Introduction
B-cell chronic lymphocytic leukemia (CLL) accounts
for about 25% of all leukemia and is the most common form of
lymphoid malignancy in western countries (1). CLL is characterized by the accumulation
of the clonal expansion and resistance to apoptosis of immature
CD5(+) B cells (2). The disease
displays a high heterogeneity in its clinical course (3,4). Staging
and prognostication of CLL is performed by two similar clinical
staging systems (5,6). However, the clinical staging systems
cannot fully reflect the high variability of CLL and predict
survival of CLL patients, as well as the response to therapy
(7). Molecular aberration plays an
important role in the initiation and progression of CLL. Several
factors that can predict the clinical course have been identified
(8–10). The most frequent deletion of genomic
DNA in CLL occurs in chromosome 13q13.4. This deletion occurs in
~50% of CLL cases and is associated with a long interval between
diagnosis and the need for treatment (11). One study was reported that an
increased DPF3 expression in CLL patients that was linked
with a significant increase of STAT5 activation in myeloid lineage
cells (granulocytes) and not in neoplastic B cells (12). However, the molecular mechanism of
these associations is still largely unknown.
miRNAs are small noncoding RNA molecules that
negatively regulate gene expression via degradation or
translational repression of their targeted mRNAs (13). miRNAs played important roles in the
pathogenesis of various human cancers and ~50% of the known human
microRNAs were located at cancer-associated regions of the genome
(14–16). In addition, miRNAs played critical
roles in the development and oncogenesis of B-cell. They were
involved in the pathogenesis of CLL (17). MiRNAs expressions were associated with
clinical characteristics of CLL, and they were likely to be served
as diagnostic and prognostic biomarkers, as well as potential
therapeutic targets in CLL (18,19).
Negrini et al revealed that miRNAs whose expression was
distinctive of cases with mutated vs. unmutated IGHV genes or cases
with 13q, 11q, and 17p deletions and trisomy 12 and miR26a,
miR532-3p, miR146-5p, and miR29c* were strongly
associated with progression-free survival in CLL (20). However, the specific regulatory
mechanism of miRNA in CLL was still needed to further explore.
In the current studies usually analyzed gene
expression by high throughput RNA sequenc, though miRNA is also
known as important in the regulation of gene expression and cancer
formation. In this study, we firstly identified the key miRNA-Gene
pairs in CLL.
In this study, differentially expressed genes (DEGs)
and miRNAs (DEMs) in CLL samples compared with normal B cell
samples were identified via bioinformatics methods. The miRNA-Gene
regulatory network in CLL was constructed. A better understanding
of the regulatory mechanism of miRNAs in CLL was obtained. Our
study may provide references for the diagnosis and therapy of
CLL.
Materials and methods
Microarray data
The miRNA expression dataset GSE62137 (21) and mRNA expression dataset GSE22529
(22) were downloaded from National
Center of Biotechnology Information (NCBI) Gene Expression Omnibus
(GEO; http://www.ncbi.nlm.nih.gov/geo/) database. The miRNA
dataset GSE62137 contained 44 samples, including 38 CLL cell
samples and 6 normal B cell samples. Some of these samples were
treated with IL-4. Our study was performed based on the untreated
samples (23 CLL cell samples and 3 normal B cell samples cultured
with nothing). MiRNA expression profile was detected via the
Agilent-021827 Human miRNA Microarray G4470C platform. The mRNA
dataset GSE22529 contained 52 samples, including 41 CLL cell
samples and 11 normal B cell samples. The microarray data we used
was detected based on GPL96 [HG-U133A] Affymetrix Human Genome
U133A Array platform.
Identification of differently
expressed miRNAs and genes
For the miRNA dataset, DEMs in CLL samples compared
with normal B cell samples were identified via the GEO2R
application of GEO. The screening threshold was FDR corrected
P<0.05 and |log2 (fold-change)|>1. For the mRNA dataset, the
raw data with CEL files were background corrected, normalized and
log2 transformed using the affy package in R. (23). If multiple probes correspond to one
gene, the mean expression value was defined as expression value.
DEGs in CLL samples compared with normal B cell samples were
identified via the limma package (24) of R. The DEGs were identified
according to the criteria of |log2 (fold-change) |>1 and FDR
corrected P<0.05.
Functional clustering analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) was a widely used
web-based tool for genomic functional annotations (25). In the present study, Gene Ontology
(GO) terms enrichment analysis were performed via DAVID. Enrichment
score >1 was used as the criteria to identify significant
functional clusters.
Screening of target genes of the
DEMs
The targets of the DEMs, as well as the miRNA-Gene
pairs, were identified based on the miRNAWalk platform. Five
databases: miRnada (26), miRDB
(27), miRWalk (28), RNA22 (29) and TargetScan (30) were used in this study to predicted the
targets of the DEMs. Only the targets that simultaneously appeared
in the five databases were screened out.
Construction of the miRNA-Gene
regulatory network
The overlaps between the DEGs and the targets of the
DEMs were selected. The miRNA-Gene regulatory network was
constructed based on the miRNA-Gene pairs, and then visualized via
Cytoscape software (version 3.4.0; www.cytoscape.org).
