Introduction
Cervical cancer is a common malignant tumor that has
a detrimental impact on the life and health of women, particularly
in developing countries. Surgery and radiotherapy are used to treat
cervical cancer at stage IIA and earlier, whereas radiotherapy
together with platinum chemotherapy sensitization constitutes the
preferred treatment strategy for locally advanced (stage IIB and
later) cervical cancer (1).
Cisplatin is a potent chemotherapeutic agent that
has been used for decades to treat a variety of cancers, including
cervical, testicular, head and neck, ovarian and lung cancer.
Cisplatin interacts directly with multiple cellular components,
including proteins, thiol-containing peptides and nucleic acids
(2). Cisplatin-DNA adducts are
responsible for the majority of the cytotoxic effects of cisplatin,
distorting the DNA double helix, which is able to inhibit DNA
replication and transcription to lead to cell cycle accumulation
and apoptosis (2,3). The intra-strand cisplatin-DNA adducts
are repaired primarily by the nucleotide excision repair pathway
(4,5).
Irradiation is the primary therapy used for the
treatment of cancer, whereas X-ray irradiation is the principal
strategy in radiotherapy. X-ray irradiation induces a number of
types of DNA lesion. DNA double-strand breaks (DSBs) are generally
regarded as the most lethal of all DNA lesions following radiation
and are repaired by two major repair pathways: Homologous
recombination (HR) and non-homologous end-joining (NHEJ) (6). The ataxiatelangiectasia mutated protein
serves a main function in HR and is only a pre-emergency DSB repair
mechanism in human cells during late S, G2 and M phases,
as the sister chromatid for replication is required as a template
for repair (7). NHEJ is present at
all times during the entire cell cycle, especially in
G0/G1 and early S phases (7,8). The known
NHEJ proteins include DNA-dependent protein kinase (DNA-PK), and
ligase IV and its cofactor X-ray cross-complementary group 4
(XRCC4). DNA-PK is a serine/threonine kinase that contains a Ku
heterodimer (Ku70 and Ku80; Ku80 is also known as Ku86) and a
DNA-PK catalytic subunit (DNA-PKcs), and it is crucial in the
maintenance of telomere stability (9,10).
The combination of cisplatin and ionizing radiation
(IR) treatment represents a common modality for treating a variety
of types of cancer, and the underlying molecular mechanism of
cisplatin-sensitizing radiotherapy has been discussed previously:
Myint et al (11) demonstrated
that clinically applicable doses of cisplatin result in the
radiosensitization of mammalian cells due to the inhibition of the
function of NHEJ. Another previous study demonstrated that the
cisplatin-IR synergistic interaction requires the DNA-PK-dependent
NHEJ pathway to join DNA DSBs, and the presence of a cisplatin
lesion in the DNA inhibits this pathway (12). In the absence of a functional NHEJ
pathway, although the cells are hypersensitive to IR, there is no
synergistic interaction with cisplatin. The function of NHEJ and
even DNA-PKcs has been identified in the combination of cisplatin
and IR; however, the function of Ku80 in this synergy remains
largely undefined (13). The aim of
the present study was to investigate the mechanism of
radiosensitization of cisplatin by inhibiting the expression of
Ku80 using the previously developed cervical carcinoma cell model
HeLa with Ku80 silencing (14).
Materials and methods
Cell line and cell culture
The human cervical adenocarcinoma cell line HeLa was
obtained from the China Center for Type Culture Collection (Wuhan,
China). The HeLa/Ku80-siRNAcell line with Ku80 silenced by stable
transfection with Ku80-targeted small interfering RNA, and it was
confirmed that Ku80 protein expression was suppressed in the
Ku80-siRNA stable cell line in our previous study (14). The cells were cultured in Dulbeccos
modified Eagles medium (DMEM; Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Invitrogen; Thermo Fisher Scientific, Inc.), 50 U/ml
penicillin and 50 µg/ml streptomycin. All cells were maintained in
a humidified 37°C incubator containing 5% CO2, fed every
2-3 days with complete medium (containing 10% FBS).
Clonogenic survival assay
Cells were plated in triplicate on 60-mm dishes at
the required density to obtain between 50 and 100 colonies/dish and
were allowed to attach for 24 h. HeLa and HeLa/Ku80-siRNA cells
were exposed to 0, 2, 3, 4, 6 and 8 Gy X-ray radiation; the cells
were cultured for between 10 and 14 days in 5% CO2 to
obtain viable colonies. Colonies were stained with 0.5 ml 0.01%
crystal violet (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
solution at room temperature for 1 h and enumerated using a light
microscope (magnification, ×40). A viable colony was defined as
having at least 50 cells after 10 days of growth. Colonies were
counted from each triplicate sample and presented as the mean ±
standard deviation (SD). The surviving fraction of treated cells
was normalized to the plating efficiency of control
(non-irradiated) cells. The cell survival ratio was obtained by
means of clone formation. A one-hit multi-target model was fitted
to the cell survival curve to calculate the dose quasithreshold
(Dq), mean lethal dose (D0) and radiosensitivity parameter (N
value). Cell survival was also plotted as a function of dose and
fitted using the linear quadratic model SF=exp(−αD-βD2),
where SF is the cell survival, D is the radiation dose, and α and β
are constants. The surviving fraction of cells at 2 Gy (SF2) was
calculated from the actual data when the cells received 2 Gy
irradiation.
MTT assay to determine the
proliferation rates of cells following exposure to cisplatin
HeLa and HeLa/Ku80-siRNA cells at the exponential
phase of growth were plated in 96-well plates at a density of
1×104 cells/well and cultured in DMEM with 0, 0.5, 2, 5,
20 and 50 µg/ml cisplatin (Sigma-Aldrich; Merck KGaA) for 4 h. The
medium was replaced with cisplatin-free medium. In total, ~10 µl (5
mg/ml) MTT (Sigma-Aldrich; Merck KGaA) was added to the wells when
these cells were treated with cisplatin for 48 h and the plates
were incubated in a 37°C incubator for 4 h. The blue formazan dye
that had formed was dissolved in 150 µl dimethyl sulfoxide
(Sigma-Aldrich; Merck KGaA). The absorbance (A) at 570 nm was
recorded using an ELISA Multiskan reader (Thermo Fisher Scientific,
Inc.). The inhibition rate was calculated as follows: Inhibition
rate (%)=(1-A/A0h) ×100%. In addition, the 50%
inhibitory concentration (IC50) was defined as the
medicine concentration at which half of all cells were killed.
