Introduction
Mitochondria serve a crucial role in the
determination of cellular life and death. Mitochondria are
essential for energy production, as most of the cellular adenosine
triphosphate (ATP) is produced by the oxidative phosphorylation
pathway in the mitochondrial matrix (1). Mitochondria are fundamental players in
numerous cellular activities, which include cellular signaling,
generation and regulation of reactive oxygen species (ROS) levels,
buffering cytosolic calcium levels and regulation of apoptosis via
the mitochondrial permeability transition pore (mtPTP) (2,3).
Mitochondrial dysfunction has been implicated in metabolic
diseases, including cancer and aging (4–6).
Therefore, mitochondrial quality control and function integrity are
essential for cellular homeostasis.
Accumulating evidence has revealed that damaged
mitochondria can be selectively removed by mitophagy, which is a
specialized form of autophagy. During mitophagy, specific
regulators, including phosphatase and tensin homolog-induced
putative kinase 1 (PINK1) and Parkin, ensure selective
sequestration of individual mitochondria as cargo (7). Mitochondrial depolarization, caused by
the loss of mitochondrial membrane potential, may trigger
mitophagy. During mitophagy, PINK1 and the E3 ubiquitin ligase
Parkin accumulate on damaged mitochondria and thereby facilitate
their segregation from the mitochondrial network. PINK1 and Parkin
then target and facilitate ubiquitination of mitochondrial proteins
that interact with various components of the core autophagy
machinery (8–10). A double-membrane structure, known as
the autophagosome, is then formed and fuses with the lysosome to
form an autolysosome, in which the clearance of mitochondria will
occur (7).
TIGAR has been determined as a
fructose-2,6-bisphosphatase that lowers glycolytic flux and
promotes antioxidant function. The reduction of glycolytic flux
promotes the pentose phosphate pathway (PPP), which generates NADPH
and increases the level of reduced glutathione, which aids to limit
ROS production (11). The
downregulation of TIGAR is linked to decreased levels of NADPH,
lower levels of reduced glutathione, and consequently an increase
in endogenous ROS (12). ROS,
including superoxide (O2−), hydrogen peroxide
(H2O2) and hydroxyl radical (OH−)
(13), can attack mitochondria and
induce mitochondrial DNA mutation and damage to cellular DNA and
proteins (6).
In the present study, it was hypothesized that the
expression of TIGAR in 5–8F nasopharyngeal carcinoma cells may
affect mitochondrial integrity and degradation. Therefore, the role
of TIGAR on mitophagy was investigated by transfecting 5–8F cells
with lentivirus-mediated small hairpin RNA (lenti-shRNA) that
targets the TIGAR gene to knockdown the expression of TIGAR. In the
present study, it was determined that the knockdown of TIGAR
initiates mitophagy that is associated with loss of mitochondrial
membrane potential and cytochrome c leakage. In addition, in the
present study, it was revealed that the knockdown of TIGAR prevents
mitochondrial degradation, as indicated by the increase in
mitochondrial mass due to swollen mitochondria.
Materials and methods
Cell lines and reagents
The 5–8F human nasopharyngeal carcinoma cell line
was obtained from the American Type Culture Collection (Manassas,
VA, USA). The cell routinely maintained in RPMI-1,640 medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (GE Healthcare Life
Sciences, Little Chalfont, UK), 100 U/ml penicillin and 100 µg/ml
streptomycin (Beyotime Institute of Biotechnology, Haimen, China)
at 37°C in 5% CO2. The cells were treated with
Cyclosporin A (CsA; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
or protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP;
Sigma-Aldrich; Merck KGaA) for 2 h in 37°C, prior to detection at a
final concentration of 5 and 10 µM, respectively.
Lenti-shRNA against TIGAR
The lenti-shRNA vector system against TIGAR was
constructed, packed, and purified by GeneChem, Inc. (Shanghai,
China) according to the protocol of the manufacturer. The shRNA
oligonucleotides were designed as sh-TIGAR
(5′-GCCAGCTTTACTGGAGAACTT-3′). A scramble sequence (sh-scramble)
was synthesized and served as a transfection control
(5′-TTACCGAGACCGTACGTAT-3′). The virus titer was measured by
GeneChem, Inc. To experimental measurement multiplicities of
infection (MOI), four groups were created: MOI 1, 10, 50 and 100,
respectively. Cells were transfected in complete medium (Gibco;
Thermo Fisher Scientific, Inc.) with 5 µg/ml polybrene (cat no.