Results
The DEMs and the DEGs
A total of 63 DEMs were identified in CLL samples
compared with normal B cell samples, including 51 downregulated
ones and 12 upregulated ones. The top 20 DEMs were listed in
Table I. For the mRNA dataset, gene
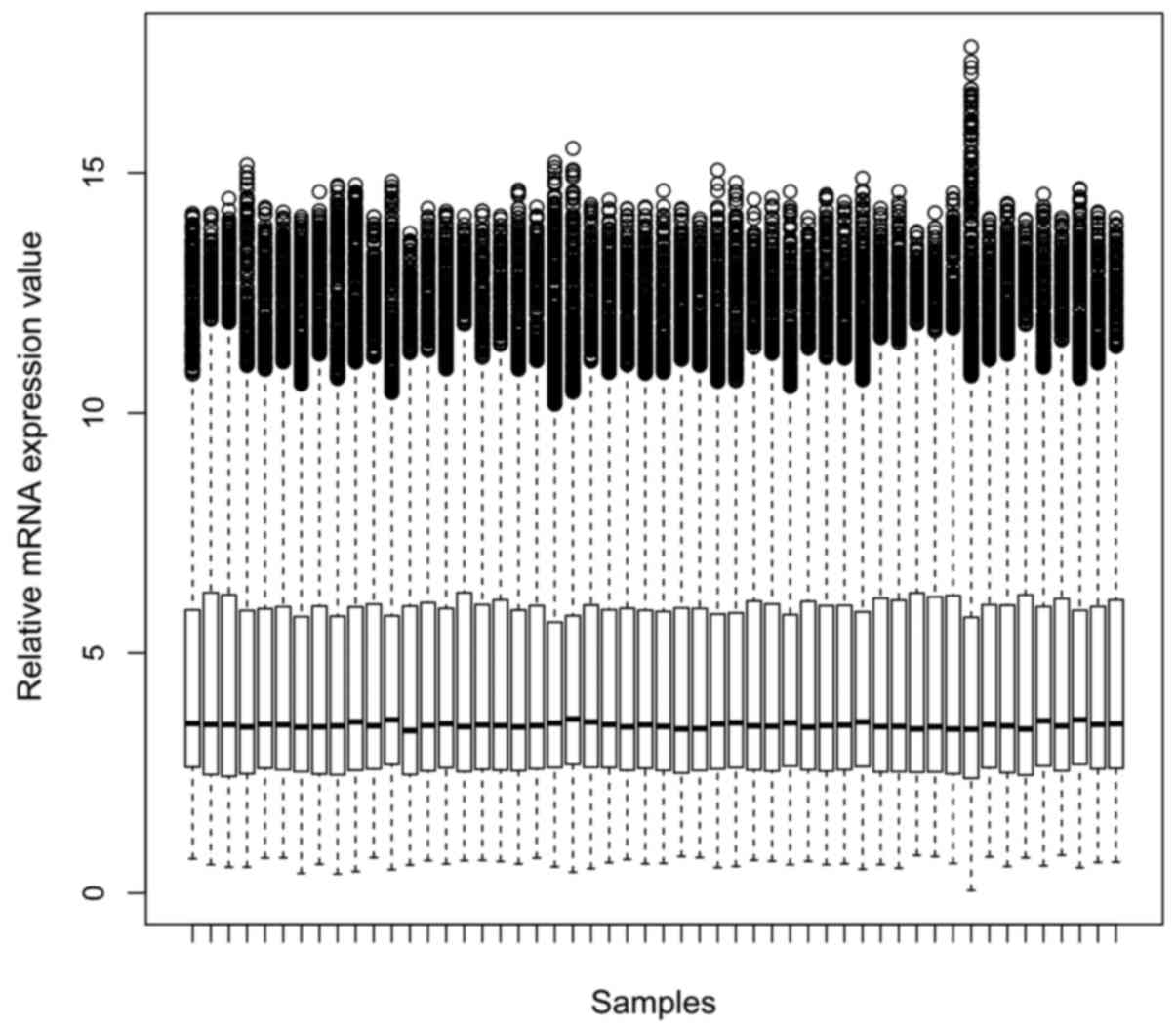
expression values of each sample after normalization were shown in
Fig. 1. 504 DEGs were identified in
CLL samples compared with normal B cell samples, including 316
downregulated ones and 188 upregulated ones. Table I showed the top 20 DEGs. Cluster
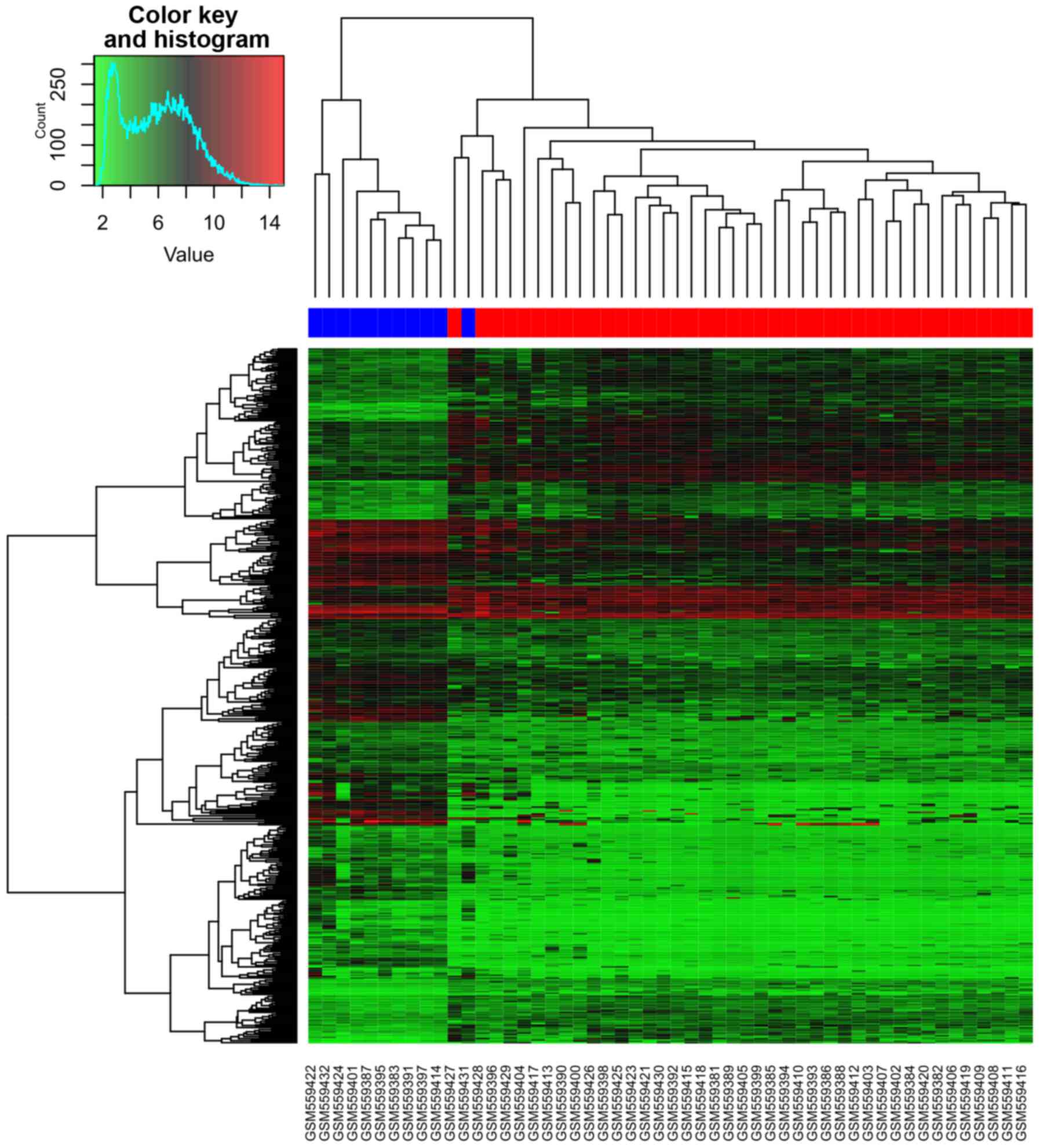
analysis of CLL samples and normal B cell samples based on the DEGs
expression values was shown in Fig.
2. From the heatmap (Fig. 2), we
found that the gene expression of 10 normal B cell samples were
distinguished from the 41 CLL samples. One normal B cell samples
was assigned to CLL samples. Accuracy rate of prediction is
98.07%.
 | Table I.The top 20 DEMs in CLL samples
compared with normal B cell samples. |
Table I.
The top 20 DEMs in CLL samples
compared with normal B cell samples.
| MiRNA ID | P-value | LogFC |
|---|
| hsa-miR-582-5p |