Cell treatment and cell cycle
analysis
The two cell lines were plated in 6-well plates at
3×105 cells/well, which corresponded to a density of
between 70 and 80% at the time of treatment. Treatment was divided
into three parts. In the first part, cells were exposed to 6 Gy 6
MV X-ray radiation (300c Gy/min). To determine the rate of
apoptosis and cell cycle distribution, cells were harvested
following irradiation for 1, 6, 12, 24, 48 and 72 h. In the second
part, cells were first cultured in DMEM with 5 µg/ml cisplatin for
4 h, then the medium was replaced with cisplatin-free medium, and
the cells were harvested for flow cytometric analysis following
treatment with cisplatin for 6, 12 and 24 h. In the third part, the
cells were exposed to X-ray radiation for 6 Gy, then treated with 5
µg/ml cisplatin (exposure for 4 h) for 23, 18 and 12 h following
irradiation for 1, 6 and 12 h. All the cells were exposed to X-ray
radiation and cisplatin for 24 h in total, prior to harvesting for
flow cytometric analysis. The harvested cells were washed with PBS
and fixed in 75% ice-cold ethanol overnight at −20°C. Following
washing in PBS again, the cells were treated with 50 µg/ml
propidium iodide and 1 mg/ml RNase A for 30 min at 37°C. Subsequent
analyses of the rate of apoptosis (sub-G1) and cell
cycle distribution were performed using CellQuest software (Version
5.1; BD Biosciences, Franklin Lakes, NJ, USA).
Microscopic imaging of
immunofluorescence staining
HeLa cells were grown on culture slides for two days
until the density increased to 50% at the time of treatment.
Following the aforementioned treatments, cells were washed in PBS,
fixed in 4% paraformaldehyde for 30 min, permeabilized in 0.5%
Triton X-100/PBS for 15 min and blocked in blocking buffer [1%
bovine serum albumin (BSA; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany)] in PBS) for 30 min. Immunostaining was performed using an
anti-phosphorylated histone H2AX (γH2AX; Ser139)
antibody (mouse anti-human, monoclonal antibody, catalog no.
05-636, EMD Millipore, Billerica, MA, USA, at a dilution of 1:500)
for 2 h at room temperature in a humidified chamber. Following
three 10-min washes, the cells were incubated with the goat
anti-mouse Alexa Fluor 568-conjugated secondary antibody
(Invitrogen; Thermo Fisher Scientific, Inc.; catalog no. A11004,
1:1,000 dilution). The DNA was stained using a mounting medium with
DAPI (Sigma-Aldrich; Merck KGaA). Immunofluorescence microscopic
imaging was performed using a LSM 510 laser-scanning confocal
microscope (Zeiss GmbH, Jena, Germany). All experiments were
performed at least three independent times.
Western blotting
For western blotting analyses, the cell pellets were
collected, and the total protein was isolated using M-PER™
mammalian protein extraction reagent (Pierce; Thermo Fisher
Scientific, Inc.). Protein concentrations were determined using a
bicinchonic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.), according to the manufacturers protocol. An
equal amount of total protein (30 µg) from each lysate was
separated by SDS-PAGE (6–10% gel). Subsequently, the separated
proteins were transferred onto nitrocellulose membranes, which were
then blocked with 5% powdered non-fat milk in Tris-buffered saline
containing Tween-20 (TBS-T) for 1 h at 37°C. Samples were incubated
with mouse anti-human antibodies against Ku80 (cat no. KuAb-2;
NeoMarkers, Inc., Portsmouth, NH, USA; at a dilution of 1:2,000),
γH2AX or β-actin (Santa Cruz Biotechnology, Inc., Dallas, TX, USA,
1:1,000 dilution) at 4°C overnight. Following washing with TBS-T
four times for 10 min each, the nitrocellulose membranes were