REVG0001; GeneChem, Inc.) and 0.5, 5, 25 and 50 µl-viral particles
respectively for 72 h at 37°C. Then, the medium was replaced and
the transfected cells were cultured with puromycin (Sigma-Aldrich;
Merck KGaA) for 3–5 days in 37°C. Subsequently, the cells with
higher survival rate and better cell state were selected using
light microscopy (Leica DMLS; Leica Microsystems) at ×200
magnification, which corresponded to a suitable MOI value. The
experiment revealed the most suitable MOI was 10. Following
transduction of 5–8F cells, colonies expressing a stable shRNA were
selected using puromycin (Sigma-Aldrich; Merck KGaA), according to
the manufacturer's protocol.
Western blotting
Total proteins from cells were extracted by
radioimmunoprecipitation buffer with protease inhibitor (Beyotime
Institute of Biotechnology, Haimen, China), then proteins (20–40 µg
per lane) were separated on an SDS-PAGE gel (separating gel, 10 or
12%; stacking gel, 5%), then proteins were transferred to a
polyvinylidene difluoride membrane (GE Healthcare, Chicago, IL,
USA). Followed by blocking with 5% milk at room temperature for 1 h
and then incubation with the following antibodies: anti-TIGAR
(1:400; cat no. sc-166290; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA); anti-GAPDH (1:1,000; cat no. 2118; Cell Signaling
Technology, Inc., Danvers, MA, USA); anti-GRP75 (1:1,000; cat no.
14887-1-AP; ProteinTech Group, Inc., Chicago, IL, USA);
anti-Cytochrome c (1:200; cat no. AC909; Beyotime Institute of
Biotechnology, Haimen, China) overnight at 4°C. The next day,
membranes were washed with TBST and incubated with goat
anti-rabbit, horseradish peroxidase-conjugated secondary antibodies
(1:3,000; cat no. SA00001-2; ProteinTech Group, Inc., Chicago, IL,
USA) for 1-2 h at room temperature. Then, the
electrochemiluminescence peroxidase substrate (catalog no.,
WBLUR0100; EMD Millipore, Billerica, Ma, USA) was used (GE
Healthcare Life Sciences) for the detection of immunoblotting on
the ChemiDoc™ MP Imaging System (Bio-Rad Laboratories,
Inc.). The densitometry was analysed by the Image Lab™
software (Version 5.2.1, Bio-Rad Laboratories, Inc.).
Immunofluorescence analysis
The cells were seeded on a coverslip in a 24-well
plate at a density of 2×104 cells/well. In brief, the
cells were fixed with 4% paraformaldehyde for 10 min at room
temperature, then blocked with 3% goat serum (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) for 1 h at room
temperature and incubated with a primary anti-TIGAR mouse antibody
(1:200; cat no. sc-166290; Santa Cruz Biotechnology, Inc.) and
anti- mitochondrial heat shock protein 70 (mtHSP70) rabbit antibody
(1:400; cat no; 14887-1-AP; ProteinTech Group, Inc.) overnight at
4°C. The cells were washed twice in phosphate-buffered saline (PBS)
and incubated with Cy3-labeled goat anti-mouse IgG (H+L) and
Cy2-labeled goat anti-rabbit IgG (H+L) antibodies (1:400; cat nos.
A0521 and A0423; Beyotime Institute of Biotechnology) for 1 h at
room temperature. Then cells were washed twice in PBS and observed
under a fluorescence microscope.
Flowcytometry
For flow cytometry experiments, the cells were
seeded in a 6-well plate at a density of 15×104
cells/well. A total of 24 h after the start of inoculation, the
medium was replaced with Hank's balanced salt solution-containing
Mito-Tracker Green (200 nM; Beyotime Institute of Biotechnology,
Haimen, China). Following incubation for 30 min at 37°C, the cells
were washed twice with PBS and resuspended in 500 µl PBS prior to
analysis by flow cytometry (FACSVerse; FACSuite™ v. 1.0.3.2942
software; BD Biosciences, Franklin Lakes, NJ, USA).
Mitochondrial membrane potential
assay
For mitochondrial membrane potential assays, the
cells were seeded in a 6-well plate at a density of
15×104 cells/well. A total of 24 h after the start of
the inoculation, the cells were stained for 30 min with 15
µmol/l5,5′, 6,6′-tetrachloro-1,1′,
3,3′-tetraethyl-imidacarbocyanineiodide (JC-1; Beyotime Institute
of Biotechnology) in cell culture media at 37°C followed by two
washes with staining buffer (Beyotime Institute of Biotechnology)
prior to analysis using a fluorescence microscope (magnification,
×400). The ratio between green and red fluorescence signals served
as a parameter for the mitochondrial membrane potential
(ΔΨm) and is independent of the mitochondrial mass.