5.32×10−17 | −8.23 |
| hsa-miR-181a |
8.86×10−04 | −8.56 |
| hsa-miR-132 |
8.86×10−04 | −6.42 |
| hsa-miR-95 |
8.86×10−04 | −4.34 |
|
hsa-miR-548c-3p |
8.86×10−04 | −3.65 |
| hsa-miR-181c |
8.86×10−04 | −3.22 |
| hsa-miR-150 |
8.86×10−04 | 1.54 |
| hsa-miR-486-5p |
8.86×10−04 | 5.65 |
| hsa-miR-451 |
8.86×10−04 | 6.43 |
| hsa-miR-144 |
8.86×10−04 | 8.56 |
| hsa-miR-28-5p |
1.81×10−03 | 1.57 |
| hsa-miR-885-3p |
1.42×10−02 | −3.2 |
|
hsa-miR-199a-3p |
1.71×10−02 | −5.62 |
| hsa-miR-155 |
1.74×10−02 | 2.00 |
| hsa-miR-126 |
2.22×10−02 | −6.29 |
| hsa-miR-29a |
2.26×10−02 | 1.27 |
| hsa-miR-21 |
2.32×10−02 | 1.42 |
| hsa-miR-202 |
2.47×10−02 | −3.53 |
| hsa-miR-29b |
3.93×10−02 | 1.15 |
|
hsa-miR-199a-5p |
3.95×10−02 | −3.42 |
Enriched functional clusters
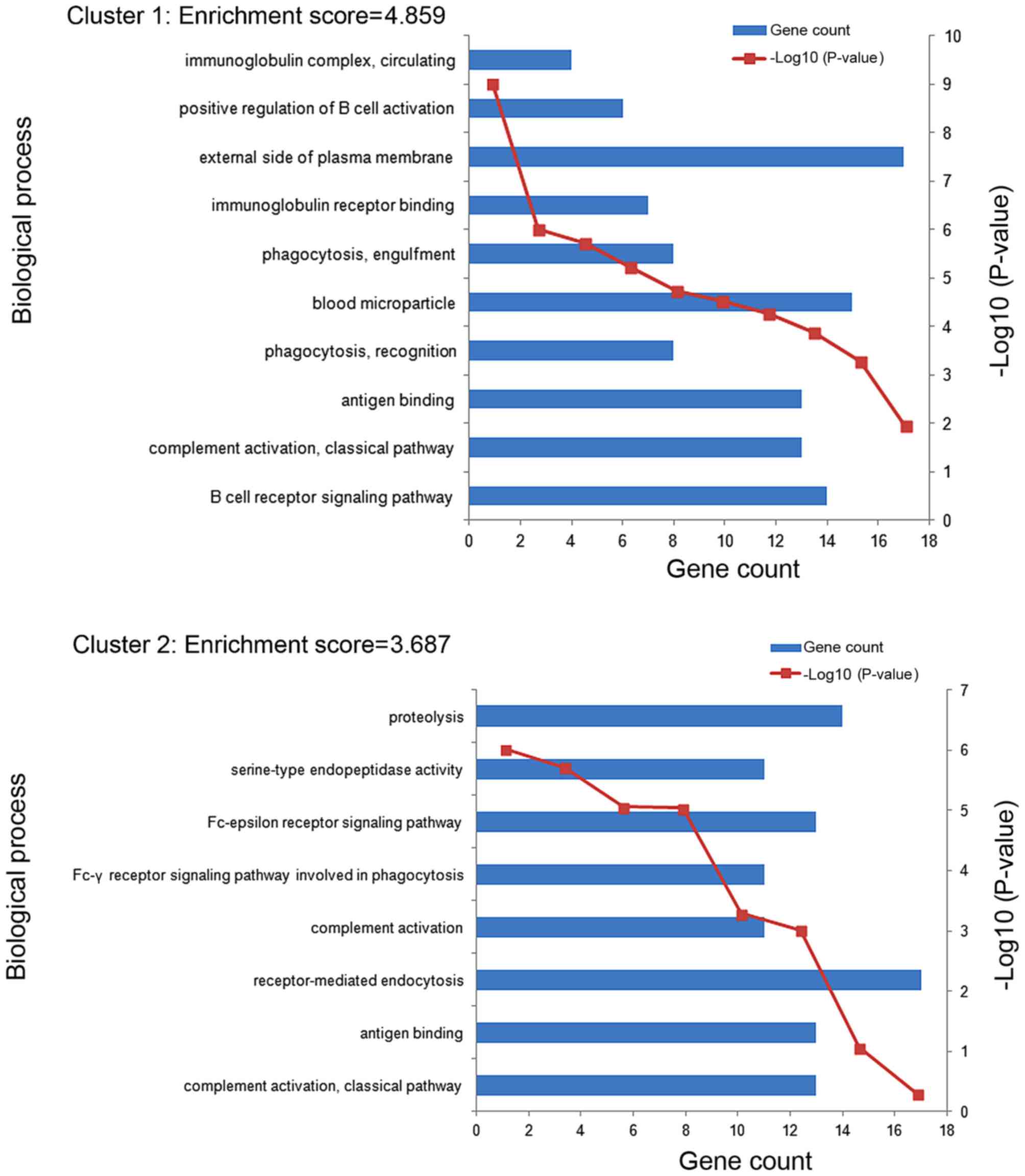
11 enriched functional clusters of the DEGs were
obtained. The enriched GO terms in the top 2 clusters (cluster 1
and cluster 2) were shown in Fig. 3.
The GO terms of clusters 1 were significantly enriched in the
process of immune response, including B cell receptor signaling