incubated with a goat anti-mouse secondary antibody (cat no.
ZB-2305; OriGene Technologies, Inc., Rockville, MD, USA; at a
dilution of 1:500) for 1 h at room temperature and washed with
TBS-T four times for 10 min each. Immunodetection was performed
using the SuperSignal West Fem to Maximum Sensitivity substrate
(Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
Results are presented as the mean ± SD of at least
three experiments, and a two-tailed unpaired Students t-test was
used to compare the statistical significance of the differences in
data from the two groups. A one-hit multi-target model
SF=1-(1-exp(-D0/D))N was fitted to the cell
survival curve to calculate Dq, D0 and N using SPSS software
(version 10.0; SPSS, Inc., Chicago, IL, USA). The linear quadratic
model SF=exp(−αD-βD2) was fitted to the cell survival
curve and α, β and SF2 were calculated from the actual data using
Sigma plot software (version 10.0; Systat Software Inc., Chicago,
IL, USA), which was also used for all statistical procedures.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Rates of apoptosis and the proportion
of HeLa cells in G2/M phase with Ku80 silencing
following 6 Gy X-ray irradiation
A dose of <4 Gy is insufficient to induce DSBs,
and a dose of >10 Gy is considered to be a lethal dose, prone to
induce cell death rather than DSBs; therefore, the majority of
researchers select a dose of between 4 and 10 Gy to irradiate
cells, and 6 Gy was considered to be a suitable dose for inducing
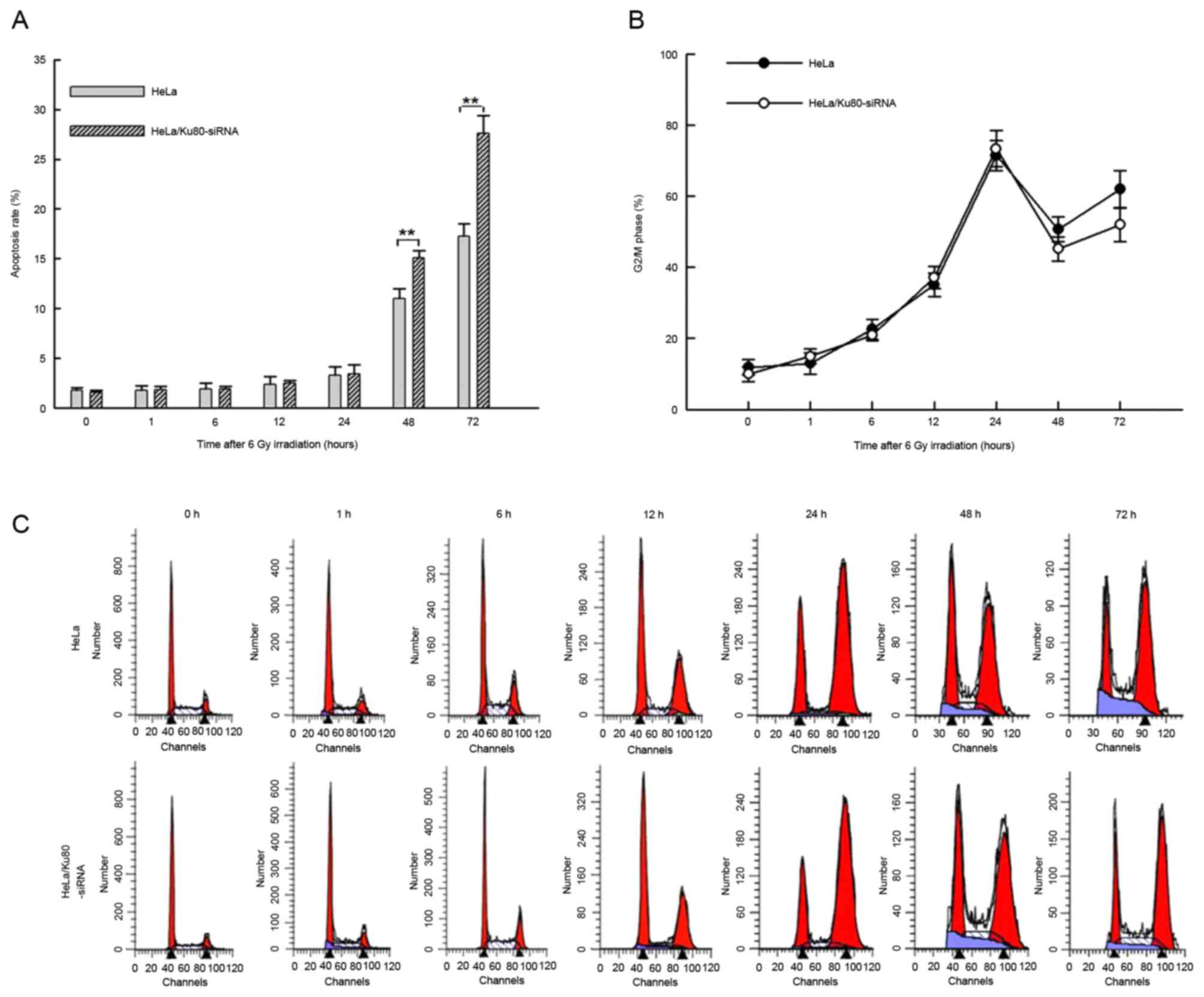
DSBs (15). Following 6 Gy IR for 1,
6, 12 and 24 h, HeLa/Ku80-siRNA and HeLa cells exhibited similar
rates of apoptosis (P>0.05), whereas at 48 and 72 h after
irradiation, the rates of apoptosis of HeLa/Ku80-siRNA cells were
increased compared with those of HeLa cells (48 h, t=6.293,
P=0.003; 72 h, t=8.282, P=0.001; Fig.
1A).
Following 6 Gy IR, the proportion of cells in
G2/M phase gradually increased at 1, 6, 12 and 24 h,
from ~15% following irradiation for 1 h to almost 70% following
irradiation for 24 h. However, no significant differences between
the two cell lines were identified at these time points (P>0.05;
Fig. 1B and C). The proportion of the
two cell lines in G2/M phase were at their peak at 24 h,
but decreased at 48 h and increased again at 72 h. The proportion
of HeLa/Ku80-siRNA cells in G2/M phase was decreased
compared with that of HeLa cells at 48 and 72 h, but no significant
difference between the two cell lines at these time points
following irradiation was identified (P>0.05; Fig. 1B and C).
Radiobiological analysis of HeLa
silencing of Ku80
SPSS and Sigma plot software were used to calculate
the radiobiological characteristics from the actual data (Table I). The SF2 and D0 values of
HeLa/Ku80-siRNA cells were decreased compared with that of HeLa
cells, which suggests that HeLa/Ku80-siRNAcells exhibited more
radiosensitivity than HeLa cells when Ku80 was silenced. The N
value and Dq value of HeLa/Ku80-siRNA cells were decreased compared
with those of the HeLa cells, indicating that when the shoulder
area of HeLa/Ku80-siRNA cells becomes smaller, the ability to
repair sub-lethal damage is weakened; in addition, the α value of
HeLa/Ku80-siRNA cells also increased significantly, which indicated
increased radiosensitivity of cells by inhibiting DNA DSB repair
induced by a single ionizing particle energy deposition. However,
the β value altered only minimally, which indicated that the
inhibition of Ku80 had little influence on the repair of DSBs
caused by double ionizing particle energy deposition.
 | Table I.Radiobiological parameters of cells
detected using a clonogenic survival assay. |
Table I.
