher Scientific, Inc.). Proteins (10 µg/sample) were
resolved through SDS-PAGE and then transferred to nitrocellulose
membranes (EMD Millipore). The membranes were blocked with 5% skim
milk for 1 h at room temperature and then western blot analysis was
performed. Following primary and secondary antibody staining,
chemiluminescence was detected using an ECL kit (Advansta Corp.,
Menlo Park, CA, USA) and the LAS-3000 imaging system (Fujifilm,
Tokyo, Japan). Primary antibodies against the following antigens
were used: Caspase-8 (pro-caspase-8 and active caspase-8; cat. no.
9746; 1:1,000), caspase-3 (pro-caspase-3 and active caspase-3; cat.
no. 9665; 1:1,000), caspase-9 (pro-caspase-9 and active caspase-9;
cat. no. 9502; 1:1,000), poly ADP-ribose polymerase (PARP; cleaved
PARP p89; cat. no. 9542; 1:1,000), β-actin (cat. no. 4967;
1:1,000), Bcl-2 (cat. no. 4967; 1:1,000), BCL2-associated X protein
(Bax; cat. no. 2772; 1:1,000), myeloid cell leukemia sequence 1
(BCL2-related; Mcl-1; cat. no. 4572; 1:1,000), BCL2
antagonist/killer 1 (Bak; cat. no. 6947; 1:1,000), X-linked
inhibitor of apoptosis protein (XIAP; cat. no. 2045; 1:1,000),
phospho (p-)AMPK (Thr174; cat. no. 2531; 1:1,000), AMPK
(cat. no. 5832; 1:1,000), eukaryotic translation initiation factor
2α (eIF2α; cat. no. 9722; 1:1,000), p-eIF2α (Ser51; cat.
no. 9721; 1:1,000), p-JNK (Thr183/Tyr185;
cat. no. 4671; 1:1,000), JNK (cat. no. 9258; 1:1,000), p-p38-MAPK
(Thr180/Tyr182; cat. no. 9211; 1:1,000),
p38-MAPK (cat. no. 9212; 1:1,000), extracellular signal-regulated
kinase 1/2 (ERK1/2; cat. no. 9102; 1:1,000), and p-ERK1/2
(Thr202/Tyr204; cat. no. 9101; 1:1,000; all
from Cell Signaling Technology, Inc., Beverly, MA, USA); protein
kinase RNA-like ER kinase (PERK; cat. no. sc-13073; 1:500), p-PERK
(Thr981; cat. no. sc-32577; 1:500), glucose-regulated
protein 78 (GRP78; cat. no. sc-1051; 1:500), and
CCAAT/enhancer-binding protein homologous protein (CHOP; cat. no.
sc-575; 1:200; all from Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA); and XIAP-associated factor 1 (XAF1; cat. no. 81353;
1:500; Abcam, Cambridge, UK). The membrane was probed with primary
antibodies overnight at 4°C, followed by the following specific
secondary antibodies; horseradish peroxidase (HRP)-conjugated goat
anti-mouse immunoglobulin G (IgG; cat. no. K0211589; 1:3,000) or
HRP-conjugated goat anti-rabbit IgG (cat. no. K0211708; 1:3,000;
both from KOMABiotech, Seoul, Korea) for 1 h at room
temperature.
Small interfering RNA (siRNA)
transfection
Experimentally verified human AMPK-siRNA duplex
(cat. no. 1121714; GCA UAU GCU GCA GGU AGA UdTdT) and negative
control-siRNA (cat. no. SN-1003) were obtained from Bioneer
Corporation (Daejeon, Korea). Cells were seeded at a concentration
of 1×105 per well in a 6-well plate and grown overnight.
Cells were treated with 100 µM Amp for 6 h and then transfected
with 200 nM siRNAs using Lipofectamine RNAiMAX Reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Cells were used for further experiments at 36 h after
transfection.
Statistical analysis
Data are presented as the mean ± standard deviation.
Statistical analyses were conducted using one-way analysis of
variance (ANOVA) using SigmaPlot software (version 10.0; Systat
Software, Inc., San Jose, CA, USA). Bonferroni post hoc analysis
was performed following one-way ANOVA for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Amp treatment of colon cancer cells
enhances the activation of ER stress and the AMPK signaling
pathway
It was first investigated whether Amp has an
anti-colon cancer effect. Annexin V/7-AAD staining and
DiOC6 staining assays were adopted to measure
cytotoxicity and Δψm, respectively, in
Amp-treated colon cancer cells. Following treatment with Amp, dose-
and time-dependent induction of apoptosis was observed in HCT-116
colon cancer cells (Fig. 1A and B;
P<0.05; DMSO-treated group vs. Amp-treated group). In addition,
Amp treatment disrupted the Δψm of HCT-116 colon cancer
cells in a dose- and time-dependent manner (Fig. 1A and B; P<0.05; DMSO-treated group
vs. Amp-treated group). The treatment of colon cancer cells
(HCT-116, HCT-8, and HT-29) with Amp also induced the cleavage and
activation of caspase-9, caspase-3, and PARP, but not of caspase-8
(Fig. 1C). These results suggested
that Amp-induced apoptosis is required for the mitochondrial
apoptosis pathway to initiate and for caspases to activate.
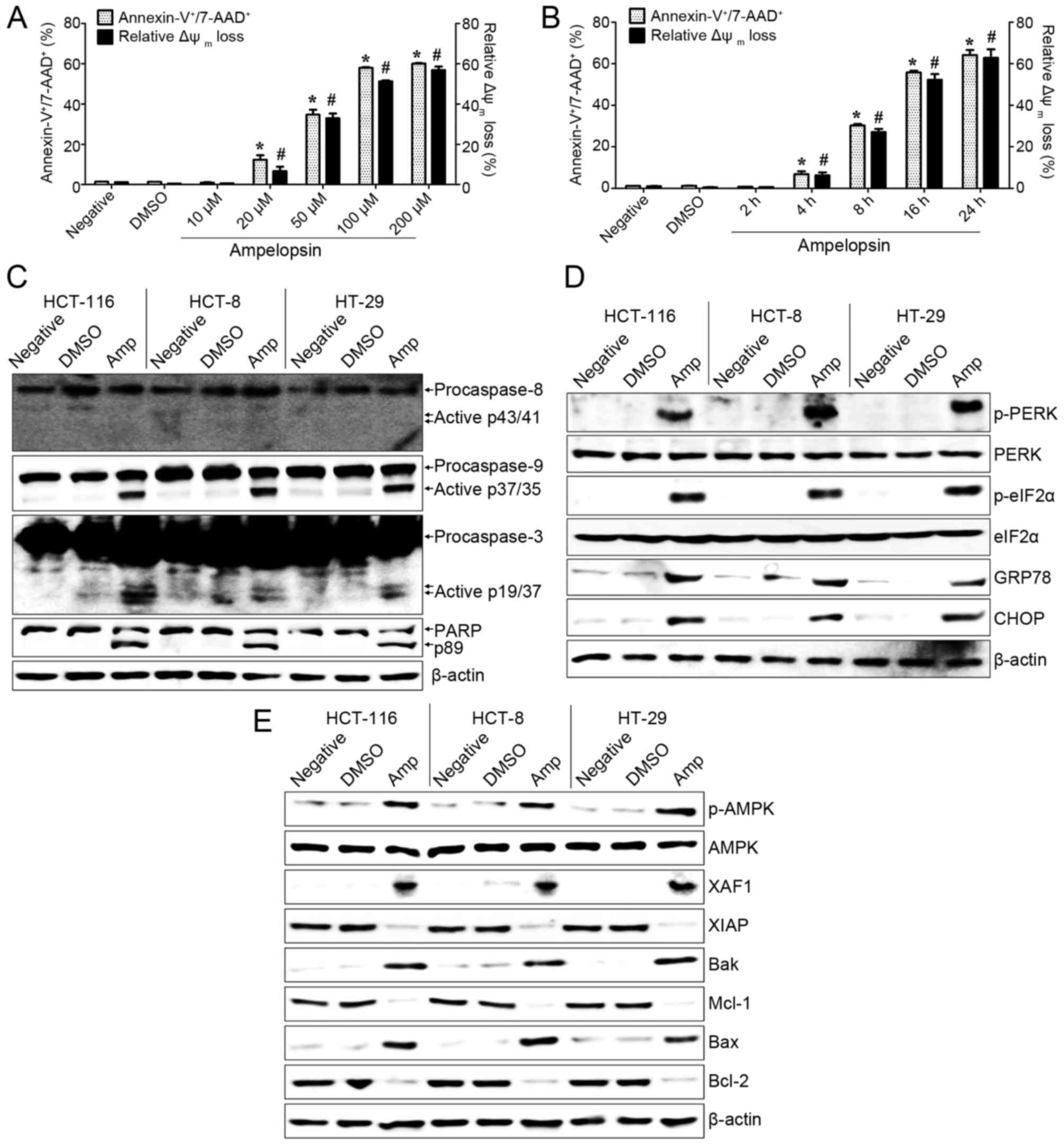 | Figure 1.Amp induces apoptosis and enhances
the activation of ER stress and AMPK signaling in colon cancer
cells. (A) HCT-116 cells were treated with 10, 20, 50, 100, or 200
µM of Amp for 24 h. (B) HCT-116 cells were treated with 100 µM of
Amp for 2, 4, 8, 16, or 24 h. To detect the degree of apoptosis,
cells were stained Annexin V-fluorescein isothiocyanate and 7-AAD
and analyzed by flow cytometry. The percentage of apoptotic cells
signifies Annexin V+/7-AAD+. To measure
disruption of Δψm, cells were stained with DiOC6.
