Introduction
As the third leading cause of cancer-associated
mortality worldwide, hepatocellular carcinoma (HCC) is one of the
most common types of cancer (1).
Although the clinical management of early-stage HCC has improved,
the prognosis of HCC remains poor owing to its high recurrence rate
(2). The prognosis of advanced HCC is
particularly poor, due in part to its refractory nature to various
anticancer drugs. An improved understanding of the pathogenesis of
this type of cancer may contribute to more effective outcomes for
the treatment of advanced HCC.
The progression of liver cirrhosis has been
demonstrated to be a primary step in the pathogenesis of HCC
(3). Chronic infections with
hepatitis B (HBV) or hepatitis C (HCV) and other major risk
factors, including alcoholic liver diseases, non-alcoholic
steatohepatitis, autoimmune hepatitis and primary biliary cirrhosis
(4), frequently cause liver
inflammation, hepatic damage and subsequently cirrhosis. It has
been speculated that the processes of HCC tumorigenesis with
cirrhosis include an accumulation of genetic alterations. Han
(5) reported that the development of
HCC is also associated with genetic aberrations (5). However, the key drivers of the
development of HCC remain unclear, and there is a requirement to
elucidate the underlying molecular mechanisms (including various
gene mutations) in the development of HCC.
Next-generation sequencing (NGS) has provided new
paradigms in many fields, including molecular biology, physiology
and medicine, that may be used to disclose the genetic basis of
various diseases (6,7). Novel genetic mutations associated with
tumorigenesis, tumor progression and metastasis have been
identified using NGS, including those in genes encoding isocitrate
dehydrogenase 1 (IDH1) in glioblastoma multiforme (8) and acute myeloid leukemia (9), chromodomain helicase DNA-binding protein
7 in small cell lung cancers (10),
glutamate metabotropic receptor 3, transformation/transcription
domain-associated protein, mitogen-activated protein kinase kinase
1/2, mitogen-activated protein kinase kinase kinase 5/9 and
phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchange
factor 2 in melanoma (11–15), Notch homolog 1 (NOTCH1) in
chronic lymphocytic leukemia (16),
splicing factor 3B subunit 1 in myelodysplasia (17,18), and
chromatin-remodeling proteins such as AT-rich-interaction domain 1A
in ovarian, kidney and gastric cancer (19,20).
Historically, the discovery of somatic mutations in
various types of cancer has been unexpected due to conventional
methods based on direct sequencing. Direct sequencing has a major
limitation regarding the identification of new somatic mutations
because of its candidate gene-based methodology. Conversely, NGS
has provided an unbiased platform to systematically discover gene
mutations and reveal the pathogenesis of various types of
cancer.
In the present study, 50 genes associated with the
development of various types of cancer were targeted, and the
association between the genetic mutations and the clinical
characteristics of HCC patients was investigated using an NGS
platform.
Materials and methods
Patients
The present study involved 57 patients (48 males, 11
females; mean age, 69.1±10.1) who had undergone surgery for HCC at
Kagawa University Hospital (Miki, Japan) between January 2001 and
March 2013. Written informed consent was provided by all patients
and the present study was conducted according to the Ethical
Guidelines for Medical and Health Research approved by the Ministry
of Health, Labour and Welfare of Japan.
Tissue samples
Cancerous and adjacent non-cancerous tissues were
collected macroscopically (3–5 mm thick sections) and immediately
frozen in liquid nitrogen following surgery. Tissues were stored at
−80°C until DNA extraction.