Radiobiological parameters of cells
detected using a clonogenic survival assay.
| Cell line | D0,
Gy | N | Dq,
Gy | SF2 | α | β |
|---|
| HeLa |
1.411±0.102 |
2.917±0.178 |
1.510±0.115 |
0.548±0.020 |
0.144±0.017 |
0.078±0.006 |
|
HeLa/Ku80-siRNA |
1.117±0.060 |
2.017±0.087 |
0.783±0.040 |
0.307±0.004 |
0.444±0.008 |
0.073±0.003 |
Rate of apoptosis of cells following 6
Gy IR and/or treatment with 5 µg/ml cisplatin
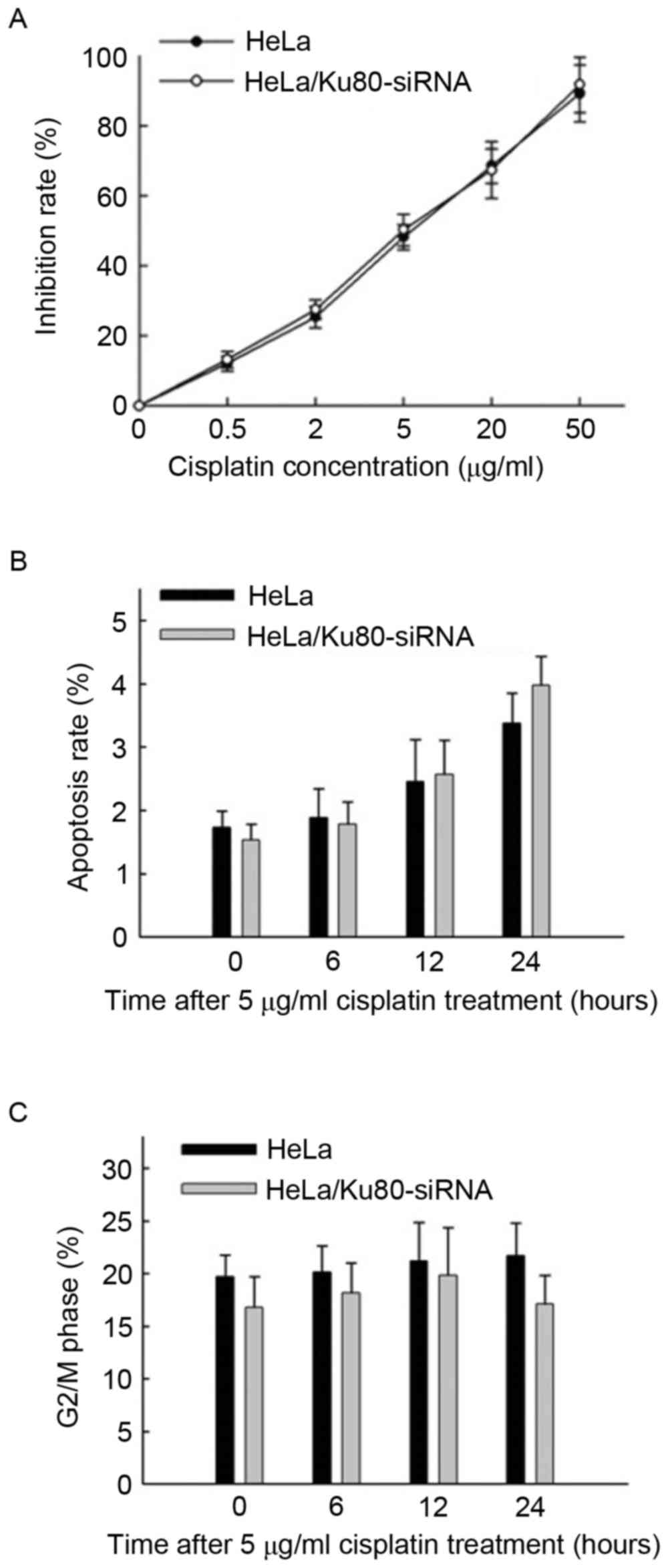
According to the MTT assay results (Fig. 2A), the IC50value of
cisplatin for the HeLa cells was (5.83±1.06) µg/ml, whereas that
for the HeLa/Ku80-siRNA cells was (5.17±0.99) µg/ml. No significant
difference in the IC50 value between the two cell lines
was identified (t=0.788, P=0.475), so the concentration of
cisplatin applied thereafter was 5 µg/ml, and the two cell lines
were exposed to cisplatin for 4 h prior to replacement with fresh
DMEM.
The rates of apoptosis of the two cell lines
increased slowly following treatment with 5 µg/ml cisplatin for 6,
12 and 24 h (Fig. 2B), and no marked
differences in cisplatin sensitivity between the two cell lines at
the different time points were identified (P>0.05).
The proportions of cells in the G2/M
phase from the two cell lines treated with 5 µg/ml cisplatin for 0,
6, 12 and 24 h were all ~20%, and no significant differences were
identified between the two cell lines at various time points
(P>0.05; Fig. 2C).
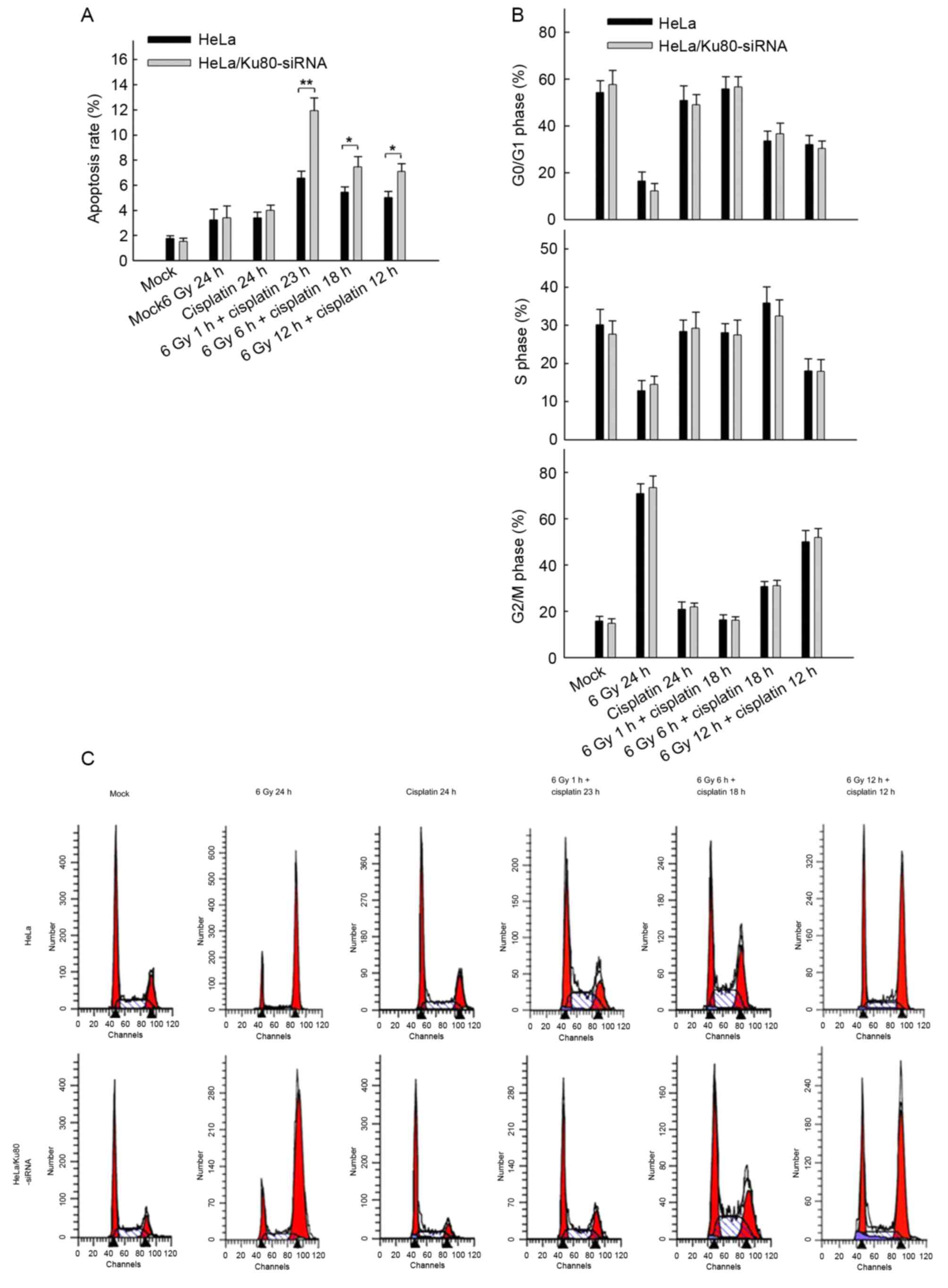
Nevertheless, following irradiation (6 Gy) and
treatment with 5 µg/ml cisplatin (4 h), the rates of apoptosis of
the two cell lines at all time points were distinct (Fig. 3A). First, when the two cell lines
received 6 Gy IR and cisplatin treatment in combination, they
exhibited increased rates of apoptosis compared with the cell lines
that only received 6 Gy IR or 5 µg/ml cisplatin alone (P<0.05).
Secondly, compared with normal HeLa cells, HeLa/Ku80-siRNAcells
exhibited increased rates of apoptosis, particularly following 6 Gy