Diminished DiOC6 fluorescence (%) indicates Δψm
disruption. Each value is presented as the mean ± standard
deviation of three determinations. (C-E) Cells were treated with
100 µM Amp for 24 h. Total cell lysates were western blotted with
antibodies against (C) caspase-8 [pro-caspase-8 and active
caspase-8 (p43/41)], caspase-9 [pro-caspase-9 and active caspase-9
(p37/35)], caspase-3 [pro-caspase-3 and active caspase-3 (p19/17)],
or PARP [(PARP and cleaved PARP (p89)]; (D) p-PERK, PERK, p-eIF2α,
eIF2α, CHOP, or GRP78; and (E) p-AMPK, AMPK, XAF1, XIAP, Bak,
Bcl-2, Bax, or Mcl-1. β-actin was used as a loading control. The
results are representative of three independent experiments.
Untreated cells (negative) and vehicle-treated cells (DMSO) were
used as the control groups. *P<0.05 and #P<0.01,
DMSO-treated cells vs. Amp-treated cells. Amp, ampelopsin; ER,
endoplasmic reticulum; AMPK, 5′ adenosine monophosphate-activated
protein kinase; 7-AAD, 7-aminoactinomycin D; Δψm,
mitochondrial membrane potential; PARP, poly ADP-ribose polymerase;
p-, phosphorylated; PERK, protein kinase RNA-like ER kinase; eIF2α,
eukaryotic translation initiation factor 2α; CHOP,
CCAAT/enhancer-binding protein homologous protein; GRP78,
glucose-regulated protein 78; XAF1, XIAP-associated factor 1; XIAP,
X-linked inhibitor of apoptosis protein; Bak, BCL2
antagonist/killer 1; Bax, BCL2-associated X protein; Mcl-1, myeloid
cell leukemia sequence 1 (BCL2-related); DMSO, dimethyl
sulfoxide. |
Subsequently, the association of ER stress with
Amp-induced colon cancer cell death was investigated. Amp-treated
colon cancer cells exhibited upregulated expression of ER
stress-associated proteins, including p-PERK, p-eIF2α, GRP78, and
CHOP (Fig. 1D). It was also examined
whether the apoptotic effect of Amp on colon cancer cells was
associated with AMPK or XAF1 expression. Expression of p-AMPK and
XAF1 in colon cancer cells was increased following treatment with
100 µM Amp for 24 h (Fig. 1E). The
expression of Bcl-2 family proteins in colon cancer cells was also
modulated by Amp treatment: Bcl-2 and Mcl-1 were easily detectable
in untreated and vehicle-treated colon cancer cells, but markedly
reduced following Amp treatment. In addition, the expression of Bak
and Bax was induced in Amp-treated colon cancer cells (Fig. 1E). These results suggest that the cell
death of Amp-treated colon cancer cells is closely associated with
ER stress-mediated apoptosis and changes in AMPK-mediated
signaling.
ER stress-dependent signaling directs
Amp-induced apoptosis in colon cancer cells
To investigate the role of ER stress in Amp-induced
apoptosis, colon cancer cells were pre-treated with salubrinal (a
selective inhibitor of eIF2α) prior to Amp treatment. Pretreatment
with salubrinal markedly inhibited the expression of p-PERK,
p-eIF2α, GRP78, and CHOP in Amp-treated colon cancer cells
(Fig. 2A). Amp-induced cleavage of
caspase-9, caspase-3, and PARP were also attenuated by pretreatment
with salubrinal in colon cancer cells (Fig. 2B). As ER stress activates JNK and
p38-MAPK (33,34), the effect of salubrinal pretreatment
on these kinases was assessed. Amp treatment of colon cancer cells
downregulated the phosphorylation of ERK, whereas it upregulated
the phosphorylation of JNK and p38-MAPK (Fig. 2C). Salubrinal pretreatment of colon
cancer cells also effectively attenuated Amp-induced
phosphorylation of JNK and p38-MAPK (Fig.
2D). Furthermore, inhibition of ER stress with salubrinal
markedly reduced the Amp-dependent phosphorylation of AMPK and
expression of XAF1 in colon cancer cells (Fig. 2D). These results indicate that ER
stress-dependent AMPK/MAPK signaling is important for Amp-mediated
apoptosis in colon cancer cells.