Next-generation sequencing
Genomic DNA was extracted from tissue samples using
the PureLink Genomic DNA Mini kit (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), according to the manufacturer's protocol. For
library construction, 10 ng DNA was amplified using the AmpliSeq
Cancer hotspot panel (version 2; Thermo Fisher Scientific, Inc.)
and Ion AmpliSeq HiFi Master Mix (Ion AmpliSeq Library kit 2.0,
Thermo Fisher Scientific, Inc.). An amplicon library was thus
generated for sequencing 2,850 hotspot mutations in 50 genes:
ABL1, AKT1, ALK, APC, ATM,
BRAF, CDH1, CDKN2A, CSF1R,
CTNNB1, EGFR, ERBB2, ERBB4,
EZH2, FBXW7, FGFR1, FGFR2,
FGFR3, FLT3, GNA11, GNAQ, GNAS,
HNF1A, HRAS, IDH1, IDH2, JAK2,
JAK3, KDR, KIT, KRAS, MET,
MLH1, MPL, NOTCH1, NPM1, NRAS,
PDGFRA, PIK3CA, PTEN, PTPN11,
RB1, RET, SMAD4, SMARCB1, SMO,
SRC, STK11, TP53 and VHL.
The amplicons were then digested, barcoded and
amplified with the Ion AmpliSeq Library kit 2.0 and Ion Xpress
barcode adapters kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. The library was then quantified using
the High Sensitivity DNA kit for the Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc., Santa Clara, CA, USA). A total of 8 pM
of each library was multiplexed and clonally amplified on Ionsphere
particles (ISPs) by emulsion polymerase chain reaction (PCR)
performed using the Ion One Touch 2 instrument with the Ion PGM
template OT2 200 kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol.
Quality control was performed using the Ionsphere
quality control kit (Thermo Fisher Scientific, Inc.) to ensure that
between 10 and 30% of template-positive ISPs were generated in the
emulsion PCR. Finally, the template ISPs were enriched, loaded onto
an Ion 318 chip and sequenced using a PGM sequencer with the Ion
PGM sequencing 200 kit (version 2), according to the manufacturer's
protocol.
Data analysis
The raw data were aligned to Human Genome version 19
(hg19) using Torrent Suite software (version 3.6.2; Thermo Fisher
Scientific, Inc.). The coverage analysis was performed using the
Coverage Analysis plugin (version 3.6; Thermo Fisher Scientific,
Inc.). Cases for which the quality was <20% and/or the average
base coverage was <500X reads and/or the frequency was <10%
were considered non-informative. Mutations were detected using the
Variant Caller plugin (version 3.6; Thermo Fisher Scientific,
Inc.). Each mutation was verified using the Integrative Genome
Viewer (IGV) from the Broad Institute (www.broadinstitute.org) (21).
Statistical analysis
Fisher's exact test (two-sided) was performed to
analyze the association between TP53, CTNNB1 and
SMARCB1 mutations, and clinicopathological parameters using
Prism 6 software (version 6.02; GraphPad Software, Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Study population
As presented in Table
I, 48 males and 11 females were characterized. Of these, 26
were <70 years old and 33 were ≥70 years old. In total, 25 of
the HCV-positive patients and 14 of the HBV-positive patients were
included, and 20 non-viral hepatitis patients were also included in
the study population. A total of 24 patients had liver cirrhosis,
and 35 patients had normal or chronic hepatitis. In addition, 47 of
the well- or moderately differentiated patients with HCC and 12 of
the poorly differentiated patients with HCC were examined. A total
of 28 stage I/II patients and 31 stage III/IV patients (TNM
classification) were characterized in the present study (Table I). TP53 mutations were
identified to be associated with age and TNM stages, and
CTNNB1 mutation was associated with viral infection
(Table I).
 | Table I.Association between
clinicopathological features and representative genetic mutations
in hepatocellular carcinoma. |
Table I.