IR for 1 h and treatment with 5 µg/ml cisplatin for 23 h. The rate
of apoptosis of HeLa/Ku80-siRNA cells was (11.92±1.02)%, which was
markedly increased compared with that of normal HeLa cells
(6.56±0.56)%, (t=7.637, P=0.002). The HeLa/Ku80-siRNA cells exposed
to 6 Gy irradiation for 6 h and treated with 5 µg/ml cisplatin for
18 h exhibited increased apoptosis compared with normal HeLa cells
that received the same treatment; their rates of apoptosis were
(7.44±0.84)% and (5.43±0.42)%, respectively (t=3.707, P=0.021).
Similar results were obtained when the two cell lines were exposed
to 6 Gy irradiation for 12 h and treated with 5 µg/ml cisplatin for
12 h. The rates of apoptosis of HeLa/Ku80-siRNA and HeLa cells were
(7.08±0.63) and (5.01±0.49)%, respectively (t=4.492, P=0.011).
These results revealed that, following Ku80 inhibition and 6 Gy IR,
the earlier the cisplatin was administered, the more apoptotic HeLa
cells appeared.
Alterations in the cell cycle
following 6 Gy IR and/or treatment with 5 µg/ml cisplatin
Fig. 3B and C present
the difference in the proportion of cells in the G2/M
phase in the two cell lines following receipt of different
treatments for 24 h. The proportion of G2/M phase cells
was 70% following a single X-ray irradiation (6 Gy) for 24 h,
whereas the proportion was only ~20% following the separate
administration of 5 µg/ml cisplatin for 24 h, similar to the cells
without any treatment, which suggested that cisplatin treatment for
24 h does not cause HeLa cells to undergo G2/M cell
cycle arrest. Following 6 Gy IR for 1 h plus cisplatin treatment
for 23 h, the proportion of G2/M phase cells remained
close to 20%. Following 6 Gy IR for 6 h plus cisplatin treatment
for 18 h, the proportion of cells in G2/M phase
increased to 30%, whereas following 6 Gy IR for 12 h plus cisplatin
treatment for 12 h, the proportion of cells in G2/M
phase further increased to ~50%. However, the differences between
the two groups were not significant (P>0.05).
