Introduction
Doxorubicin (DOX) is one of the most widely used and
successful antitumor drugs; however, it has a number of serious
side effects, including hematopoietic suppression, nausea,
vomiting, extravasation and alopecia, of which cardiac toxicity has
been the major concern with its use in cancer treatment (1). Cardiac toxicity may be present following
DOX chemotherapy as symptoms ranging from asymptomatic
electrocardiography-changes to pericarditis and decompensated
cardiomyopathy (2). The mechanisms by
which DOX causes cardiac toxicity have been widely investigated
(2).
Cardiac toxicity from DOX involves changes in the
high-energy phosphate pool, endothelin-1 levels and disturbances of
myocardial adrenergic signaling (2).
An increase in cardiac oxidative stress is also associated with
DOX-induced cardiomyopathy through damage, including lipid
peroxidation, reduced antioxidant levels and fewer sulfhydryl
groups (3). Previous studies have
established that DOX-induced myocardial apoptosis performs a role
in the development of heart failure (4,5). The
DOX-induced cardiac toxicity causes myofibrillar deterioration
through cardiomyocyte apoptosis (6),
endothelial cell apoptosis (7) and
intracellular calcium dysregulation (8). The level of apoptosis can be measured by
monitoring caspase-3, one of the major executors of apoptosis
(9).
It has been suggested that dysregulation of
autophagy performs an important role in DOX-induced cardiotoxicity;
however, the detailed mechanisms were less well understood
(10). DOX-induced cardiomyocyte
autophagy may be protective or detrimental depending on the dosage
of DOX (11). However, it is now
speculated that cardiomyocyte autophagy may be one of the major
mechanisms of cardiotoxicity in itself (12,13).
Autophagy is an essential process for optimal
cellular function and survival, where damaged or unwanted proteins
and organelles are removed from the cell (14). Autophagy may be stimulated in order to
protect the cell from stress stimuli or, alternatively, to
contribute to cell death (10).
Cardiac autophagy is a maladaptive response to hemodynamic stress
(15). An increased number of
autophagosomes have been observed in cardiac tissues from patients
with aortic stenosis (16) and
dilated cardiomyopathy (17).
Autophagy serves a role in cardiomyocyte salvage (18) and it is essential for cardioprotection
when induced by ischemic preconditioning (19,20). It
also performs distinct roles during ischemia and reperfusion. It
may be protective during ischemia, whereas detrimental during
reperfusion (21). The Beclin 1-Vps34
complex was pivotal in driving autophagy, which is required for
autophagosome formation; autophagy related 14 (ATG14L) enhances
vacuolar protein sorting 34 (Vps34) activity in a Beclin
1-dependent manner, thus positively regulating autophagy, while
Rubicon suppresses autophagosome maturation through inhibiting
Vps34 activity (22–24). There are a number of ways of measuring
autophagy. As microtubule-associated protein 1A/1B-light chain 3
(LC3)-II is consistently kept in the membrane of the autophagosome
until it fuses with the lysosomal membrane, LC3-II is used as a
marker of autophagosomes (25). The
formation of the autophagosome membrane is followed by its
subsequent fusion with the lysosome via lysosomal-associated
membrane proteins (LAMP1 and LAMP2), as well as Rab7 and UV
radiation resistance-associated gene (3). Co-localization of LC3-II and LAMP
reflects the fusion of autophagosome and lysosomal, that is to say,
autophagosome maturation (26).
Finally, nucleoporin 62 (p62) can be used to monitor autophagic
flux, since total cellular expression levels of p62 are inversely
associated with autophagic activity (27).