Association between
clinicopathological features and representative genetic mutations
in hepatocellular carcinoma.
|
|
| TP53 |
| CTNNB1 |
| SMARCB1 |
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| Characteristic | n | Wild-type | Mutant | P-value | Wild-type | Mutant | P-value | Wild-type | Mutant | P-value |
|---|
| Sex |
|
|
| 0.513 |
|
| 0.077 |
|
| 0.573 |
|
Male | 48 | 24 | 24 |
| 28 | 20 |
| 43 | 5 |
|
|
Female | 11 | 7 | 4 |
| 10 | 1 |
| 11 | 0 |
|
| Age, years |
|
|
| 0.019 |
|
| 0.102 |
|
| 0.372 |
|
<70 | 26 | 9 | 17 |
| 20 | 6 |
| 25 | 1 |
|
|
≥70 | 33 | 22 | 11 |
| 18 | 15 |
| 29 | 4 |
|
| Viral
infection |
|
|
| 0.228 |
|
| 0.006 |
|
| 0.952 |
|
HCV | 25 | 16 | 9 |
| 14 | 11 |
| 23 | 2 |
|
|
HBV | 14 | 5 | 9 |
| 14 | 0 |
| 13 | 1 |
|
|
NBNC | 20 | 10 | 10 |
| 10 | 10 |
| 18 | 2 |
|
| Fibrosis stage |
|
|
| 0.597 |
|
| 0.18 |
|
| 0.309 |
| F0, F1,
F2, F3 | 35 | 17 | 18 |
| 20 | 15 |
| 30 | 5 |
|
| F4 | 24 | 14 | 10 |
| 18 | 6 |
| 24 | 0 |
|
| Histological
grade |
|
|
| 0.999 |
|
| 0.182 |
|
| 0.573 |
|
WD/MD | 47 | 25 | 22 |
| 28 | 19 |
| 42 | 5 |
|
| PD | 12 | 6 | 6 |
| 10 | 2 |
| 12 | 0 |
|
| TNM |
|
|
| 0.037 |
|
| 0.576 |
|
| 0.639 |
|
I/II | 28 | 16 | 12 |
| 17 | 11 |
| 26 | 2 | 25 |
|
III/IV | 31 | 9 | 22 |
| 21 | 10 |
| 28 | 3 |
|
Mutation profiling by NGS
A total of 118 samples (59 liver tissues including
both cancer and adjacent normal tissues) were sequenced for the