High-performance liquid chromatography
analysis (HPLC)
HPLC (Agilent Technologies, Inc., Santa Clara, CA,
USA) was performed to determine the levels of ATP. The reverse
phase consisted of a C18 column (4.6×150 mm; particle size, 3.5 m).
The mobile phase comprised buffer A (15 mM
KH2PO4) and buffer B and methanol (90:10
ratio). A total of 20 µl sample or standard was injected into the
column. The flow rate was 0.5 ml/min, and column temperature was
25°C. The samples were analyzed by ultra violet detection at 254
nm.
Transmission electron microscopy
The cells were pre-fixed with a mixed solution of 3%
glutaraldehyde, then post-fixed in 1% osmium tetroxide, dehydrated
in a series of acetone (50, 70, 80 and 90% acetone each for 15 min
then 100% twice for20 min), infiltrated and embedded in EM bed812
(Electron Microscopy Sciences, Inc., Hatfield, PA, USA) at 37°C for
12 h, 45°C for 12 h and 60°C for 24 h. The semi-thin sections (5-µm
thick) were stained with methylene blue and ultra-thin sections
(50-nm thick) were cut using a diamond knife, stained with uranyl
acetate for 30 min followed by lead citrate for 10 min at room
temperature. The sections were examined using a transmission
electron microscope (Hitachi, Ltd., Tokyo, Japan; H-600IV).
Statistical analysis
All experiments were performed at least in