Previous studies have shown that there is cross talk
between autophagic and apoptotic pathways, including inducer and
signaling pathways (28,29). Autophagy protein 5 (Atg5), which was
previously characterized as a protein specifically required for
autophagy, can directly lead to cell death by activating the
apoptotic pathway without the activation of autophagic pathways
(30,31). Furthermore, B cell lymphoma-2 (Bcl-2)
and B cell lymphoma-extra-large, the well-characterized
anti-apoptotic proteins, appear to be important modulators of
autophagy through binding and inhibiting Beclin 1 (32). Preservation of zinc finger
transcription factor GATA-binding protein-4 (GATA-4) has been shown
to protect cardiomyocytes from DOX-induced toxicity by inhibiting
apoptosis and autophagy through regulating expression of Bcl-2
positively, and Beclin-1 negatively (5,33).
However, the cross talk between autophagy and apoptosis is complex.
In certain conditions, autophagy can suppress apoptosis, and in
others, autophagy can induce apoptosis (34). The interaction of autophagy and
apoptosis in the background of DOX-induced cardiac toxicity
requires further exploration.
Our previous study provided the first evidence that
thrombopoietin (TPO) is a protective factor against DOX-induced
cardiotoxicity, since it was demonstrated that TPO reduced
DOX-induced apoptosis of H9C2 cells (35). TPO is a cytokine produced by liver and
kidney and regulates the production of platelets. It promotes
megakaryocytic/platelet lineage, angiogenesis and inhibits
apoptosis (36,37). Other studies are in agreement with the
present results (38,39). A study reported that erythropoietin
(EPO), which is homogenous with TPO, regulates autophagy via the
protein kinase B (Akt)/mechanistic target of rapamycin signaling
pathway (40). However, the
regulation of TPO on autophagy has not been reported. TPO shares
homology with EPO (41) and their
function is also similar. Since EPO is also a heart protective
factor (42,43), it was proposed that TPO may also
regulate autophagy.
Therefore, in the present study, the hypothesis that
TPO may perform protective roles in cardiac cells against
DOX-induced autophagy and apoptosis was investigated. The research
model was established in H9C2 cells, the cardiomyocyte cell line
that is often used in studies on cardiomyocyte function (44), and was used in our previous study
(35). Bafilomycin A1 (BFA) treatment
was used as a positive control for autophagy inhibition, since BFA
is a specific inhibitor of vacuolar type H+-ATPase in cells that
may effectively hinder fusion between autophagosomes and lysosomes
(45).
Materials and methods
Cell culture
Rat myoblast H9C2 cell line (American Type Culture
Collection; cat. no. CRL-1446) was purchased from the Chinese
Academy of Sciences (Shanghai, China) and maintained in Dulbecco's
modified Eagle's medium (Invitrogen; Thermo Fisher Scientific.
Inc., Waltham, MA, USA) supplemented with 10% fetal calf serum
(Invitrogen; Thermo Fisher Scientific, Inc.), 2 mM glutamine, 100
IU penicillin and 100 µg/ml streptomycin (46). Cells were cultured at 37°C in a
humidified atmosphere with 5% CO2.
Drug treatments
TPO (3SBio Inc., Shenyang, China) was dissolved in
saline to make a stock solution of 25 µg/ml and then diluted to
make a final concentration of 10 ng/ml for subsequent experiments.
DOX (Sangon Biotech Co., Ltd., Shanghai, China) was dissolved in
saline to make a stock solution of 25 mg/ml and then diluted to
make a final concentration of 5 µg/ml for subsequent experiments.
This dose was used with reference to our previous study, where
different doses of DOX and their effects on H2C9 cells were
investigated (35). BFA (Sangon
Biotech Co., Ltd.) was dissolved in saline to make a stock solution
of 100 µg/ml and then diluted to make a final concentration of 10
nM for autophagy inhibition. Cells were pretreated with TPO for 36
h, and then treated with DOX for 24 h, or treated with DOX for 24
h, and then with BFA for 6 h.