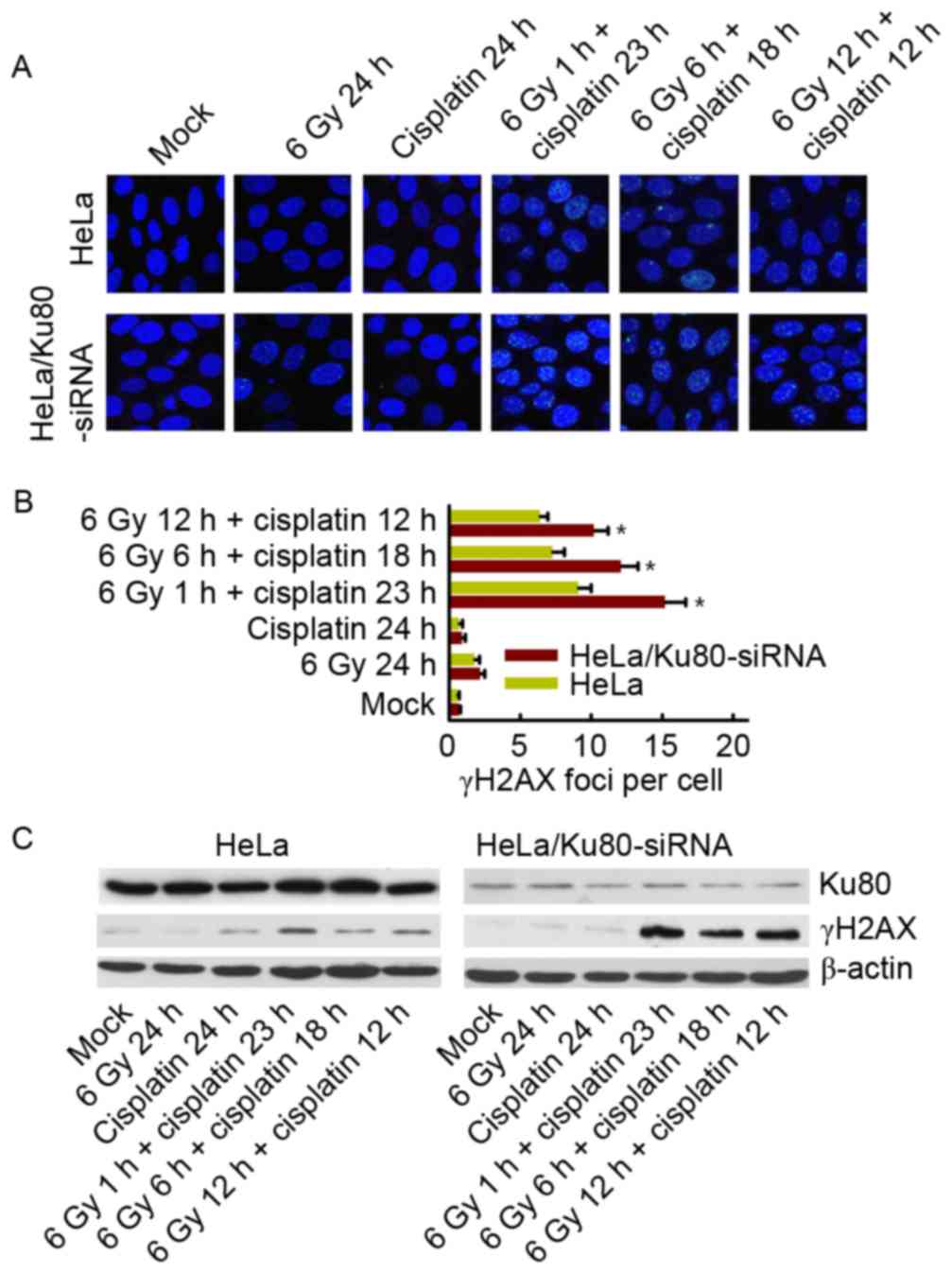
γH2AX phosphorylation in cells exposed
to 6 Gy IR and/or cisplatin
At 24 h after treatment with 6 Gy IR and/or
cisplatin, alterations in γH2AX phosphorylation were observed.
There was no difference between the two cell lines following 6 Gy
IR alone and cisplatin treatment alone; the two cell lines had few
γH2AX foci remaining. However, when treated with 6 Gy IR and
cisplatin, HeLa cells with Ku80 inhibition exhibited an increased
number of remaining γH2AX foci, particularly following 6 Gy IR for
1 h plus cisplatin for 23 h, as presented in Fig. 4A and B. Western blotting also revealed
the same results (Fig. 4C).
Discussion
The aim of the present study was to investigate the
effect of Ku80 on cisplatin radiosensitization in cervical cancer.
It was identified that irradiation in combination with cisplatin is
able to promote the apoptosis of HeLa cells with the inhibition of
Ku80, and the earlier cisplatin was administered following
irradiation (with in 6 h), the more apoptotic cells were induced.
This suggests that Ku80 may be a potent molecular target of
cisplatin radiosensitization, and the present study also provides
evidence of the appropriate timing for cisplatin to sensitize
radiotherapy.
DSBs are generally regarded as the most lethal of
all DNA lesions following radiation; it is known that the NHEJ
pathway serves a crucial function in repairing DSBs during
radiation in mammalian cells. When DSBs are produced, the Ku
heterodimer, which has a high affinity for DNA, is able to bind
preferentially to the free DNA ends, possibly to prevent nuclease
attack; subsequently, the conformation of Ku changes, allowing it
to interact with the DNA-PKcs-Artemis complex. Following
autophosphorylation, DNA-PKcs recruits the XRCC4-ligase IV complex
to process the DNA ends and to initiate re-ligation to form a
single DNA molecule (16). Ku80 is a
major gene for DNA lesion repair. In the present study, the HeLa
cell line with stable Ku80 inhibition using RNAi technology was
used to investigate the alterations in radiobiological
characteristics following IR. The results indicated that the rate
of apoptosis of HeLa/Ku80-siRNA cells following 6 Gy 6 MV X-ray
irradiation at 48 and 72 h increased, whereas the SF2 and
D0 values obtained from clonogenic survival assays
decreased. These results suggested that the HeLa cells with Ku80
inhibition become more radiosensitive, similar to the results of
previous studies (17–19).
When DNA is damaged by radiation, the cell will
initiate a cell cycle checkpoint signal transduction pathway.
Individual cells arrest in G1, S or G2/M
phase to repair DNA lesions. Following DNA damage, there are three
possible outcomes: i) Repair following injury and re-entry into the
normal cell cycle; ii) apoptosis, which occurs when DNA damage is
too severe and the cells cannot repair; and iii) tumor formation,
in which cells proceed beyond the repair of damaged DNA and
re-entry into the cell cycle (20).
In the present study, the cell cycle of HeLa cells with Ku80
silenced and normal HeLa cells was analyzed. Following X-ray
irradiation alone, the two cell lines appeared to be arrested in
G2/M phase, and the number of cells arrested in
G2/M phase gradually increased to reach a maximum at 24
h after irradiation, with a slight decline at 48 h and a second
peak at 72 h. It was observed that the proportion of cells in
G2/M phase at 72 h following 6 Gy irradiation was
increased compared with cells without irradiation. One possibility
is that 6 Gy is not a lethal dose which induces DSBs rather than
cell death. Upon irradiation, cells are prone to arrest in
G2/M phase to repair the DNA lesions and accumulate
mostly at 24 h. These cells gradually passed the G2/M
arrest point and, following the entire duration of the cell cycle,
became arrested in G2/M phase again at 72 h for
detection and repair of the DNA lesions. However, a number of cells
were not able to overcome the first G2/M arrest and
resulted in apoptosis, whereas other cells repaired their DNA
lesions completely and entered the normal cell cycle. Therefore,
the number of G2/M phase cells at 72 h decreased.