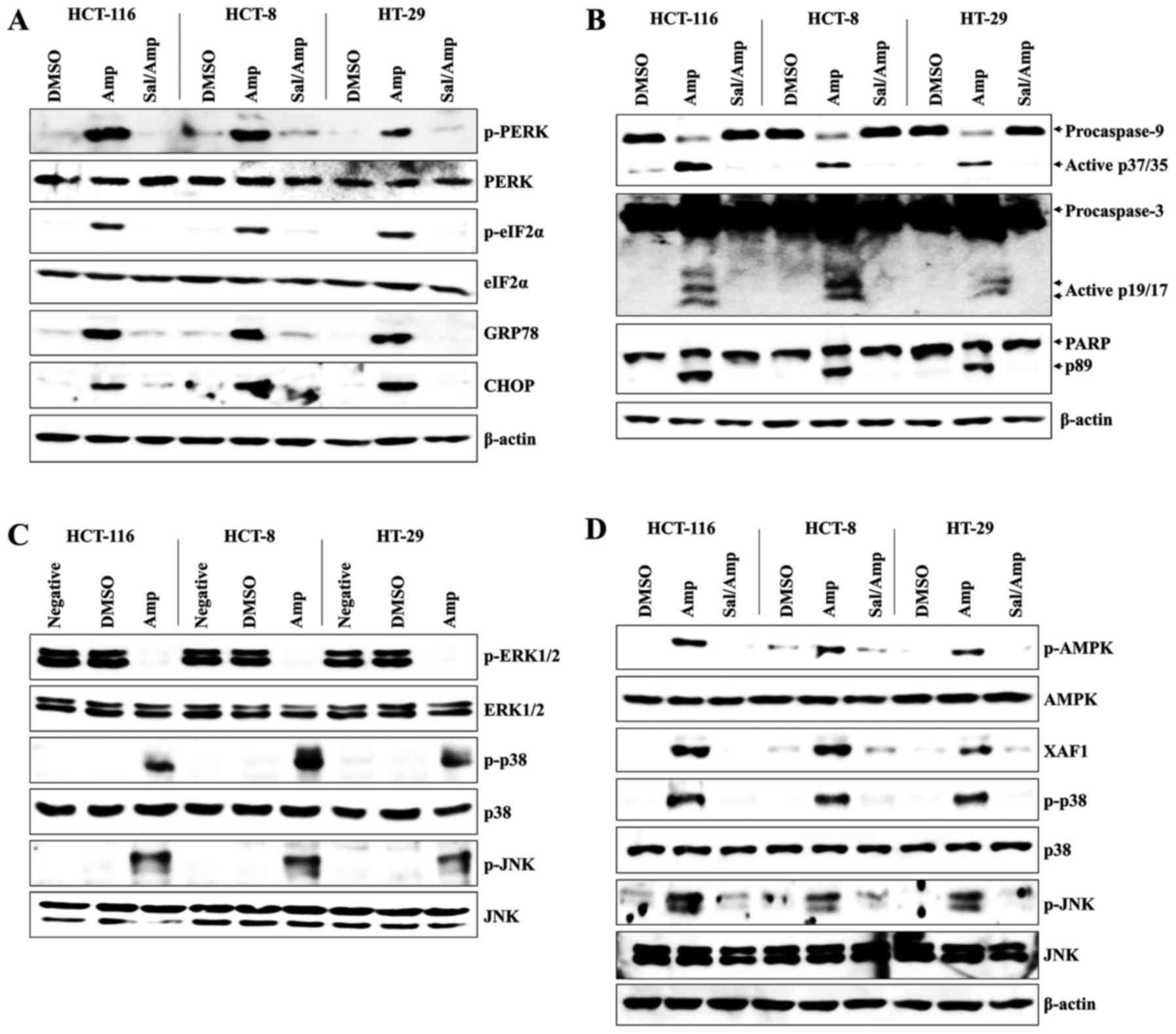 | Figure 2.ER stress signaling is closely
related to Amp-induced apoptosis of colon cancer cells. (A, B and
D) Cells were pretreated with 2 µM salubrinal for 1 h and then
treated with 100 µM Amp or DMSO at 37°C for 24 h. Total protein was
extracted from cell lysates and blotted for (A) p-PERK, PERK,
p-eIF2α, eIF2α, CHOP, or GRP78 protein; (B) caspase-8
[pro-caspase-8 and active caspase-8 (p43/41)], caspase-9
[pro-caspase-9 and active caspase-9 (p37/35)], caspase-3
[pro-caspase-3 and active caspase-3 (p19/17)] or PARP [PARP and
cleaved PARP (p89)]; (D) and p-AMPK, AMPK, XAF1, p-JNK, JNK,
p-p38-MAPK or p38-MAPK protein. β-actin was used as a loading
control. (C) Cells were treated with 100 µM Amp for 24 h. Western
blotting for p-ERK1/2, ERK1/2, p-p38 MAPK, p38 MAPK, p-JNK, or JNK
was performed. The results are representative of three independent
experiments. Vehicle-treated cells (DMSO) were used as the control
group. ER, endoplasmic reticulum; Amp, ampelopsin; DMSO, dimethyl
sulfoxide; p-, phosphorylated; PERK, protein kinase RNA-like ER
kinase; eIF2α, eukaryotic translation initiation factor 2α; CHOP,
CCAAT/enhancer-binding protein homologous protein; GRP78,
glucose-regulated protein 78; PARP, poly ADP-ribose polymerase;
AMPK, 5′ adenosine monophosphate-activated protein kinase; XAF1,
XIAP-associated factor 1; JNK, c-Jun N-terminal protein kinase;
p38-MAPK, p38 mitogen-activated protein kinase; ERK1/2,
extracellular signal-regulated kinase 1/2; Sal, salubrinal. |
ER stress triggers AMPK/MAPK-dependent
apoptosis signaling in Amp-treated colon cancer cells
Expression of AMPK was downregulated by siRNA to
study the association with ER stress in the Amp-induced apoptosis
of colon cancer cells. Although AMPK-knockdown in HCT-116 and HT-29
cells produced no change in the Amp-dependent induction of the ER
stress-related proteins p-PERK and CHOP, the levels of XAF1, p-p38,
and p-JNK were markedly reduced in the two cell lines (Fig. 3A). AMPK knockdown also noticeably
suppressed the activation (cleavage) of caspase-9, caspase-3 and
PARP (Fig. 3B). In addition,
downregulation of AMPK with siRNA in HCT-116 and HT-29 colon cancer
cells reduced the Amp-induced expression of the apoptotic Bcl-2
family proteins Bak and Bax, while enhancing the expression of the
anti-apoptotic Bcl-2 family proteins Bcl-2 and Mcl-1 (Fig. 3B). These results demonstrate that AMPK
is one of the downstream signaling targets of ER stress and
mediates Amp-induced colon cancer cell apoptosis.