pathway (GO:0050853), complement activation classical pathway
(GO:0006958), antigen binding (GO:0003823) and so on. The GO terms
of clusters 2 were mainly enriched in the process of immune
recognition receptors includes complement activation classical
pathway (GO:0006958), antigen binding (GO:0003823),
receptor-mediated endocytosis (GO:0006898) and so on. The most
enriched GO term in these 2 clusters involve an immune
response.
The miRNA-gene regulatory network
A total of 405 miRNA-Gene regulatory pairs were
identified via the miRNAWalk platform. The miRNA-Gene regulatory
pairs contained 351 target genes of the DEMs, including 9 overlaps
with the DEGs. These overlaps were regulated by 10 DEMs, and then
10 miRNA-Gene regulatory pairs were obtained. Afterwards, a
regulatory network between these overlaps and the DEMs were
constructed and visualized (Fig. 4).
As seen in Fig. 4, 9 target genes
differentially expressed (TRAM2, BASP1, PRDM4, DGKG, TOP1,
JOSD1, NOTCH2, AKAP12, CNTN6) are regulated by 9 different
miRNAs (hsa-miR-138b, hsa-miR-352, hsa-miR-1, hsa-miR-302b,
hsa-miR-326, hsa-miR-136, hsa-miR-181c, hsa-miR-145,
hsa-miR-150).