AmpliSeq Hotspot Cancer panel (version 2). In order to determine
the appropriate variants, variations were retrieved that were
present only in the cancerous region of each patient and absent
from the normal portion of the same individual.
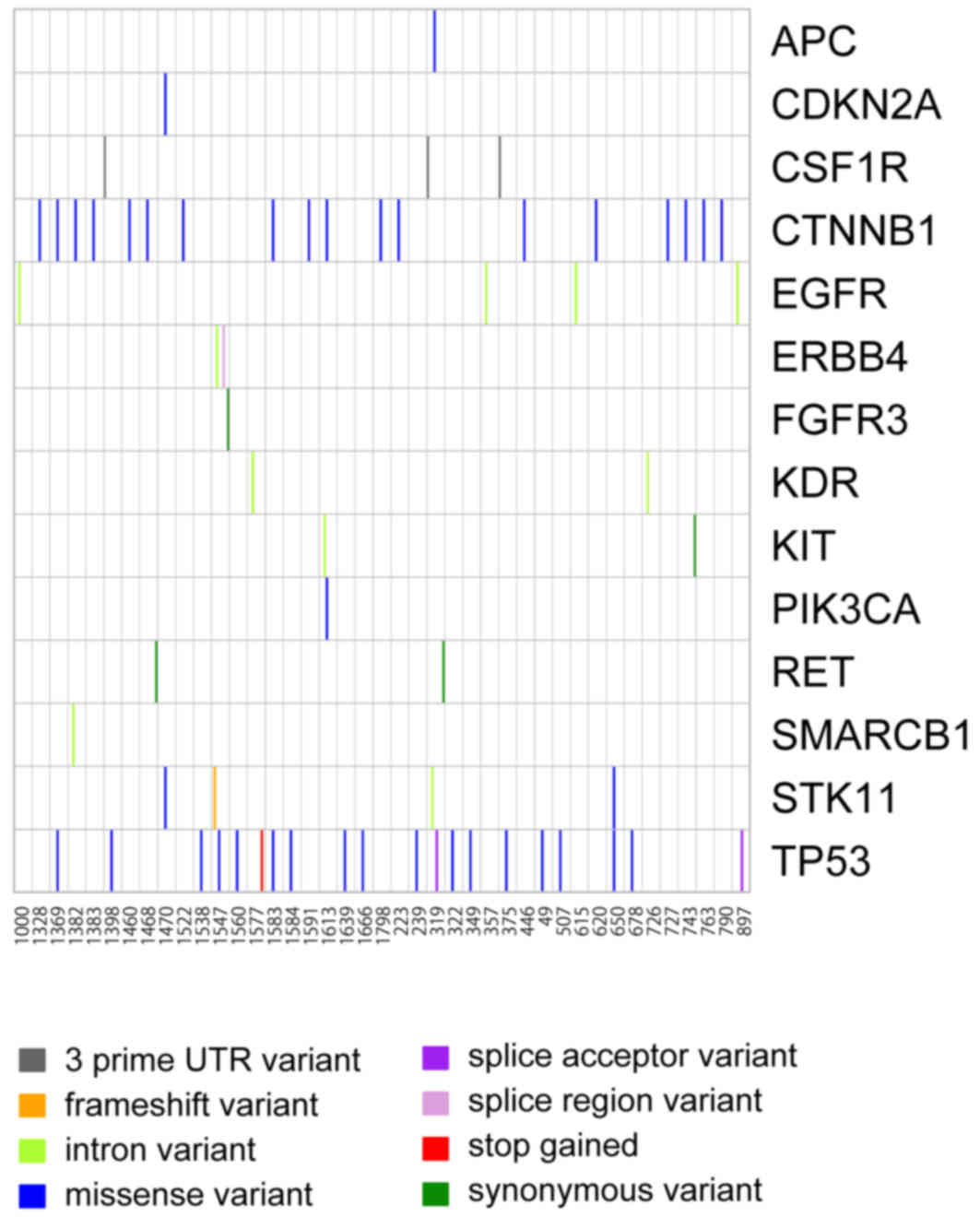
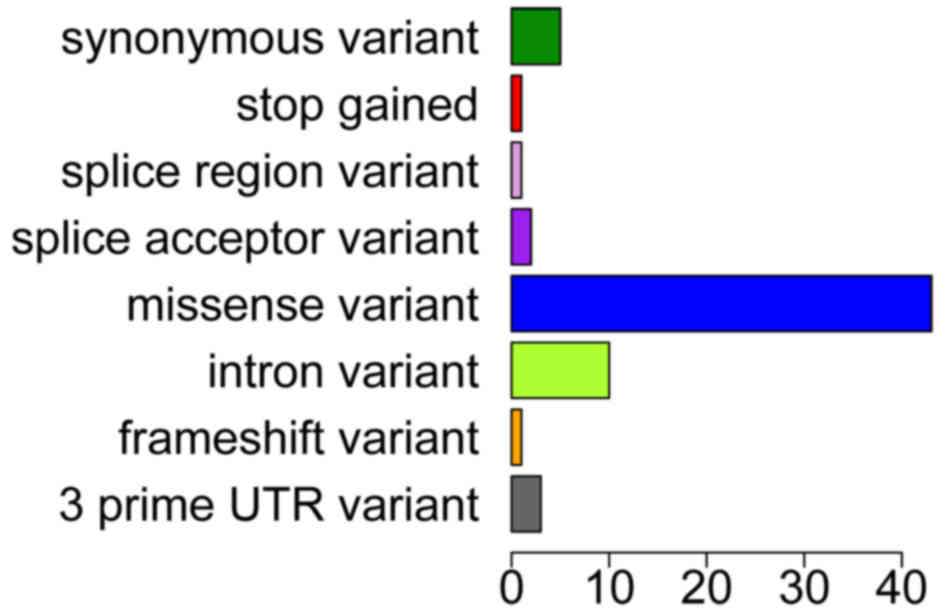
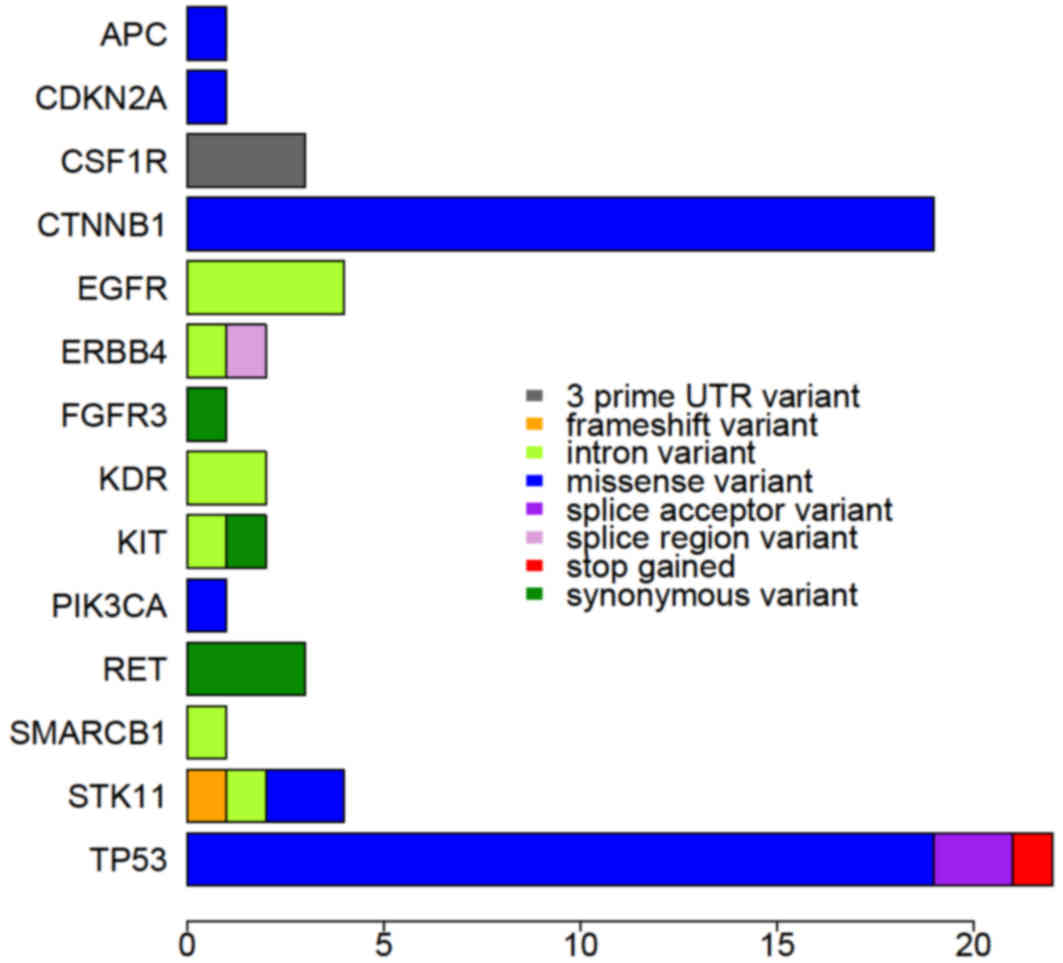
In total, 14 of the 50 genes (28%) revealed any
mutation, and the number of missense variants was the highest in
several variants (Fig. 1), including
synonymous variant, intron variant and frameshift variant (Fig. 2). The most common somatic mutations
identified were in genes TP53 (35.6%) and CTNNB1
(30.5%), and the most frequent variants of those genes were
missense variants (Fig. 3).