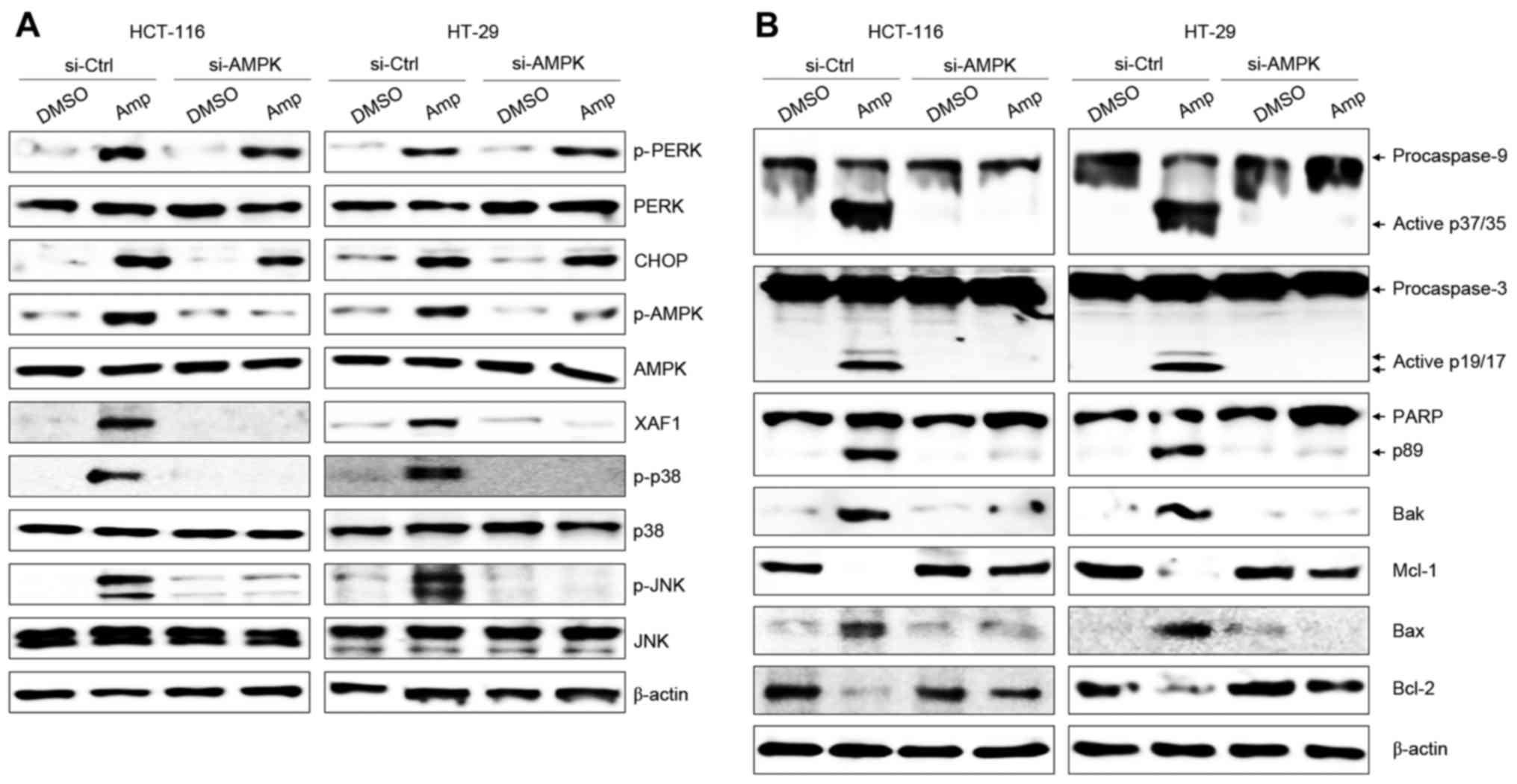 | Figure 3.ER stress triggers AMPK-dependent
apoptosis signaling in Amp-treated colon cancer cells. Cells were
treated with 100 µM Amp for 6 h then transfected with 200 nM
si-AMPK using the Lipofectamine RNAiMAX reagent according to the
manufacturer's protocol. Cells were used for further experiments 36
h after transfection. Whole-cell lysates were subjected to western
blotting using (A) p-PERK, PERK, CHOP, p-AMPK, AMPK, XAF1,
p-p38-MAPK, p38-MAPK, p-JNK, or JNK, and (B) caspase-9
[pro-caspase-9 and active caspase-9 (p37/35)], caspase-3
[pro-caspase-3 and active caspase-3 (p19/17)] or PARP [PARP and
cleaved PARP (p89)], Bcl-2, Bax, Bak, or Mcl-1 antibodies. β-actin
was used as a loading control. The results are representative of
three independent experiments. Vehicle-treated cells (DMSO) were
used as the control group. ER, endoplasmic reticulum; AMPK, 5′
adenosine monophosphate-activated protein kinase; Amp, ampelopsin;
p-, phosphorylated; PERK, protein kinase RNA-like ER kinase; CHOP,
CCAAT/enhancer-binding protein homologous protein; XAF1,
XIAP-associated factor 1; p38-MAPK, p38 mitogen-activated protein
kinase; JNK, c-Jun N-terminal protein kinase; PARP, poly ADP-ribose
polymerase; Bax, BCL2-associated X protein; Bak, BCL2
antagonist/killer 1; Mcl-1, myeloid cell leukemia sequence 1
(BCL2-related); DMSO, dimethyl sulfoxide; si-Ctrl, control small
interfering RNA; si-AMPK, small interfering RNA targeting AMPK. |
MAPK activation leads to the
expression of XAF1 in Amp-treated colon cancer cells
The roles of JNK and p38-MAPK activation in ER
stress and AMPK expression were investigated using SB203580, an
inhibitor of p38 phosphorylation, and SP600125, an inhibitor of
JNK. HCT-116 and HT-29 cells were pretreated with either SB203580
or SP600125, followed by Amp treatment. Although the Amp-induced
p-PERK, CHOP, and AMPK expression levels were unaffected in the two
cell lines by either SB203580 or SP600125, each of these inhibitors
led to a marked attenuation of Amp-mediated XAF1 expression
(Fig. 4A). In addition, SB203580 and
SP600125 effectively prevented Amp-induced activation and cleavage
of caspase-9, caspase-3, and PARP. The levels of the anti-apoptotic
proteins Bcl-2 and Mcl-1 were also restored by MAPK inhibition
(Fig. 4B). These results reveal a
critical role for JNK and p38-MAPK-dependent XAF1 induction in
Amp-treated colon cancer cells.
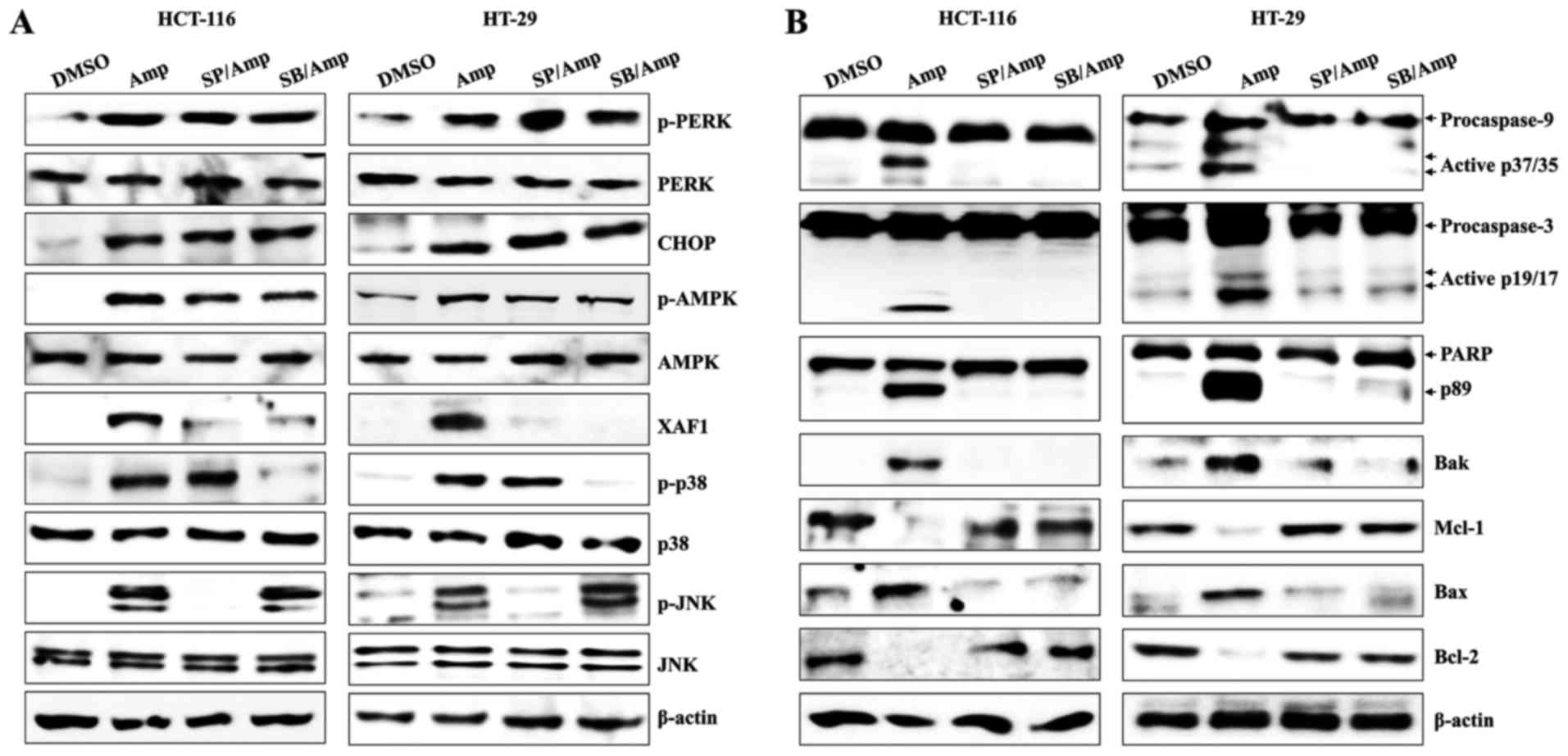 | Figure 4.MAPK activation leads to apoptosis of
Amp-treated colon cancer cells. Cells were pretreated with 25 µM
SP600125 or 10 µM SB203580 at 37°C for 1 h and then treated with
100 µM Amp or DMSO (control) at 37°C for 24 h. Total protein was
extracted from cell lysates, and western blotting for (A) p-PERK,
PERK, CHOP, p-AMPK, AMPK, XAF1, p-p38-MAPK, p38-MAPK, p-JNK, or JNK
protein, and (B) caspase-9 [pro-caspase-9 and active caspase-9
(p37/35)], caspase-3 [pro-caspase-3 and active caspase-3 (p19/17)],