triplicate. The results are expressed as the mean ± standard
deviation. Comparisons of flow cytometry results were performed
using two-way analysis of variance and post-hoc Tukey's test.
Comparisons of ATP content were performed using a paired Student's
t-test. Statistical analysis was performed using SPSS 16.0 software
(SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 5.0 (GraphPad
Software, Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
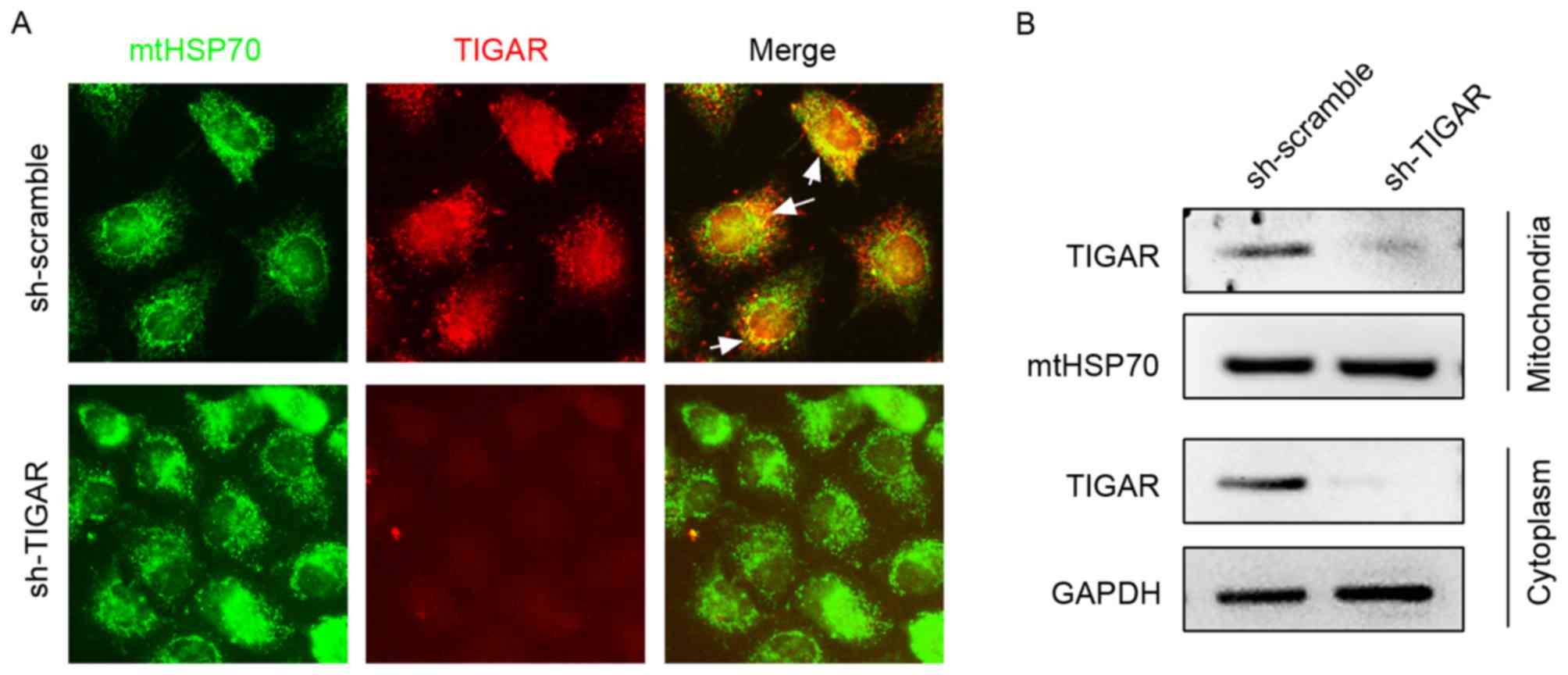
TIGAR localizes to mitochondria in
5-8F cells
In the present study, the expression pattern of
TIGAR in 5-8F cells was detected using immunofluorescence staining,
which demonstrated its partial localization on mitochondria marked
by mtHSP70 (Fig. 1A). To confirm
mitochondrial and cytoplasmic localization, mitochondrial and
cytoplasmic proteins were separated and subjected to western blot
analysis (Fig. 1B). To investigate
the function of TIGAR on mitochondrial function, lentivirus was
used to introduce shRNA (lenti-shRNA), which targets the TIGAR gene
into 5-8F cells, and consequently marked reduction in the levels of
TIGAR was observed in the mitochondria and cytoplasm, which
strongly supported mitochondrial and cytoplasmic localization of
TIGAR (Fig. 1).
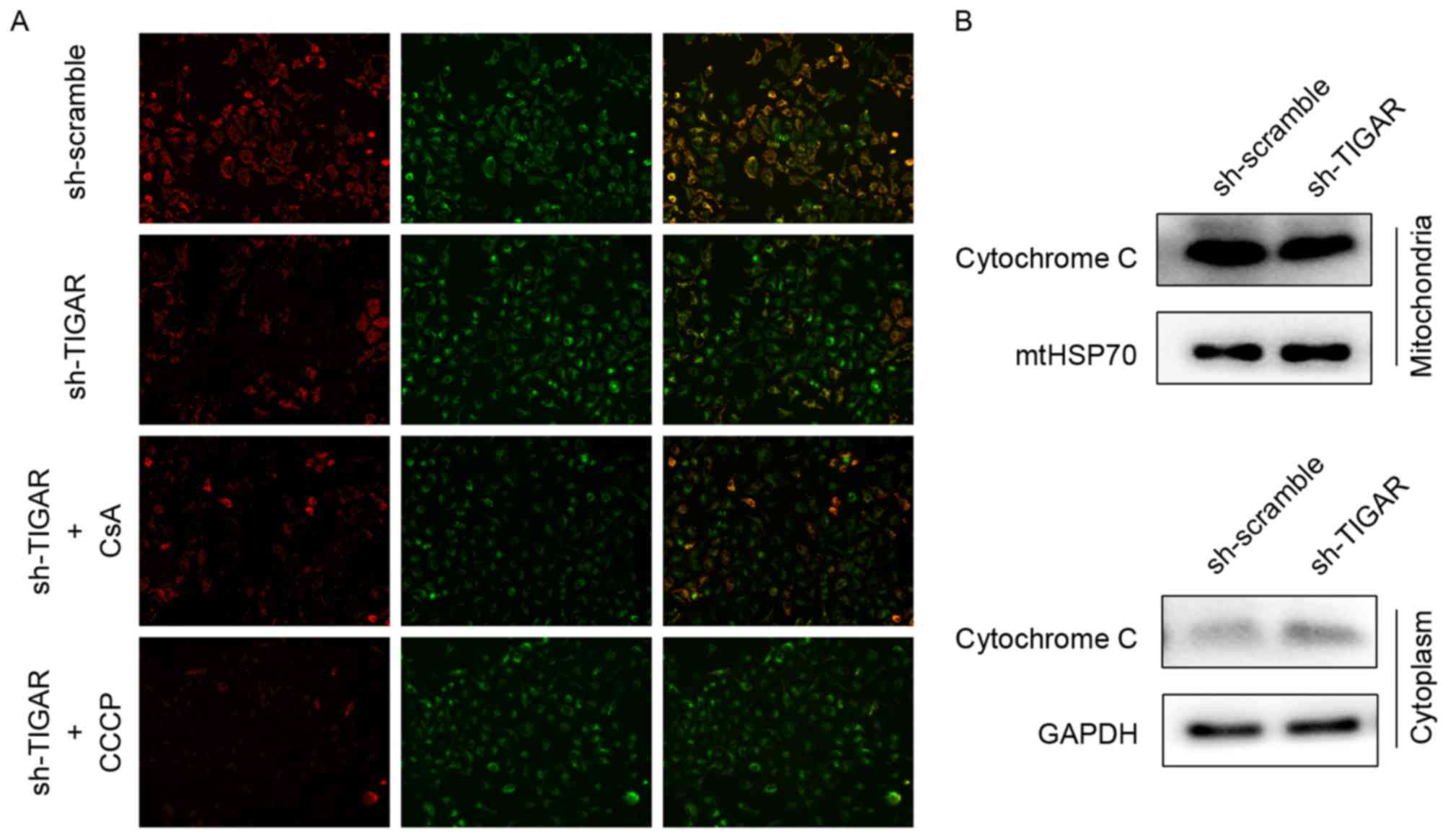