Somatic mutations in CSF1R (5.1%),
EGFR (6.8%), RET (5.1%) and STK11 (6.1%) genes
were also identified in the HCC patients, although these genes
exhibited various types of variant at low frequency (Fig. 3). Frameshift variant in STK11,
splice acceptor variant in TP53, splice region variant in
ERBB4 and stop-gained variant in TP53 were
specifically determined, and these variants may alter the
transcription of those genes.
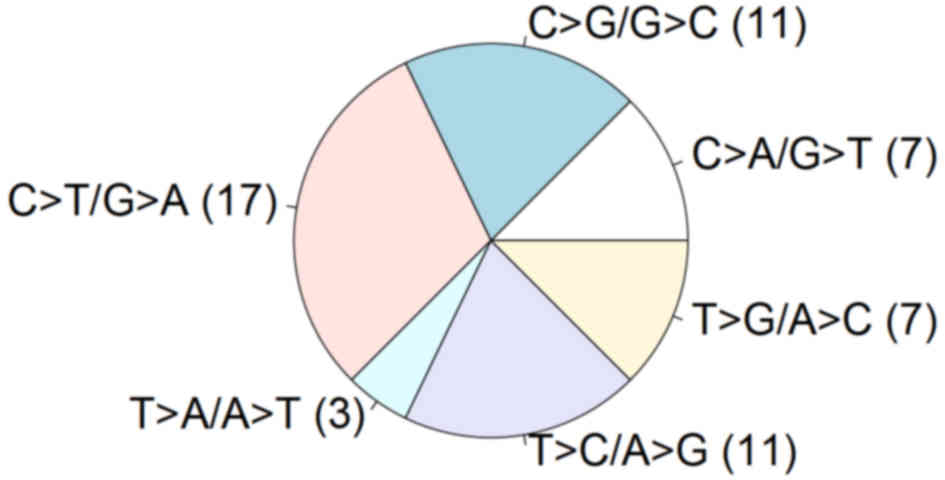
The mutation spectrum revealed C:G>T:A
transitions (50%), which were the most abundant alteration
(22), and other transversions
including C:G>G:C (19.6%), T:A>C:G (19.6%), C:G>A:T
(12.5%), T:A>G:C (12.5%) and T:A>A:T (5.4%) (Fig. 4). This spectrum pattern differs from
that in other solid tumors (22).
Clinicopathological features and
genetic mutations
The p53 pathway was identified to be the most
frequently altered in HCC in the present study. The presence of
TP53-inactivating mutation (35.6%) and CDKN2A
mutations (1.7%) revealed the significance of the p53 pathway
during the development of HCC. Indeed, TP53 mutations in the
tumors at advanced stages were significantly more frequent than
those in the tumors at early stages (P=0.037; Table I).
The Wnt/β-catenin pathway was identified as the
second most frequently altered pathway in HCC. This was evident by
the presence of CTNNB1 (30.5%) and APC (1.7%)
mutations. Although no association between the CTNNB1
mutations and clinicopathological features, including sex, viral
infection, fibrosis stage, histological grade and TNM
classification was identified to be significant in the patients
with HCC, age (<70 and ≥70 years) was identified to be
significantly associated with CTNNB1 mutations (P=0.019;
Table I).
Discussion
The accumulation of genetic alterations is required
during the development of HCC. Although advances in investigative
techniques have exposed genetic deviations in HCC, including
genetic mutations in CTNNB1 and TP53 (5), the overall picture of genetic
alterations during the tumorigenesis of HCC remains unclear.
The oncogene CTNNB1 has been identified as
one of the most commonly mutated genes in HCC (23). Overall, CTNNB1 is mutated in
nearly 30% of HCC, and the mutation frequency alters by different
etiologies. In the present study, no CTNNB1 mutation was
observed in the HBV-associated HCC, although CTNNB1
mutations were observed in 44 and 50% of the HCV-associated HCC and
non-virus-associated HCC, respectively. Guichard et al
(24) identified that the frequency
of CTNNB1 mutation is only 11% in HBV-associated HCC
compared with 40% in HCC of other etiologies (24). These results support those of the
present study that the CTNNB1 mutation was less predominant
in HBV-associated hepatocarcinogenesis.