Furthermore, the proportion of HeLa/Ku80-siRNA cells in
G2/M arrest was decreased compared with that of HeLa
cells at 48 and 72 h after irradiation, presumably due to the
suppression of Ku80 and NHEJ repair pathway inhibition. These
processes result in DNA damage that is beyond repair and more cells
that undergo apoptosis, which is consistent with the increased rate
of apoptosis of HeLa/Ku80-siRNA cells at 48 and 72 h after 6 Gy
IR.
Cisplatin is one of the most widely used
chemotherapy agents to treat cancer, which is able to enhance the
local control ratio and decrease the rate of metastasis in
combination with radiotherapy in cervical cancer; however, its
underlying molecular mechanism of action is not fully understood
(21). The present study identified
that, compared with normal cells, HeLa cells lacking Ku80
expression exhibited no significant difference in the proliferation
inhibition and rate of apoptosis when 5 µg/ml cisplatin was
administered alone. This may be due to the lower level of DSBs
caused by this concentration of cisplatin, which requires only
limited or no NHEJ repair. Therefore, the use of cisplatin alone in
Ku80-silenced HeLa cells did not significantly increase the rates
of proliferation and apoptosis. Furthermore, the two cell lines
treated with X-ray irradiation and 5 µg/ml cisplatin (exposed for 4
h) in combination within 24 h were investigated. Following the
administration of 6 Gy 6 MV X-ray irradiation alone or 5 µg/ml
cisplatin (exposed for 4 h) alone for 24 h, whether or not the Ku80
protein was suppressed, the rate of apoptosis of the two cell lines
did not increase. However, the results were markedly different
following treatment with IR and cisplatin together. First, the rate
of apoptosis of cells following combination therapy was
significantly increased compared with that following X-ray
irradiation or cisplatin alone. Secondly, it was identified that
the earlier cisplatin was administered after X-ray irradiation
(e.g. 1 h after irradiation), the higher the rate of apoptosis was
achieved in HeLa cells with Ku80 inhibited. Additionally, the
alterations in the cell cycle revealed that the earlier cisplatin
was administered following irradiation, the more cells were
arrested in G0/G1 and S phase; therefore,
fewer cells were arrested in G2/M phase. A non-lethal
dose of irradiation was able to arrest the cell cycle in
G2/M phase to repair the DNA damage and induce cancer
cell survival and radioresistance, which is the main focus of the
present study. Whereas cisplatin treatment prior to irradiation was
able to prevent DNA damage repair and decrease the proportion of
cells in G2/M phase, particularly in DSB repair
activity-impaired HeLa cells with Ku80 suppression, and it was
identified that the earlier cisplatin was administered, the greater
the number of cells killed.
The phosphorylation of H2AX at Ser139 is
a critical event in the series of early responses to IR-induced DNA
DSBs (22). Because of its essential
function in the DSB response, γH2AX is considered to be a sensitive
biomarker for DSBs. In the present study, microscopic imaging of
immunofluorescence staining and Western blotting were used to
observe the change in γH2AX following various treatments. The
results identified that, 24 h after treatment with IR and
cisplatin, HeLa cells with Ku80 inhibition exhibited more γH2AX
foci, particularly in the group with 6 Gy IR for 1 h plus cisplatin
for 23 h. These results suggested that the earlier cisplatin was
administered following irradiation, the more DSBs remained. In this
regard, preliminary studies have also provided some explanations:
One demonstrated that Ku proteins (including Ku80 and Ku70)
participate in cisplatin-DNA adduct repair (23); two other studies have identified that
cisplatin exhibits its sensitizing effect due to the inhibition of
the NHEJ pathway and by preventing DNA-PKcs protein
phosphorylation, which resulted in a decrease in DSB repair
(11,12). A previous study also identified that
cell killing by cisplatin occurs in a cell-autonomous manner by
means of the formation of platinum-DNA adducts that, if not removed
by DNA repair, inhibit transcription and replication, and damaged
cells are able to transmit a death signal to neighboring cells
(24). This signal produced within
the cell is damaged by the kinase function of the Ku70, Ku80 and
DNA-PK complex, and is conveyed to the recipient cell by direct
cell-to-cell communication through gap junctions. However, these
results only partially explained why cisplatin was able to
sensitize cells to irradiation and did not explain why the earlier
cisplatin is administered, the stronger the killing effect
following X-ray irradiation. We hypothesize that the DNA damage of
HeLa cells induced by X-ray irradiation with cisplatin formed the
cisplatin-damaged DNA adducts to promptly repair the DNA damage
(such as DSBs) in G1 and S phase, as the vast majority
of DNA damage produced by irradiation is repaired within 6 h of
radiotherapy (9). Therefore, the
earlier cisplatin is administered within 6 h, the greater the
number of DSBs held in G1 and S phase for repair. The
lack of Ku80 in HeLa cells with a damaged NHEJ repair pathway
resulted in a larger proportion of apoptotic cells compared with
normal HeLa cells. However, this hypothesis requires further
investigation at the molecular level.
The results of the present study suggest that when
patients receive either radiotherapy alone or combined cisplatin
and radiotherapy, Ku80 was able to become a latent target for
molecular therapeutics. A previous study demonstrated that Ku80 is
highly expressed in a variety of tumor tissues, including cervical
cancer (25). A number of studies not
only noted that increased expression of Ku proteins indicated a
poor prognosis in cervical cancer, head and neck cancer,
hypopharyngeal cancer, lung cancer and other tumors, but also
clinically demonstrated that cisplatin in combination with
radiation is the principal treatment for these tumors (26–29).
Therefore, inhibiting Ku80 may become a new therapeutic approach to
promote the effective treatment of Ku80-expressingtumors (such as
cervical cancer) combined with radiation and cisplatin. More
importantly, the results of the present study suggest that
cisplatin is able to maximize tumor killing as soon as possible
after radiation and provides evidence of the appropriate timing for
the administration of cisplatin to sensitize radiotherapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81372434 and
81372664).