PARP [PARP and cleaved PARP (p89)], Bcl-2, Bax, Bak or Mcl-1
protein were performed. β-actin was used as a loading control. The
results are representative of three independent experiments. MAPK,
mitogen-activated protein kinase; Amp, ampelopsin; DMSO, dimethyl
sulfoxide; p-, phosphorylated; PERK, protein kinase RNA-like
endoplasmic reticulum kinase; CHOP, CCAAT/enhancer-binding protein
homologous protein; AMPK, 5′ adenosine monophosphate-activated
protein kinase; XAF1, XIAP-associated factor 1; JNK, c-Jun
N-terminal protein kinase; PARP, poly ADP-ribose polymerase; Bax,
BCL2-associated X protein; Bak, BCL2 antagonist/killer 1; Mcl-1,
myeloid cell leukemia sequence 1 (BCL2-related); SP, SP600125; SB,
SB203580. |
Amp-induced ROS promote the ER
stress/AMPK-mediated apoptosis of colon cancer cells
ROS are implicated in a variety of biological
functions and are an upstream signal activating AMPK in
epigallocatechin-3-gallate-mediated colon cancer cell death
(35). In addition, Amp treatment
reportedly triggers ROS generation in breast cancer cells (29). To assess the effect of ROS on ER
stress-induced apoptosis in Amp-treated colon cancer cells, the ROS
scavenger NAC was used. Amp treatment induced ROS production in
HCT-116 cells in a time-dependent manner (P<0.05; non-treated
cells vs. Amp-treated cells; P<0.01; non-treated cells vs.
Amp-treated cells; Fig. 5A).
Pretreatment with NAC efficiently quenched the Amp-induced
generation of ROS (P<0.01; Amp-treated cells vs. cells
co-treated with NAC and Amp; Fig.
5B), attenuated Amp-dependent induction of p-PERK, CHOP, p-AMPK
and XAF1, and reduced the activation of JNK and p38-MAPK by Amp
(Fig. 5C). NAC pretreatment also
suppressed the Amp-induced cleavage and activation of caspase-9,
caspase-3, and cleaved PARP in colon cancer cells (Fig. 5D). ROS levels in Amp-treated colon
cancer cells were unaffected by salubrinal (P<0.01; DMSO-treated
cells vs. Amp-treated cells; Fig.
5E), AMPK siRNA (P<0.01; DMSO-treated cells vs. Amp-treated
cells; Fig. 5F), SB203580, or
SP600125 (P<0.01; DMSO-treated cells vs. Amp-treated cells;
Fig. 5G). These results suggest that
ROS generated by Amp treatment are responsible for initiating the
ER stress-mediated apoptosis pathway in colon cancer cells.
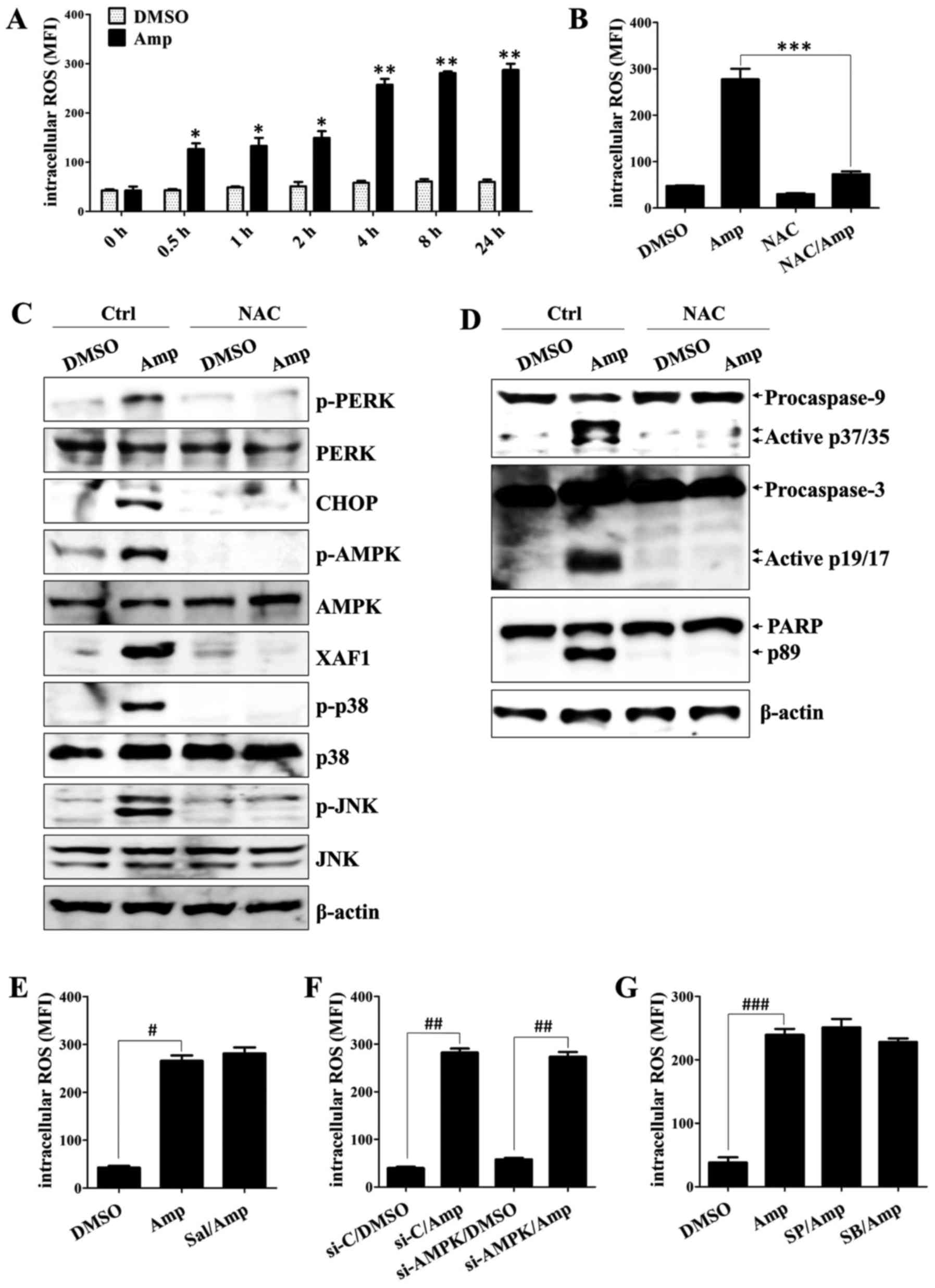 | Figure 5.Amp promotes ER stress/AMPK-mediated
apoptosis through ROS generation in colon cancer cells. (A, B and
E-G) Cells were pretreated with 10 µM
2′,7′-dichlorodihydro-fluorescein diacetate for 30 min and then
treated with 100 µM Amp or DMSO at 37°C for (A) 0–24 h, or (B and
E-G) 8 h. The values in the 2′,7′-dichlorofluorescein histograms
indicate the MFI. (B-D) To quench ROS, cells were pretreated with
10 mM NAC for 1 h. Cells were then washed with PBS and treated with
100 µM Amp for (B) 8 h or (C and D) 24 h. (B) The effect of NAC on
Amp-induced ROS production was measured using flow cytometry.