Knockdown of TIGAR leads to loss of ΔΨ
min 5-8F cells
Decreased ΔΨm can be detected by ΔΨm-dependent
mitochondrial fluorescent probe, JC-1, and is a sensitive indicator
of mitochondrial damage and dysfunction. In normal conditions, JC-1
aggregates to mitochondria and reflects red fluorescence, whereas
it transfers to the cytoplasm as monomers and reflects green
fluorescence when the ΔΨm is decreased. Therefore, ΔΨm can be
reflected by the transition in fluorescence. Upon knockdown of
TIGAR in 5-8F cells, the ratio of red to green fluorescence
decreased, indicating a reduction in ΔΨm. CsA, acalcineurin
inhibitor that has the ability to maintain ΔΨm, was added to rescue
the reduction in ΔΨm. The CCCP was used as a positive control to
reduce ΔΨm in the present study (Fig.
2A).
When ΔΨm is reduced, the mtPTPs open, which leads to
the leakage of mitochondrial content, including cytochrome c
(14). In the present study, the
levels of cytochrome c following ΔΨm reduction in respective
mitochondrial and cytoplasmic proteins were analyzed by western
blot analysis. Knockdown of TIGAR led to increased levels of
cytochrome c in the cytoplasmic fraction compared with control
group, suggesting a mitochondrial leak (Fig. 2B).
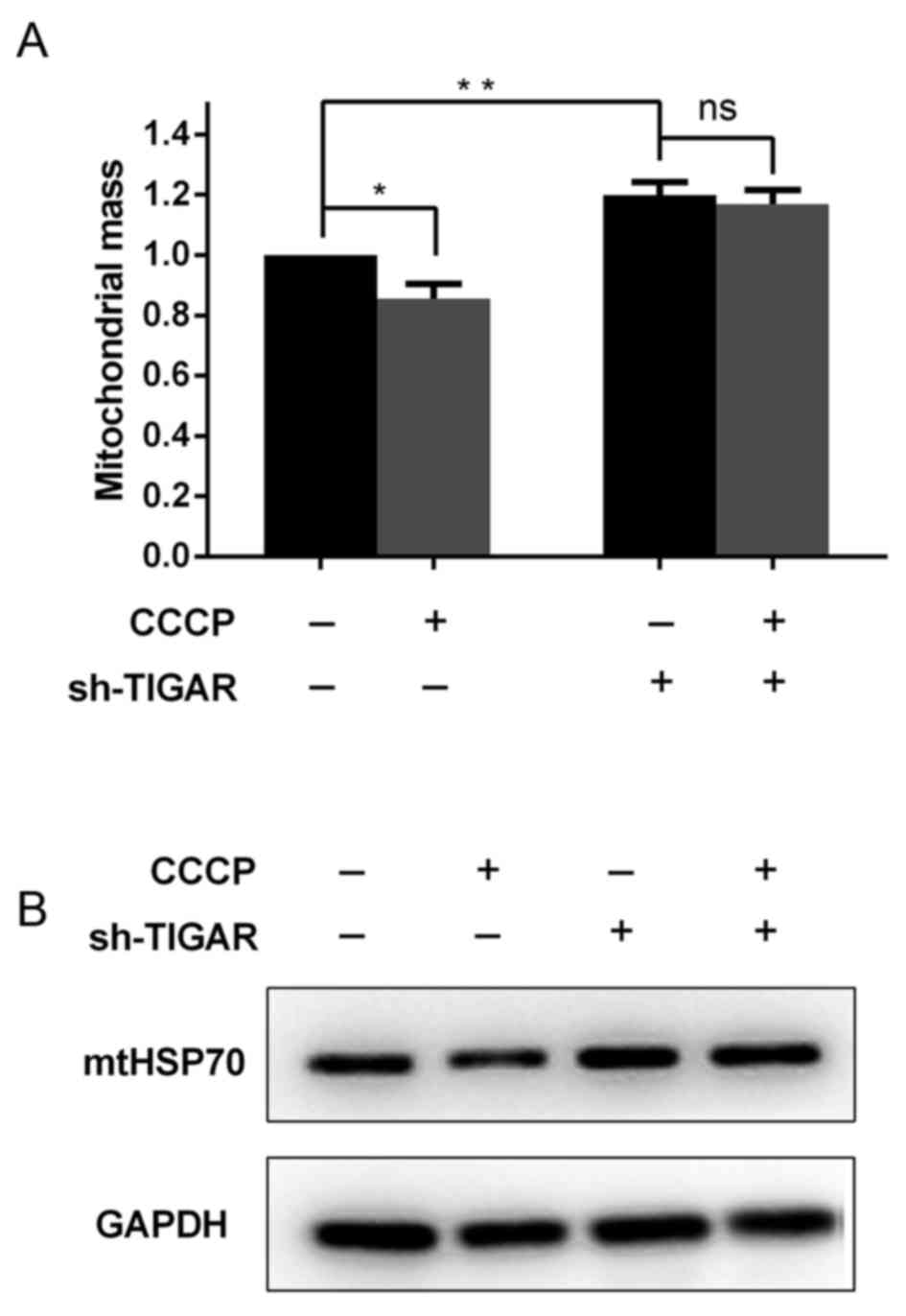
Knockdown of TIGAR increases