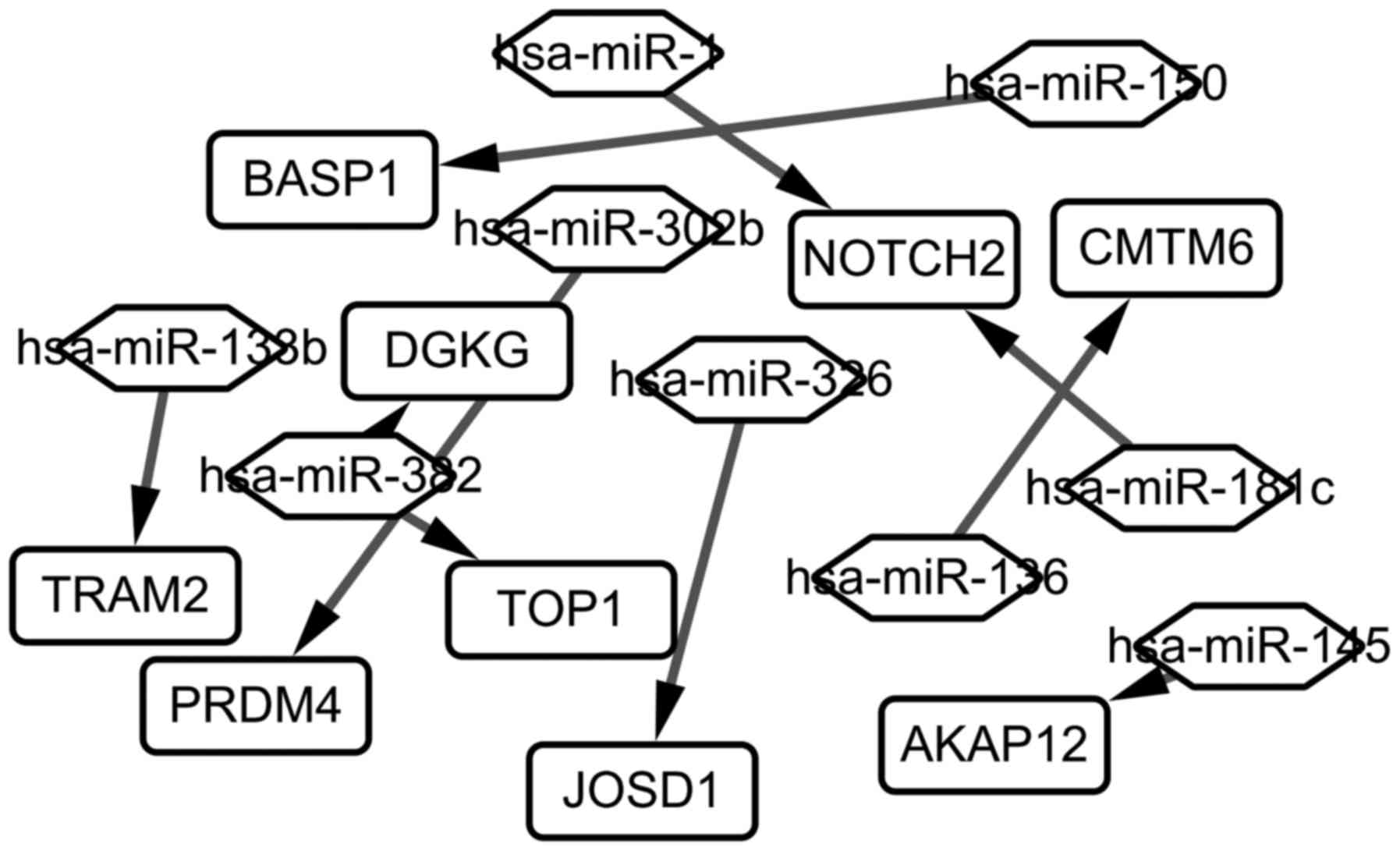 | Figure 4.The miRNA-Gene regulatory network in
chronic lymphocytic leukemia. The rectangle represents the gene,
and the rhombus represents the miRNA. Has, homo sapiens;
miRNA, microRNA; Notch2, neurogenic locus notch homolog protein 2;
PRDM4, PR/SET domain 4; AKAP12, A-kinase anchoring protein 12;
BASP1, brain abundant membrane attached signal protein 1; DGKG,
diacylglycerol kinase γ; CMTM6, CKLF like MARVEL transmembrane
domain containing 6; TRAM2, translocation associated membrane
protein 2; TOP1, topoisomerase (DNA) I; JOSD1, josephin domain
containing 1. |
Discussion
CLL is the most common form of leukemia in western
countries with the incidence of approximately 1 out of 100,000
patients per year (31), but little
is known about its initiation and progression (32). MiRNAs play critical roles in B-cell
oncogenesis by regulating the expression of many genes, and they
are involved in the pathogenesis of CLL. In this study, a
miRNA-Gene regulatory network in CLL was constructed via
bioinformatics methods, which would help us make a better
understanding of the molecular mechanism of CLL.
In the enriched GO terms in cluster 1,
immunoglobulin complex, circulating and positive regulation of B
cell activation were most closely related to the initiation and
progression of CLL. While, in the enriched GO terms in cluster 2,
proteolysis and serine-type endopeptidase activity had the most
closely relationship with CLL. The mutational status of
immunoglobulin heavy-chain variable-region (IgVH) genes
in the leukemic cells of CLL was an important prognostic factor in
the disease. Approximately 50–70% of patients with CLL had somatic
hypermutation in IgVH genes of the leukemic cells
(9,33). In addition, immunoglobulin could also
be used as a therapeutic agent for CLL, and intravenous
immunoglobulin G may alter the response of CLL cells to
chemotherapy (34). B cell activation
was an important initiation factor of CLL. CLL was characterized by
the progressive accumulation of clonal mature B cells in the blood,
bone marrow, and secondary lymphoid organs (35). B cell receptor signaling represented
one of the central pathways to enhance survival and proliferation
in CLL (10). One research showed
that differences existed in the activation of B-CLL cells in
vivo, and these patterns were correlated with disease activity
(36). Although the importance of
protein proteolysis in biological functions was frequently unclear,
new technologies had started to unravel the critical role of
clipping in cellular homeostasis and disease (37,38).