It is also well known that TP53, which is a
tumor suppressor, is frequently mutated and inactivated in HCC
(24). Interestingly, in contrast
with the CTNNB1 mutation, a TP53 mutation was
detected in 64% of the HBV-associated HCC in the present study,
although TP53 mutations were detected in 36 and 50% of the
HCV-associated HCC and non-virus-associated HCC. This result was
confirmed by independent studies reporting that TP53 occurs
more frequently in HBV-associated HCC with a frequency between 30
and 40% compared with nearly 20% in HCV-associated HCC (24,25). In
addition, TP53 mutation was exclusive from CTNNB1
mutation in HBV-associated HCC (24).
This result indicates that TP53 mutation may be a key factor
in HBV-associated HCC.
Single point somatic mutations contain both a
transition and a transversion. Various types of mutation spectrum
have been found in various tumor with different rates of
transitions and transversions (22).
In the present study, C:G>T:A transition (50%), C:G>G:C
transversion (19.6%), T:A>C:G transition (19.6%), C:G>A:T
transversion (12.5%), T:A>G:C (12.5%) transversion, and
T:A>A:T transversion (5.4%) were detected in the HCC. A number
of studies have demonstrated that somatic mutation patterns are
significantly distinct from the expected spectrum (24,26–28). In
HCC, the C:G>T:A transition, T:A>C:G transition and
C:G>A:T transversion are common mutations; however, in the
present study, the C:G>G:C transversion was observed at a high
frequency. This suggests that the C:G>G:C transversion may be
associated with the etiology of HCC.
The use of NGS in the present study also revealed
mutations associated with HCC that had not been clearly determined,
including STK11, CSF1R and RET. In those
mutations, STK11 mutations were critical, since 75% of those
mutations were frameshift variants and missense variants. A
previous genetic analysis of the STK11 gene demonstrated
that this mutation may serve a role in tumor progression in a
subset of HCC, protecting from p53-dependent apoptosis (29). Huang et al (30) identified that decreased expression of
STK11 is associated with poor prognosis. These results
indicate that STK11, which has a lower mutation frequency,
may be critical for a subset of HCC.
In conclusion, the use of NGS in the present study
identified a number of novel gene mutations in HCC, including
established mutations and disproved mutations. These results
provide new insight into and improved understanding of the
etiology, and the development, of HCC. Further investigations
including whole exome sequencing are required to fully elucidate
genetic mutations in HCC.
References
|
1
|
Morishita A and Masaki T: miRNA in
hepatocellular carcinoma. Hepatol Res. 45:128–141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Venook AP, Papandreou C, Furuse J and de
Guevara LL: The incidence and epidemiology of hepatocellular
carcinoma: A global and regional perspective. Oncologist. 15 Suppl
4:S5–S13. 2010. View Article : Google Scholar
|
|
3
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han ZG: Functional genomic studies:
Insights into the pathogenesis of liver cancer. Annu Rev Genomics
Hum Genet. 13:171–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meyerson M, Gabriel S and Getz G: Advances
in understanding cancer genomes through second-generation
sequencing. Nat Rev Genet. 11:685–696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shendure J and Ji H: Next-generation DNA
sequencing. Nat Biotechnol. 26:1135–1145. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mardis ER, Ding L, Dooling DJ, Larson DE,
McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath
SD, et al: Recurring mutations found by sequencing an acute myeloid
leukemia genome. N Engl J Med. 361:1058–1066. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pleasance ED, Stephens PJ, O'Meara S,
McBride DJ, Meynert A, Jones D, Lin ML, Beare D, Lau KW, Greenman
C, et al: A small-cell lung cancer genome with complex signatures
of tobacco exposure. Nature. 463:184–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berger MF, Hodis E, Heffernan TP, Deribe
YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E,
Ghosh P, et al: Melanoma genome sequencing reveals frequent PREX2
mutations. Nature. 485:502–506. 2012.PubMed/NCBI
|
|
12
|
Nikolaev SI, Rimoldi D, Iseli C, Valsesia
A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, et
al: Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2
mutations in melanoma. Nat Genet. 44:133–139. 2012. View Article : Google Scholar
|
|
13
|
Prickett TD, Wei X, Cardenas-Navia I, Teer
JK, Lin JC, Walia V, Gartner J, Jiang J, Cherukuri PF, Molinolo A,
et al: Exon capture analysis of G protein-coupled receptors
identifies activating mutations in GRM3 in melanoma. Nat Genet.