mitochondrial mass by preventing mitochondrial degradation
In addition to the reduction in ΔΨm, it was further
investigated in the present study whether mitochondrial mass was
affected in TIGAR-knocked down cells. For these analyses,
Mito-tracker Green, a dye that stains mitochondria independent of
the ΔΨm, was used as aforementioned. Notably, it was indicated that
the knockdown of TIGAR was able to increase the mitochondrial mass,
regardless of the reduced ΔΨm (Fig.
3A). To confirm this result, whole cell lysates were extracted
from 5-8F cells to determine the levels of the mitochondrial
housekeeping protein, mtHSP70, which can reflect the mitochondrial
mass in these cells. It was demonstrated that in TIGAR-knocked down
cells, the level of mtHSP70 increased compared with the control
cells (Fig. 3B), which was associated
with the increase in mitochondrial mass observed (Fig. 3A).
To investigate whether degradation of damaged
mitochondria was affected in TIGAR-knocked down cells, CCCP was
added to 5-8F cells to induce mitochondrial damage and degradation.
Following a 2 h incubation with CCCP, mitochondrial mass was
significantly decreased in the control cells, whereas in
TIGAR-knocked down cells mitochondrial mass was maintained
(Fig. 3A and B). This suggested that
TIGAR serves a crucial role in mitochondrial degradation.