Specific histone H2A proteolysis as disruption of the histone code
was reported to drive hematopoietic cells in lymphomagenesis and
result in lymphoid malignancies (39). Furthermore, matriptase was reported to
be highly upregulated in CLL, which promoted cancer cell invasion
(40). Serine-type endopeptidase also
played critical roles in CLL. A serine endopeptidase, kallikrein B1
(KLKB1) was reported to be overexpressed in CLL, and its expression
could be served as a novel molecular biomarker for the diagnosis of
CLL (41).
The miRNA-Gene regulatory network contained 9 DEGs;
they were BASP1, NOTCH2, CMTM6, DGKG, TRAM2, TOP1, PRDM4,
JOSD1 and AKAP12. DAVID is a comprehensive system of
biological function annotation information for large-scale genes,
so it is too little to perform the DAVID analysis on this 9
overlapping DEGs. According to the European Molecular Biology
Laboratory (EMBL) database (http://www.ebi.ac.uk/), we found 9 DEGs were enriched
the following GO terms. BASP1, NOTCH2, TOP1, PRDM4, AKAP12
and JOSD1 were included in the biological process of protein
binding (GO:0005515) which child terms included antigen binding,
immunoglobulin receptor binding. JOSD1 was aslo included in
the biological process of proteolysis (GO:0006508). JOSD1,
NOTCH2 and AKAP12 wwere included in the biological
process of hydrolase activity (GO:0016787), receptor activity
(GO:0004872), regulation of protein kinase C signaling
(GO:0090036), respectively, which were closely related to
complement activation, classical pathway, positive regulation of B
cell activation. TRAM2 and DGKG were respectively
included in the biological process of integrall component of
menbrane (GO:001601), intracellular signal transduction (0035556)
which were closely related to signal transduction. To sum up, the
GO terms of 9 DEGs were associated with the GO terms of all DEGs.
Many of these genes were associated with the pathobiology of CLL.
For example, NOTCH2 was a member of the NOTCH gene family.
The Notch gene family encodes transmembrane receptors that modulate
differentiation, proliferation and apoptotic programs in response
to extracellular ligands expressed on neighboring cells (42). Enforced expression of NOTCH1IC in bone
marrow stem cells caused T-cell leukemia in mice, indicating a
causative role for NOTCH family in T-cell oncogenesis (43). The NOTCH2 oncogene was reported
to be overexpressed in B-CLL cells, and was also related to the
failure of apoptosis characteristic for this disease. Deregulation
of NOTCH2 signaling was involved in the aberrant expression
of CD23 in B-CLL (44). The
overexpression of CD23 was one of the hallmarks of B-CLL
cells (45). PRDM4 was a
highly conserved member of the PRDM family (46). One study reported that PRDM4
could control proliferation and differentiation, which played
critical roles in tumorigenesis (47). Another study reported that PRDM4
protein mapped to a tumor suppressor locus on human chromosome and
could affect the processes of ovarian, gastric, and pancreatic
cancers (48). AKAP12, which
functioned as a kinase scaffold protein and as a dynamic regulator
of the b2-adrenergic receptor complex, was one of the A-kinase
anchoring proteins (49).
AKAP12 played an important role in regulating cell cycle,
cytokinesis cell adhesion, signaling, and oncogenic suppression
(50). AKAP12 expression was
closely related to tumorigenesis. The downregulation of
AKAP12 expression had been reported in human prostate
cancers in vivo, suggesting that the inactivation of
AKAP12 expression was associated with oncogenesis (51). In gastric cancer, AKAP12 may
function as an important negative regulator of the survival pathway
(49). In addition, AKAP12
expression could also be served as a predictor for survival in CLL
(52).
In conclusion, miRNAs played a critical role in
regulating the process of CLL. They could affect CLL by regulating
the processes of immunoreaction and protein degradation. Genes such
as NOTCH2, PRDM4 and AKAP12 proved be their
regulating targets in CLL. These DEGs which were related to CLL,
could potentially serve as biomarkers for detection, prognosis,
monitoring and predicting therapeutic responses in CLL. However,
further studies were still needed to confirm our results.
Acknowledgements
We would like to thank all the members of our
research group for their enthusiastic participation in the present
study.
References
|
1
|
Di Bernardo MC, Crowther-Swanepoel D,
Broderick P, Webb E, Sellick G, Wild R, Sullivan K, Vijayakrishnan
J, Wang Y, Pittman AM, et al: A genome-wide association study
identifies six susceptibility loci for chronic lymphocytic
leukemia. Nat Genet. 40:1204–1210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reddy KS: Chronic lymphocytic leukaemia
profiled for prognosis using a fluorescence in situ hybridisation
panel. Br J Haematol. 132:705–722. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Byrd JC, Stilgenbauer S and Flinn IW:
Chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ
Program. 1–183. 2004.PubMed/NCBI
|
|
4
|
Cramer P and Hallek M: Prognostic factors
in chronic lymphocytic leukemia-what do we need to know? Nat Rev
Clin Oncol. 8:38–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Binet JL, Auquier A, Dighiero G, Chastang
C, Piguet H, Goasguen J, Vaugier G, Potron G, Colona P, Oberling F,
et al: A new prognostic classification of chronic lymphocytic
leukemia derived from a multivariate survival analysis. Cancer.
48:198–206. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rai KR, Sawitsky A, Cronkite EP, Chanana
AD, Levy RN and Pasternack BS: Clinical staging of chronic
lymphocytic leukemia. Blood. 1975;46(2):219-234. Blood.