References
|
1
|
Petera J, Sirak I, Beranek M, Vosmik M,
Drastikova M, Paulikova S and Soumarova R: Molecular predictive
factors of outcome of radiotherapy in cervical cancer. Neoplasma.
58:469–475. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kartalou M and Essigmann JM: Mechanisms of
resistance to cisplatin. Mutat Res. 478:23–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu X, Fan W, Xu S and Zhou Y:
Sensitization to the cytotoxicity of cisplatin by transfection with
nucleotide excision repair gene xeroderma pigmentosun group a
antisense RNA in human lung adenocarcinoma cells. Clin Cancer Res.
9:5874–5879. 2003.PubMed/NCBI
|
|
5
|
Guggenheim ER, Xu D, Zhang CX, Chang PV
and Lippard SJ: Photoaffinity isolation and identification of
proteins in cancer cell extracts that bind to platinum-modified
DNA. Chembiochem. 10:141–157. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang C and Lees-Miller SP: Detection and
repair of ionizing radiation-induced DNA double strand breaks: New
developments in nonhomologous end joining. Int J Radiat Oncol Biol
Phys. 86:440–449. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Collis SJ, DeWeese TL, Jeggo PA and Parker
AR: The life and death of DNA-PK. Oncogene. 24:949–961. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burma S and Chen DJ: Role of DNA-PK in the
cellular response to DNA double-strand breaks. DNA Repair (Amst).
3:909–918. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lieber MR, Ma Y, Pannicke U and Schwarz K:
Mechanism and regulation of human non-homologous DNA end-joining.
Nat Rev Mol Cell Biol. 4:712–720. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goodwin JF and Knudsen KE: Beyond DNA
repair: DNA-PK function in cancer. Cancer Discov. 4:1126–1139.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Myint WK, Ng C and Raaphorst GP: Examining
the non-homologous repair process following cisplatin and radiation
treatments. Int J Radiat Biol. 78:417–424. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boeckman HJ, Trego KS and Turchi JJ:
Cisplatin sensitizes cancer cells to ionizing radiation via
inhibition of nonhomologous end joining. Mol Cancer Res. 3:277–285.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sears CR and Turchi JJ: Complex
cisplatin-double strand break (DSB) lesions directly impair
cellular non-homologous end-joining (NHEJ) independent of
downstream damage response (DDR) pathways. J Biol Chem.
287:24263–24272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhuang L, Yu SY, Huang XY, Gao QL, Xiong H
and Leng Y: Effect of Ku80 expression inhibition by RNA
interference on proliferation of cervical carcinoma cell line HeLa.
Ai Zheng. 26:252–257. 2007.PubMed/NCBI
|
|
15
|
Jayakumar S, Bhilwade HN, Pandey BN,
Sandur SK and Chaubey RC: The potential value of the neutral comet
assay and the expression of genes associated with DNA damage in
assessing the radiosensitivity of tumor cells. Mutat Res.
748:52–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Valerie K and Povirk LF: Regulation and
mechanisms of mammalian double-strand break repair. Oncogene.
22:5792–5812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Williams GJ, Hammel M, Radhakrishnan SK,
Ramsden D, Lees-Miller SP and Tainer JA: Structural insights into
NHEJ: Building up an integrated picture of the dynamic DSB repair
super complex, one component and interaction at a time. DNA Repair
(Amst). 17:110–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gullo C, Au M, Feng G and Teoh G: The
biology of Ku and its potential oncogenic role incancer. Biochim
Biophys Acta. 1765:223–234. 2006.PubMed/NCBI
|
|
19
|
Zhuang L, Cao Y, Xiong H, Gao Q, Cao Z,
Liu F, Qiu H, Yu S and Huang X: Suppression of DNA-PKcs and Ku80
individually and in combination: Different effects of radiobiology
in HeLa cells. Int J Oncol. 39:443–451. 2011.PubMed/NCBI
|
|
20
|
Melo J and Toczyski D: A unified view of
the DNA-damage checkpoint. Curr Opin Cell Biol. 14:237–245. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wieringa HW, van der Zee AG, de Vries EG
and van Vugt MA: Breaking the DNA damage response to improve
cervical cancer treatment. Cancer Treat Rev. 42:30–40. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fillingham J, Keogh MC and Krogan NJ:
GammaH2AX and its role in DNA double-strand break repair. Biochem
Cell Biol. 84:568–577. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Turchi JJ and Henkels K: Human Ku
autoantigen binds cisplatin-damaged DNA but fails to stimulate
human DNA-activated protein kinase. J Biol Chem. 271:13861–13867.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jensen R and Glazer PM:
Cell-interdependent cisplatin killing by Ku/DNA-dependent protein
kinase signaling transduced through gap junctions. Proc Natl Acad
Sci USA. 101:pp. 6134–6139. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moll U, Lau R, Sypes MA, Gupta MM and
Anderson CW: DNA-PK, the DNA-activated protein kinase, is
differentially expressed in normal and malignant human tissues.
Oncogene. 18:3114–3126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Harima Y, Sawada S, Miyazaki Y, Kin K,
Ishihara H, Imamura M, Sougawa M, Shikata N and Ohnishi T:
Expression of Ku80 in cervical cancer correlates with response to
radiotherapy and survival. Am J Clin Oncol. 26:e80–e85. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pavón MA, Parreño M, León X, Sancho FJ,
Céspedes MV, Casanova I, Lopez-Pousa A, Mangues MA, Quer M,
Barnadas A and Mangues R: Ku70 predicts response and primary tumor
recurrence after therapy in locally advanced head and neck cancer.
Int J Cancer. 123:1068–1079. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hayashi J, Sakata KI, Someya M, Matsumoto
Y, Satoh M, Nakata K, Hori M, Takagi M, Kondoh A, Himi T and
Hareyama M: Analysis and results of Ku and XRCC4 expression in
hypopharyngeal cancer tissues treated with chemoradiotherapy. Oncol
Lett. 4:151–155. 2012.PubMed/NCBI
|
|
29
|
Ma Q, Li P, Xu M, Yin J, Su Z, Li W and
Zhang J: Ku80 is highly expressed in lung adenocarcinoma and
promotes cisplatin resistance. J Exp Clin Cancer Res. 31:992012.
View Article : Google Scholar : PubMed/NCBI
|