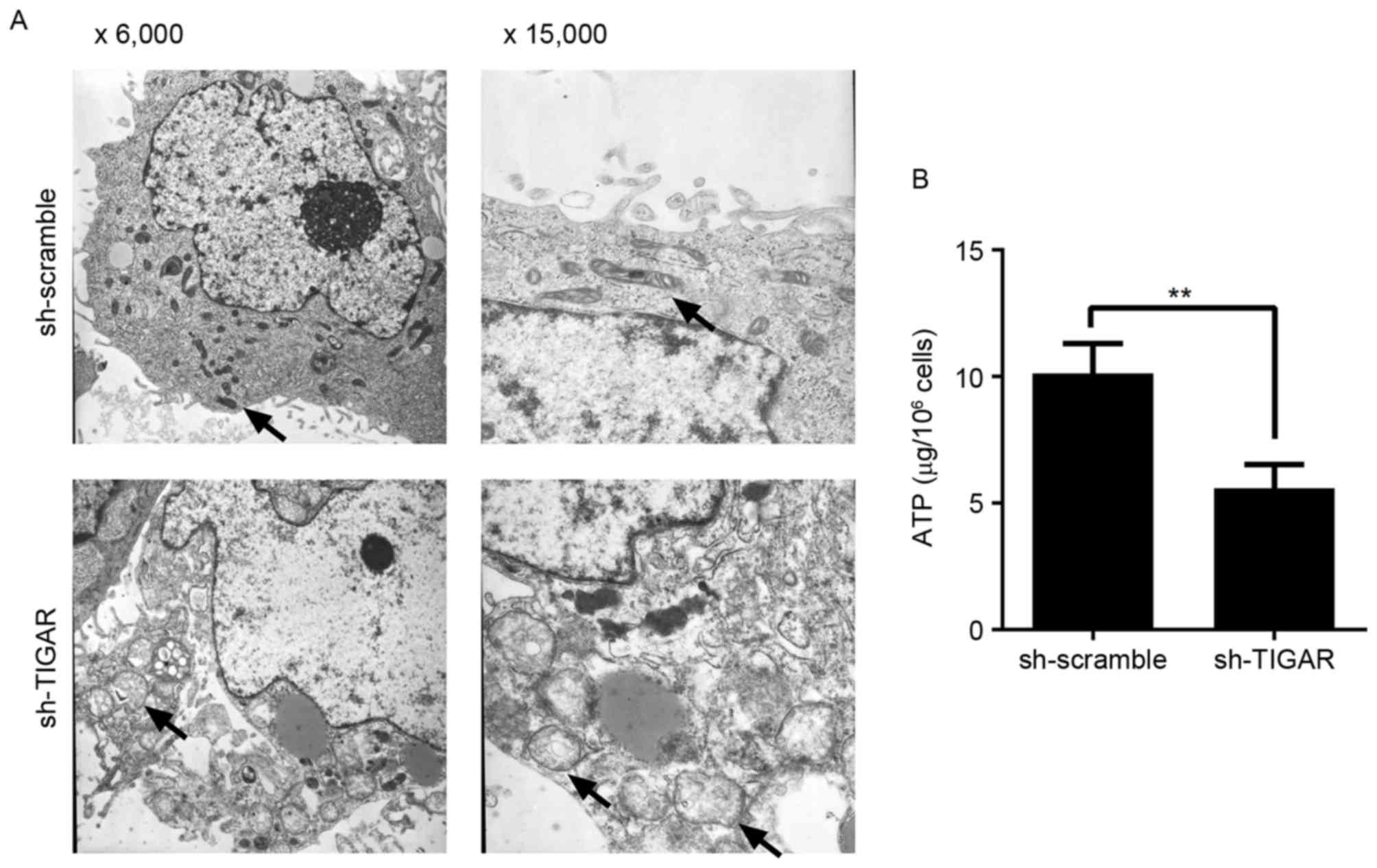
Knockdown of TIGAR affects
mitochondrial structure and physiological function
In the present study, reduction in ΔΨm was able to
prevent degradation of mitochondria in 5-8F cells. In addition, to
detect the integrity of mitochondria, the ultrastructure of
mitochondria was analyzed by transmission electron microscopy. It
was demonstrated that in TIGAR-knocked down cells, the mitochondria
had an abnormal appearance and were observed to exhibit
characteristics, including mitochondrial swelling, crista collapse
and vacuolization (Fig. 4A).
Furthermore, the ability to produce ATP was affected in these
cells, indicating physiological dysfunction (Fig. 4B).
Discussion
TIGAR has the ability to limit fructose-2,
6-bisphosphatase levels, which results in a shift from glycolysis
to PPP (11). NADPH, as the product
of the PPP pathway, serves an important role in ROS scavenging
(11). As expected, a number of
previous studies have reported that the knockdown of TIGAR is able
to decrease NADPH production and reduce glutathione, thus
contributing to ROS accumulation (12,15,16).
Simultaneously, dysregulated mitochondrial activity resulted in
production of ROS as a by-product (17,18). In
the present study, it was detected that the loss of TIGAR leads to
increased ROS levels (data not shown), which may cause irreversible
damage to bio macromolecules and organelles (6).
Mitochondria are the source and target of ROS.
Mitochondrial quality (structural and functional integrity) is
essential for cellular function. Damaged mitochondria should be
cleared in time, otherwise they may lead to metabolic diseases,
including cancer (5). In the present
study, notable reduction in ΔΨm and cytochrome c leakage in
TIGAR-knocked down cells was observed, which suggested the
initiation of mitophagy (19,20). Of note, instead of clearance of
mitochondria, an increase in mitochondrial mass was observed. For
the degradation of mitochondria, a series of events is required to
occur: i) Mitochondrial damage associated with membrane potential
reduction; ii) an isolation membrane encircles damaged mitochondria
to form double-membrane vesicles known as autophagosomes and iii)
fusion with lysosome to digest its contents (21). TIGAR may serve an unknown role in the
final two steps of the process. However, this is beyond the scope
of the present study and requires further investigation.