128:21092016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kay NE, O'Brien SM, Pettitt AR and
Stilgenbauer S: The role of prognostic factors in assessing
‘high-risk’ subgroups of patients with chronic lymphocytic
leukemia. Leukemia. 21:1885–1891. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rassenti LZ, Huynh L, Toy TL, Chen L,
Keating MJ, Gribben JG, Neuberg DS, Flinn IW, Rai KR, Byrd JC, et
al: ZAP-70 compared with immunoglobulin heavy-chain gene mutation
status as a predictor of disease progression in chronic lymphocytic
leukemia. N Engl J Med. 351:893–901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Damle RN, Wasil T, Fais F, Ghiotto F,
Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J,
et al: Ig V gene mutation status and CD38 expression as novel
prognostic indicators in chronic lymphocytic leukemia. Blood.
94:1840–1847. 1999.PubMed/NCBI
|
|
10
|
Hamblin TJ, Davis Z, Gardiner A, Oscier DG
and Stevenson FK: Unmutated Ig V(H) genes are associated with a
more aggressive form of chronic lymphocytic leukemia. Blood.
94:1848–1854. 1999.PubMed/NCBI
|
|
11
|
Calin GA, Ferracin M, Cimmino A, Di Leva
G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, et
al: A MicroRNA signature associated with prognosis and progression
in chronic lymphocytic leukemia. N Engl J Med. 353:1793–1801. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Theodorou M, Speletas M, Mamara A,
Papachristopoulou G, Lazou V, Scorilas A and Katsantoni E:
Identification of a STAT5 target gene, Dpf3, provides novel
insights in chronic lymphocytic leukemia. PLoS One. 8:e761552013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lim LP, Lau NC, Garrett-Engele P, Grimson
A, Schelter JM, Castle J, Bartel DP, Linsley PS and Johnson JM:
Microarray analysis shows that some microRNAs downregulate large
numbers of target mRNAs. Nature. 433:769–773. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M
and Croce CM: Human microRNA genes are frequently located at
fragile sites and genomic regions involved in cancers. Proc Natl
Acad Sci USA. 101:pp. 2999–3004. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Michael MZ, O' Connor SM, van Holst
Pellekaan NG, Young GP and James RJ: Reduced accumulation of
specific microRNAs in colorectal neoplasia. Mol Cancer Res.
1:882–891. 2003.PubMed/NCBI
|
|
16
|
Takamizawa J, Konishi H, Yanagisawa K,
Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y,
et al: Reduced expression of the let-7 microRNAs in human lung
cancers in association with shortened postoperative survival.
Cancer Res. 64:3753–3756. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fernando TR, Rodriguez-Malave NI and Rao
DS: MicroRNAs in B cell development and malignancy. J Hematol
Oncol. 5:72012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marton S, Garcia MR, Robello C, Persson H,
Trajtenberg F, Pritsch O, Rovira C, Naya H, Dighiero G and Cayota
A: Small RNAs analysis in CLL reveals a deregulation of miRNA
expression and novel miRNA candidates of putative relevance in CLL
pathogenesis. Leukemia. 22:330–338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferracin M, Zagatti B, Rizzotto L,
Cavazzini F, Veronese A, Ciccone M, Saccenti E, Lupini L, Grilli A,
De Angeli C, et al: MicroRNAs involvement in fludarabine refractory
chronic lymphocytic leukemia. Mol Cancer. 9:1232010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Negrini M, Cutrona G, Bassi C, Fabris S,
Zagatti B, Colombo M, Ferracin M, D'Abundo L, Saccenti E, Matis S,
et al: microRNAome expression in chronic lymphocytic leukemia:
Comparison with normal B-cell subsets and correlations with
prognostic and clinical parameters. Clin Cancer Res. 20:41412014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ruiz-Lafuente N, Alcaraz-García MJ,
Sebastián-Ruiz S, García-Serna AM, Gómez-Espuch J, Moraleda JM,
Minguela A, García-Alonso AM and Parrado A: IL-4 up-regulates
MiR-21 and the MiRNAs hosted in the CLCN5 gene in chronic
lymphocytic leukemia. PLoS One. 10:e01249362015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gutierrez A Jr, Tschumper RC, Wu X,
Shanafelt TD, Eckel-Passow J, Huddleston PM III, Slager SL, Kay NE
and Jelinek DF: LEF-1 is a prosurvival factor in chronic
lymphocytic leukemia and is expressed in the preleukemic state of
monoclonal B-cell lymphocytosis. Blood. 116:2975–2983. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma N and Gao X: β-actin is predicted as
one of the potential targets of miR-145: Choose internal control
gene in verification of microRNA target. Carcinogenesis.
34:2362013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang X: miRDB: A microRNA target
prediction and functional annotation database with a wiki
interface. RNA. 14:1012–1017. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dweep H, Sticht C, Pandey P and Gretz N:
miRWalk-database: Prediction of possible miRNA binding sites by
‘walking’ the genes of three genomes. J Biomed Inform. 44:839–847.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rigoutsos I, Miranda K and Huynh T: rna22:
A unified computational framework for discovering miRNA precursors,
localizing mature miRNAs, identifying 3′ UTR target-islands and
determining the targets of mature-miRNAs. IBM Corporation; Yorktown
Heights, NY: 2007
|
|
30
|
Edris B: A comparison of the Oligomap and
TargetScan algorithms for miRNA target analysis. PhD
dissertationStanford University Publication no. Bmi231. Stanford,
CA: 2011
|
|
31
|
Pfeil AM, Imfeld P, Pettengell R, Jick SS,
Szucs TD, Meier CR and Schwenkglenks M: Trends in incidence and
medical resource utilisation in patients with chronic lymphocytic
leukaemia: Insights from the UK Clinical Practice Research Datalink
(CPRD). Ann Hematol. 94:421–429. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fabbri G and Dalla-Favera R: The molecular
pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer.