Whole-cell lysates were subjected to western blotting using (C)
p-PERK, PERK, CHOP, p-AMPK, AMPK, XAF1, p-p38-MAPK, p38-MAPK,
p-JNK, and JNK antibodies, and (D) caspase-9 [pro-caspase-9 and
active caspase-9 (p37/35)], caspase-3 [pro-caspase-3 and active
caspase-3 (p19/17)] or PARP [PARP and cleaved PARP (p89)]
antibodies. β-actin was used to normalize protein content. (E) To
block ER stress, cells were pretreated with salubrinal (2 µM) for 1
h and then treated with 100 µM Amp for 8 h. (F) To prevent AMPK
activation, cells were treated with 100 µM Amp for 6 h, then
transfected with 200 nM si-AMPK using the Lipofectamine RNAiMAX
Reagent according to the manufacturer's protocol. Cells were used
for further experiments 36 h after transfection. (G) To inhibit the
JNK or p38-MAPK cascade, cells were pretreated with 25 µM SP600125
or 10 µM SB203580 at 37°C for 1 h and then treated with 100 µM Amp
or DMSO for 8 h. The results are representative of three
independent experiments. *P<0.05 and **P<0.01, untreated
cells vs. Amp-treated cells; ***P<0.01, Amp-treated cells vs.
cells co-treated with NAC and Amp; #P<0.01,
DMSO-treated cells vs. Amp-treated cells; ##P<0.01,
DMSO-treated cells vs. Amp-treated cells; ###P<0.01,
DMSO-treated cells vs. Amp-treated cells. Vehicle-treated cells
(DMSO) were used as the control group. Amp, ampelopsin; ER,
endoplasmic reticulum; AMPK, 5′ adenosine monophosphate-activated
protein kinase; ROS, reactive oxygen species; DMSO, dimethyl
sulfoxide; NAC, N-acetyl-L-cysteine; p-, phosphorylated; PERK,
protein kinase RNA-like ER kinase; CHOP, CCAAT/enhancer-binding
protein homologous protein; XAF1, XIAP-associated factor 1;
p38-MAPK, p38-mitogen-activated protein kinase; JNK, c-Jun
N-terminal protein kinase; PARP, poly ADP-ribose polymerase; MFI,
mean fluorescence intensity; Sal, salubrinal; Ctrl, control;
si-AMPK, small interfering RNA targeting AMPK; si-C, control small
interfering RNA; SP, SP600125; SB, SB203580. |
Discussion
Amp, a major component of Ampelopsis
grossedentata, is reported to possess not only important
pharmacological activities, such as anti-inflammatory and
hepatoprotective properties (25,26), but
also anti-cancer activity. The anti-tumor activity of Amp is
associated with the induction of apoptosis (27–30) and
the inhibition of metastasis (36).
Amp causes ER stress, resulting in the inhibition of cell growth
and induction of apoptosis in human breast cancer cells (29). However, the anti-tumor effects of Amp
on colon cancer have not been studied previously, and its
underlying mechanism of action remains to be investigated. ER
stress, initiated by various stress conditions, results in the
release the Ca2+ from the ER, leading to CaMKKβ-mediated
AMPK activation (12,14). Severe ER stress triggers apoptotic
cell death by inducing CHOP, activating caspase-12, and disrupting
the balance between pro-apoptotic and anti-apoptotic members of the
Bcl-2 family (13,37). Amp-mediated ROS activate ER stress and
promote signaling pathways that lead to breast cell death (29). However, the association between ER
stress and AMPK activation in apoptotic signaling is not fully
understood. The present study demonstrated that Amp activates a
pro-apoptotic signaling pathway in colon cancer cells by promoting
ROS generation, which causes ER stress. Amp-induced ER stress
subsequently triggers the activation of AMPK to induce
XAF1-mediated apoptosis through the JNK/p38-MAPK signaling pathway
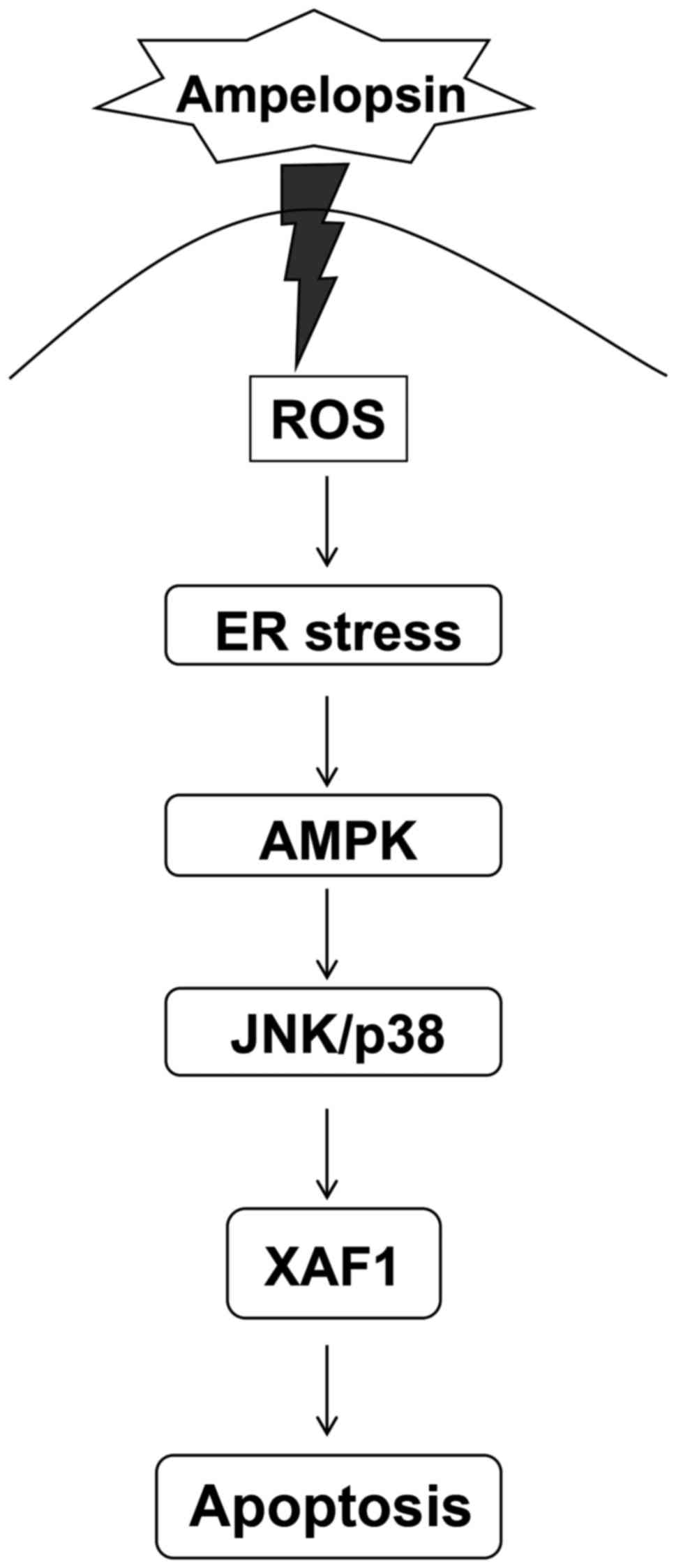
(Fig. 6).