In the present study, RNA interference was employed
to investigate the potential effects of TIGAR on mitochondrial
function in 5-8F cells. In the present study, it was revealed that
TIGAR is localized on mitochondria. In addition, the knockdown of
TIGAR resulted in reduced ΔΨm and leakage of cytochrome c
from the mitochondria to the cytoplasm. The mass of mitochondria
was further determined, which was unexpectedly increased in
TIGAR-knocked down cells. This increase in mitochondrial mass was
associated with abnormal mitochondrial characteristics, including
mitochondrial swelling, crista collapse and vacuolization. In
addition, mitochondrial physiological dysfunction was demonstrated
as indicated by the reduction in ATP production. In conclusion,
TIGAR affects mitochondrial integrity and degradation in 5-8F
cells.
Acknowledgements
The present study was supported by the Scientific
Research Foundation of the Education Department of Sichuan Province
(grant no. 15ZA0163), the Affiliated Hospital of Southwest Medical
University Foundation (grant no. 201519) and the Southwest Medical
University Foundation (grant no. 20130388).
References
|
1
|
Rich PR: The molecular machinery of
Keilin's respiratory chain. Biochem Soc Trans. 31:1095–1105. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wallace DC, Fan W and Procaccio V:
Mitochondrial energetics and therapeutics. Annu Rev Pathol.
5:297–348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: More than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mammucari C and Rizzuto R: Signaling
pathways in mitochondrial dysfunction and aging. Mech Ageing Dev.
131:536–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goldman SJ, Taylor R, Zhang Y and Jin S:
Autophagy and the degradation of mitochondria. Mitochondrion.
10:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wallace DC: A mitochondrial paradigm of
metabolic and degenerative diseases, aging and cancer: A dawn for
evolutionary medicine. Annu Rev Genet. 39:359–407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ashrafi G and Schwarz TL: The pathways of
mitophagy for quality control and clearance of mitochondria. Cell
Death Differ. 20:31–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun Y, Vashisht AA, Tchieu J, Wohlschlegel
JA and Dreier L: Voltage-dependent anion channels (VDACs) recruit
Parkin to defective mitochondria to promote mitochondrial
autophagy. J Biol Chem. 287:40652–40660. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawajiri S, Saiki S, Sato S, Sato F,
Hatano T, Eguchi H and Hattori N: PINK1 is recruited to
mitochondria with parkin and associates with LC3 in mitophagy. FEBS
Lett. 584:1073–1079. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vives-Bauza C, Zhou C, Huang Y, Cui M, de
Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al:
PINK1-dependent recruitment of Parkin to mitochondria in mitophagy.
Proc Natl Acad Sci USA. 107:pp. 378–383. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bensaad K, Tsuruta A, Selak MA, Vidal MN,
Nakano K, Bartrons R, Gottlieb E and Vousden KH: TIGAR, a
p53-inducible regulator of glycolysis and apoptosis. Cell.
126:107–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bensaad K, Cheung EC and Vousden KH:
Modulation of intracellular ROS levels by TIGAR controls autophagy.
EMBO J. 28:3015–3026. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mercurio F and Manning AM: NF-kappaB as a
primary regulator of the stress response. Oncogene. 18:6163–6171.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hough MA, Silkstone G, Worrall JA and
Wilson MT: NO binding to the proapoptotic cytochrome c-cardiolipin
complex. Vitam Horm. 96:193–209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yin L, Kosugi M and Kufe D: Inhibition of
the MUC1-C oncoprotein induces multiple myeloma cell death by
down-regulating TIGAR expression and depleting NADPH. Blood.
119:810–816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ye L, Zhao X, Lu J, Qian G, Zheng JC and
Ge S: Knockdown of TIGAR by RNA interference induces apoptosis and
autophagy in HepG2 hepatocellular carcinoma cells. Biochem Biophys
Res Commun. 437:300–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eng C, Kiuru M, Fernandez MJ and Aaltonen
LA: A role for mitochondrial enzymes in inherited neoplasia and
beyond. Nat Rev Cancer. 3:193–202. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction and aging. Rejuvenation Res.
8:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|