43:1119–1126. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stark MS, Woods SL, Gartside MG, Bonazzi
VF, Dutton-Regester K, Aoude LG, Chow D, Sereduk C, Niemi NM, Tang
N, et al: Frequent somatic mutations in MAP3K5 and MAP3K9 in
metastatic melanoma identified by exome sequencing. Nat Genet.
44:165–169. 2012. View
Article : Google Scholar
|
|
15
|
Wei X, Walia V, Lin JC, Teer JK, Prickett
TD, Gartner J, Davis S; NISC Comparative Sequencing Program, ;
Stemke-Hale K, Davies MA, et al: Exome sequencing identifies GRIN2A
as frequently mutated in melanoma. Nat Genet. 43:442–446. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Puente XS, Pinyol M, Quesada V, Conde L,
Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz
M, et al: Whole-genome sequencing identifies recurrent mutations in
chronic lymphocytic leukaemia. Nature. 475:101–105. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Papaemmanuil E, Cazzola M, Boultwood J,
Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS,
Hellstrom-Lindberg E, Gambacorti-Passerini C, et al: Somatic SF3B1
mutation in myelodysplasia with ring sideroblasts. N Engl J Med.
365:1384–1395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yoshida K, Sanada M, Shiraishi Y, Nowak D,
Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et
al: Frequent pathway mutations of splicing machinery in
myelodysplasia. Nature. 478:64–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang K, Kan J, Yuen ST, Shi ST, Chu KM,
Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al: Exome sequencing
identifies frequent mutation of ARID1A in molecular subtypes of
gastric cancer. Nat Genet. 43:1219–1223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y,
Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et
al: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thorvaldsdottir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Greenman C, Stephens P, Smith R, Dalgliesh
GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C,
et al: Patterns of somatic mutation in human cancer genomes.
Nature. 446:153–158. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li M, Zhao H, Zhang X, Wood LD, Anders RA,
Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, et al:
Inactivating mutations of the chromatin remodeling gene ARID2 in
hepatocellular carcinoma. Nat Genet. 43:828–829. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang Z, Jhunjhunwala S, Liu J, Haverty
PM, Kennemer MI, Guan Y, Lee W, Carnevali P, Stinson J, Johnson S,
et al: The effects of hepatitis B virus integration into the
genomes of hepatocellular carcinoma patients. Genome Res.
22:593–601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Totoki Y, Tatsuno K, Yamamoto S, Arai Y,
Hosoda F, Ishikawa S, Tsutsumi S, Sonoda K, Totsuka H, Shirakihara
T, et al: High-resolution characterization of a hepatocellular
carcinoma genome. Nat Genet. 43:464–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim CJ, Cho YG, Park JY, Kim TY, Lee JH,
Kim HS, Lee JW, Song YH, Nam SW, Lee SH, et al: Genetic analysis of
the LKB1/STK11 gene in hepatocellular carcinomas. Eur J Cancer.
40:136–141. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang YH, Chen ZK, Huang KT, Li P, He B,
Guo X, Zhong JQ, Zhang QY, Shi HQ, Song QT, et al: Decreased
expression of LKB1 correlates with poor prognosis in hepatocellular
carcinoma patients undergoing hepatectomy. Asian Pac J Cancer Prev.
14:1985–1988. 2013. View Article : Google Scholar : PubMed/NCBI
|