AMPK activation promotes catabolic processes,
resulting in the inhibition of lipid, glycogen, and protein
synthesis, leading to the concomitant inhibition of cell growth and
proliferation (1,2). Metformin, which activates AMPK, induces
ER stress-mediated apoptosis of acute lymphoblastic leukemia
(16). These reports are consistent
with results that metabolic stress impairs protein-processing
capacity to induce the UPR in the ER (38). When UPR in the ER is persistent and
unresolved, cell survival processes are subverted by pro-apoptotic
signaling pathways (39). When PERK
is activated by stressful conditions, it phosphorylates eIF2α to
attenuate protein synthesis, and upregulated eIF2α regulates CHOP
expression, thereby mediating apoptosis (40). Although Amp-induced AMPK activity
reportedly protects endothelial cells through autophagy (31), those studies also indicate that AMPK
activation may be associated with the induction of ER stress to
induce its anti-cancer effect (31,41). In
accordance with those results, the results of the present study
indicated that ER stress in Amp-treated colon cancer cells
regulates the activation of AMPK. Salubrinal, an inhibitor of ER
stress, effectively prevented the phosphorylation of AMPK and
inhibited the apoptosis of colon cancer cells. Knockdown of AMPK
with siRNA, however, did not attenuate ER stress, indicating that
ER stress precedes AMPK activation, and also that increases in
intracellular Ca2+ concentrations released from the ER
likely activate AMPK.
XAF1 suppresses cancer through two principal
mechanisms. To enhance apoptosis, XAF1 interacts directly with
endogenous XIAP, suppressing its anti-caspase activity (21). XAF1 also localizes to the mitochondria
and disrupts the Δψm, leading to cancer cell death and
suppression of tumor growth (42–44). XAF1
expression is upregulated by the inhibition of ERK signaling
(24). The nuclear translocation of
XAF1 and Bax are dependent on the phosphorylation of p38-MAPK in
melphalan-induced apoptosis of Epstein-Barr virus-transformed B
cells (45). The present study showed
that Amp treatment of colon cancer cells could induce apoptosis in
a dose-dependent manner, increase the expression of XAF1 and
pro-apoptotic Bcl-2 family proteins (Bak and Bax), and increase the
levels of phosphorylated JNK and p38-MAPK. Amp-induced XAF1
expression and apoptosis were inhibited by salubrinal, SB203580
(p38-MAPK inhibitor), or SP600125 (JNK inhibitor), as well as by
the knockdown of AMPK. These results suggest an essential function
for ER stress-mediated JNK/p38-MAPK signaling in the control of
XAF1 expression in Amp-induced apoptosis in colon cancer cells.
ROS exert significant effects on multiple cellular
functions (46). ROS cause ER stress,
which results in the generation of more ROS (45,47).
Although Amp-mediated ROS trigger ER stress/AMPK activation and
JNK/p38-MAPK signaling, leading to apoptosis in colon cancer cells,
it is important to consider that Amp is not necessarily pro-ROS in
all circumstances (25,48). In a previous study of
lipopolysaccharide-activated macrophages, low doses of Amp
effectively suppressed ROS generation and the release of
inflammatory cytokines (25).
Low-dose Amp has also been demonstrated to inhibit phosphoinositide
3-kinase activation without attenuating MAPK (ERK, JNK and
p38-MAPK) activation (25), and
Amp-mediated ROS reduction has been identified to trigger the
apoptosis of hepatocellular carcinoma cells (48). These results indicate that effective
chemotherapeutic doses will need to be carefully determined and
that mechanism(s) of action must be elucidated within each dose
range and for each cell type to mitigate the risk of pleiotropic
effects. Taken together, the results of the present study suggested
that the ER stress-mediated AMPK signaling pathway could induce the
apoptosis of ampelopsin-exposed colon cancer cells and that the
AMPK-mediated apoptosis signaling pathway with ampelopsin could be
a promising strategy for improving the clinical outcomes of
patients with colon cancer.
Acknowledgements
The present study was supported by the Basic Science
Research Program of the Ministry of Education (grant no.
NRF-2015R1D1A1A01056672) and the Ministry of Science, ICT and
Future Planning (grant no. NRF-2015R1C1A2A01053732) through the
National Research Foundation of the Republic of Korea.
References
|
1
|
Hardie DG and Carling D: The AMP-activated
protein kinase-fuel gauge of the mammalian cell? Eur J Biochem.
246:259–273. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kemp BE, Mitchelhill KI, Stapleton D,
Michell BJ, Chen ZP and Witters LA: Dealing with energy demand: The
AMP-activated protein kinase. Trends Biochem Sci. 24:22–25. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan HX, Xiong Y and Guan KL: Nutrient
sensing, metabolism, and cell growth control. Mol Cell. 49:379–387.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shaw RJ, Bardeesy N, Manning BD, Lopez L,
Kosmatka M, DePinho RA and Cantley LC: The LKB1 tumor suppressor
negatively regulates mTOR signaling. Cancer Cell. 6:91–99. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Laderoute KR, Calaoagan JM, Chao WR, Dinh
D, Denko N, Duellman S, Kalra J, Liu X, Papandreou I, Sambucetti L
and Boros LG: 5′-AMP-activated protein kinase (AMPK) supports the
growth of aggressive experimental human breast cancer tumors. J
Biol Chem. 289:22850–22864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kefas BA, Cai Y, Ling Z, Heimberg H, Hue
L, Pipeleers D and Van de Casteele M: AMP-activated protein kinase
can induce apoptosis of insulin-producing MIN6 cells through
stimulation of c-Jun-N-terminal kinase. J Mol Endocrinol.
30:151–161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okoshi R, Ozaki T, Yamamoto H, Ando K,
Koida N, Ono S, Koda T, Kamijo T, Nakagawara A and Kizaki H:
Activation of AMP-activated protein kinase induces p53-dependent
apoptotic cell death in response to energetic stress. J Biol Chem.
283:3979–3987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim HS, Wannatung T, Lee S, Yang WK, Chung
SH, Lim JS, Choe W, Kang I, Kim SS and Ha J: Quercetin enhances
hypoxia-mediated apoptosis via direct inhibition of AMPK activity
in HCT116 colon cancer. Apoptosis. 17:938–949. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen MB, Zhang Y, Wei MX, Shen W, Wu XY,
Yao C and Lu PH: Activation of AMP-activated protein kinase (AMPK)
mediates plumbagin-induced apoptosis and growth inhibition in
cultured human colon cancer cells. Cell Signal. 25:1993–2002. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schröder M: Endoplasmic reticulum stress
responses. Cell Mol Life Sci. 65:862–894. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Malhotra JD and Kaufman RJ: The
endoplasmic reticulum and the unfolded protein response. Semin Cell
Dev Biol. 18:716–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, et al: Control of macroautophagy by calcium,
calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell.