16:145–162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fais F, Ghiotto F, Hashimoto S, Sellars B,
Valetto A, Allen SL, Schulman P, Vinciguerra VP, Rai K, Rassenti
LZ, et al: Chronic lymphocytic leukemia B cells express restricted
sets of mutated and unmutated antigen receptors. J Clin Invest.
102:1515–1525. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Besa EC: Use of intravenous immunoglobulin
in chronic lymphocytic leukemia. Am J Med. 76:209–218. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhong Y, El-Gamal D, Dubovsky JA, Beckwith
KA, Harrington BK, Williams KE, Goettl VM, Jha S, Mo X, Jones JA,
et al: Selinexor suppresses downstream effectors of B-cell
activation, proliferation and migration in chronic lymphocytic
leukemia cells. Leukemia. 28:1158–1163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tötterman TH, Carlsson M, Funderud S,
Simonsson B, Oberg G and Nilsson K: Chronic B-lymphocytic
leukemia-expression of B cell activation markers in relation to
activity of the disease. Nouv Rev Fr Hematol. 30:279–281.
1988.PubMed/NCBI
|
|
37
|
Doucet A, Butler GS, Rodríguez D, Prudova
A and Overall CM: Metadegradomics: Toward in vivo quantitative
degradomics of proteolytic post-translational modifications of the
cancer proteome. Mol Cell Proteomics. 7:1925–1951. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rogers LD and Overall CM: Proteolytic
post-translational modification of proteins: Proteomic tools and
methodology. Mol Cell Proteomics. 12:3532–3542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taylor KH, Briley A, Wang Z, Cheng J, Shi
H and Caldwell CW: Aberrant epigenetic gene regulation in lymphoid
malignancies. Semin Hematol. 50:38–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gao L, Liu M, Dong N, Jiang Y, Lin CY,
Huang M, Wu D and Wu Q: Matriptase is highly upregulated in chronic
lymphocytic leukemia and promotes cancer cell invasion. Leukemia.
27:1191–1194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Adamopoulos PG, Kontos CK, Papageorgiou
SG, Pappa V and Scorilas A: KLKB1 mRNA overexpression: A novel
molecular biomarker for the diagnosis of chronic lymphocytic
leukemia. Clin Biochem. 48:849–854. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pear WS, Aster JC, Scott ML, Hasserjian
RP, Soffer B, Sklar J and Baltimore D: Exclusive development of T
cell neoplasms in mice transplanted with bone marrow expressing
activated Notch alleles. J Exp Med. 183:2283–2291. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hubmann R, Schwarzmeier JD, Shehata M,
Hilgarth M, Duechler M, Dettke M and Berger R: Notch2 is involved
in overexperssion of CD23 in B-cell chronic lymphocytic leukemia.
Blood. 99:3742–3747. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lopez-Matas M, Rodriguez-Justo M, Morilla
R, Catovsky D and Matutes E: Quantitative expression of CD23 and
its ligand CD21 in chronic lymphocytic leukemia. Haematologica.
85:1140–1145. 2000.PubMed/NCBI
|
|
46
|
Bogani D, Morgan MA, Nelson AC, Costello
I, McGouran JF, Kessler BM, Robertson EJ and Bikoff EK: The PR/SET
domain zinc finger protein Prdm4 regulates gene expression in
embryonic stem cells but plays a nonessential role in the
developing mouse embryo. Mol Cell Biol. 33:3936–3950. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chittka A, Nitarska J, Grazini U and
Richardson WD: Transcription factor positive regulatory domain 4
(PRDM4) recruits protein arginine methyltransferase 5 (PRMT5) to
mediate histone arginine methylation and control neural stem cell
proliferation and differentiation. J Biol Chem. 287:42995–43006.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang XH and Huang S: PFM1 (PRDM4), a new
member of the PR-domain family, maps to a tumor suppressor locus on
human chromosome 12q23-q24.1. Genomics. 61:319–325. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Choi MC, Jong HS, Kim TY, Song SH, Lee DS,
Lee JW, Kim TY, Kim NK and Bang YJ: AKAP12/Gravin is inactivated by
epigenetic mechanism in human gastric carcinoma and shows growth
suppressor activity. Oncogene. 23:7095–7103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Akakura S and Gelman IH: Pivotal role of
AKAP12 in the regulation of cellular adhesion dynamics: Control of
cytoskeletal architecture, cell migration and mitogenic signaling.
J Signal Transduct. 2012:5291792012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xia W, Unger P, Miller L, Nelson J and
Gelman IH: The Src-suppressed C kinase substrate, SSeCKS, is a
potential metastasis inhibitor in prostate cancer. Cancer Res.
61:5644–5651. 2001.PubMed/NCBI
|
|
52
|
van't Veer MB, Brooijmans AM, Langerak AW,
Verhaaf B, Goudswaard CS, Graveland WJ, van Lom K and Valk PJ: The
predictive value of lipoprotein lipase for survival in chronic
lymphocytic leukemia. Haematologica. 91:56–63. 2006.PubMed/NCBI
|