25:193–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsu YC and Ip MM: Conjugated linoleic
acid-induced apoptosis in mouse mammary tumor cells is mediated by
both G protein coupled receptor-dependent activation of the
AMP-activated protein kinase pathway and by oxidative stress. Cell
Signal. 23:2013–2020. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leclerc GM, Leclerc GJ, Kuznetsov JN,
DeSalvo J and Barredo JC: Metformin induces apoptosis through
AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts.
PLoS One. 8:e744202013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin YC, Wu MH, Wei TT, Lin YC, Huang WC,
Huang LY, Lin YT and Chen CC: Metformin sensitizes anticancer
effect of dasatinib in head and neck squamous cell carcinoma cells
through AMPK-dependent ER stress. Oncotarget. 5:298–308.
2014.PubMed/NCBI
|
|
18
|
Calle EE, Rodriguez C, Walker-Thurmond K
and Thun MJ: Overweight, obesity, and mortality from cancer in a
prospectively studied cohort of U.S. adults. N Engl J Med.
348:1625–1638. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ambrosini G, Adida C and Altieri DC: A
novel anti-apoptosis gene, survivin, expressed in cancer and
lymphoma. Nat Med. 3:917–921. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fong WG, Liston P, Rajcan-Separovic E, St
Jean M, Craig C and Korneluk RG: Expression and genetic analysis of
XIAP-associated factor 1 (XAF1) in cancer cell lines. Genomics.
70:113–122. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liston P, Fong WG, Kelly NL, Toji S,
Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW and Korneluk
RG: Identification of XAF1 as an antagonist of XIAP anti-Caspase
activity. Nat Cell Biol. 3:128–133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang L, Cao Z, Yan H and Wood WC:
Coexistence of high levels of apoptotic signaling and inhibitor of
apoptosis proteins in human tumor cells: Implication for cancer
specific therapy. Cancer Res. 63:6815–6824. 2003.PubMed/NCBI
|
|
23
|
Wang J, Peng Y, Sun YW, He H, Zhu S, An X,
Li M, Lin MC, Zou B, Xia HH, et al: All-trans retinoic acid induces
XAF1 expression through an interferon regulatory factor-1 element
in colon cancer. Gastroenterology. 130:747–758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu LF, Wang J, Zou B, Lin MC, Wu YL, Xia
HH, Sun YW, Gu Q, He H, Lam SK, et al: XAF1 mediates apoptosis
through an extracellular signal-regulated kinase pathway in colon
cancer. Cancer. 109:1996–2003. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qi S, Xin Y, Guo Y, Diao Y, Kou X, Luo L
and Yin Z: Ampelopsin reduces endotoxic inflammation via repressing
ROS-mediated activation of PI3K/Akt/NF-κB signaling pathways. Int
Immunopharmacol. 12:278–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murakami T, Miyakoshi M, Araho D, Mizutani
K, Kambara T, Ikeda T, Chou WH, Inukai M, Takenaka A and Igarashi
K: Hepatoprotective activity of tocha, the stems and leaves of
Ampelopsis grossedentata, and ampelopsin. Biofactors. 21:175–178.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ni F, Gong Y, Li L, Abdolmaleky HM and
Zhou JR: Flavonoid ampelopsin inhibits the growth and metastasis of
prostate cancer in vitro and in mice. PLoS One. 7:e388022012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang B, Dong S, Cen X, Wang X, Liu X,
Zhang H, Zhao X and Wu Y: Ampelopsin sodium exhibits antitumor
effects against bladder carcinoma in orthotopic xenograft models.
Anticancer Drugs. 23:590–596. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou Y, Shu F, Liang X, Chang H, Shi L,
Peng X, Zhu J and Mi M: Ampelopsin induces cell growth inhibition
and apoptosis in breast cancer cells through ROS generation and
endoplasmic reticulum stress pathway. PLoS One. 9:e890212014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou Y, Liang X, Chang H, Shu F, Wu Y,
Zhang T, Fu Y, Zhang Q, Zhu JD and Mi M: Ampelopsin-induced
autophagy protects breast cancer cells from apoptosis through
Akt-mTOR pathway via endoplasmic reticulum stress. Cancer Sci.
105:1279–1287. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang X, Zhang T, Shi L, Kang C, Wan J,
Zhou Y, Zhu J and Mi M: Ampelopsin protects endothelial cells from
hyperglycemia-induced oxidative damage by inducing autophagy via
the AMPK signaling pathway. Biofactors. 41:463–475. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rosenkranz AR, Schmaldienst S, Stuhlmeier
KM, Chen W, Knapp W and Zlabinger GJ: A microplate assay for the
detection of oxidative products using
2′,7′-dichlorofluorescin-diacetate. J Immunol Methods. 156:39–45.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nishitoh H, Saitoh M, Mochida Y, Takeda K,
Nakano H, Rothe M, Miyazono K and Ichijo H: ASK1 is essential for
JNK/SAPK activation by TRAF2. Mol Cell. 2:389–395. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hwang JT, Ha J, Park IJ, Lee SK, Baik HW,
Kim YM and Park OJ: Apoptotic effect of EGCG in HT-29 colon cancer
cells via AMPK signal pathway. Cancer Lett. 247:115–121. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng HQ and Liu DY: Anti-invasive and
anti-metastatic effect of ampelopsin on melanoma. Ai Zheng.
22:363–367. 2003.(In Chinese). PubMed/NCBI
|
|
37
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kaufman RJ: Stress signaling from the
lumen of the endoplasmic reticulum: Coordination of gene
transcriptional and translational controls. Genes Dev.
13:1211–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao L and Ackerman SL: Endoplasmic
reticulum stress in health and disease. Curr Opin Cell Biol.
18:444–452. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim A, Im M and Ma JY: Ethanol extract of
Remotiflori radix induces endoplasmic reticulum stress-mediated
cell death through AMPK/mTOR signaling in human prostate cancer
cells. Sci Rep. 5:83942015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang J, He H, Yu L, Xia HH, Lin MC, Gu Q,
Li M, Zou B, An X, Jiang B, et al: HSF1 down-regulates XAF1 through
transcriptional regulation. J Biol Chem. 281:2451–2459. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tu SP, Sun YW, Cui JT, Zou B, Lin MC, Gu
Q, Jiang SH, Kung HF, Korneluk RG and Wong BC: Tumor suppressor
XIAP-Associated factor 1 (XAF1) cooperates with tumor necrosis
factor-related apoptosis-inducing ligand to suppress colon cancer
growth and trigger tumor regression. Cancer. 116:1252–1263. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Straszewski-Chavez SL, Visintin IP,
Karassina N, Los G, Liston P, Halaban R, Fadiel A and Mor G: XAF1
mediates tumor necrosis factor-alpha-induced apoptosis and X-linked
inhibitor of apoptosis cleavage by acting through the mitochondrial
pathway. J Biol Chem. 282:13059–13072. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Park GB, Kim YS, Kim D, Kim S, Lee HK, Cho
DH, Lee WJ and Hur DY: Melphalan-induced apoptosis of
EBV-transformed B cells through upregulation of TAp73 and XAF1 and
nuclear import of XPA. J Immunol. 191:6281–6291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rigoulet M, Yoboue ED and Devin A:
Mitochondrial ROS generation and its regulation: Mechanisms
involved in H(2)O(2) signaling. Antioxid Redox Signal. 14:459–468.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang J, Lam GY and Brumell JH: Autophagy
signaling through reactive oxygen species. Antioxid Redox Signal.
14:2215–2231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu B, Tan X, Liang J, Wu S, Liu J, Zhang
Q and Zhu R: A reduction in reactive oxygen species contributes to
dihydromyricetin-induced apoptosis in human hepatocellular
carcinoma cells. Sci Rep. 4:70412014. View Article : Google Scholar : PubMed/NCBI
|