Western blot analysis
Cells were lysed in 200 µl of Laemmli buffer
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The protein
concentration was determined by the Bradford method. The protein
was then boiled in 1X loading buffer for 5 min and electrophoresed
on 12% SDS-PAGE. Total protein (30 µg) was loaded per lane and
electrophoretically transferred to a polyvinylidene fluoride
membrane. The membrane was blocked for 2 h at 37°C with 5% skimmed
milk powder. The membranes were incubated with primary antibodies
overnight at 4°C. Following three washes with TBST, the immobilized
protein samples were then incubated with horseradish peroxidase
secondary antibodies (cat. no. A0208; Beyotime Institute of
Biotechnology, Shanghai, China; A0208, dilution, 1:1,000) for 1 h
at 4°C, and the protein complex was detected using the enhanced
chemiluminescence substrate system (Pierce Biotechnology, Inc.,
Rockford, IL, USA). In particular, the blotting and imaging of
Bcl-2 and GATA-4 were performed with a full-automatic western blot
system (Protein Simple, San Jose, CA, USA).
The following primary antibodies were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA): p62 (cat. no.
5114, dilution, 1:1,000), Beclin-1 (cat. no. 3495, dilution,
1:1,000) and caspase-3 (cat. no. 9665, dilution, 1:1,000). The
following primary antibodies were purchased from Abcam (Cambridge,
UK): LAMP1 (cat. no. ab24170, dilution, 1:1,000), Bcl-2 (cat. no.
ab59348, dilution, 1:500) and GATA-4 (cat. no. ab134057, dilution,
1:500). LC3 was purchased from Thermo Fisher Scientific, Inc. (cat.
no. PA1-16930, dilution, 1:500). β-actin (cat. no. 20536-1-AP,
dilution, 1:1,000) and GAPDH (cat. no. 10494-1-AP, dilution,
1:1,000) antibodies were purchased from Proteintech Group
(Rosemont, IL, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from H9C2 cells, using RNA
extraction buffer E-Z 96 total RNA kit (Omega Bio-Tek, Norcross,
GA, USA), according to the manufacturer's protocol. The RT kit was
RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Temperature protocol was 42°C.
The sequences of all forward and reverse primers,
including the sequence of the reference gene primer are presented
below: R-GAPDH forward, 5′-GCTCTCTGCTCCTCCCTGTTCT-3′ and reverse,
5′-GCCAAATCCGTTCACACCGACCT-3′; R-LC3 forward,
5′-GCACTACGGCTGCTCTTTATAC-3′ and reverse,
5′-CACAGATCTTCAACAGCACAGT-3′; R-Beclin 1 forward,
5′-CAGGATGGTGTCTCTCGAAGA-3′ and reverse,
5′-CCCCGATCAGAGTGAAGCTAT-3′; R-ATG14L forward,
5′-CCGAACAATGGGGACTACTCT-3′ and reverse,
5′-TGGTGTAGGCAGGGTTGTTAT-3′; R-Rubicon forward,
5′-TCCCAGTTCAGTTCACGTGA-3′ and reverse, 5′-TGTAGGAAGCAGTGCGAGAA-3′;
R-Caspase-3 forward, 5′-TGGACTGCGGTATTGAGACA-3′ and reverse,
5′-GCGCAAAGTGACTGGATGAA-3′; R-Bcl-2 forward,
5′-AACTCTTCAGGGATGGGGTG-3′ and reverse, 5′-CACAGAGCGATGTTGTCCAC-3′;
and R-GATA-4 forward, 5′-CTAAACCTTACTGGCCGTAGC-3′ and reverse,
5′-GGGAGAAACAGCGTAAATGA-3′.
RT-qPCR analysis for autophagy-associated genes was
performed using an ABI StepOnePlus™ sequence detection
system (Thermo Fisher Scientific, Inc.). The LightCycler
amplification of PCR products was detected with SYBR-Green I dye
(Roche Diagnostics, Basel, Switzerland). PCR cycling conditions
were 95°C for 10 min and 40 cycles of 95°C for 15 sec, 60°C for 30
sec, and 72°C for 15 sec, followed by a final melting curve program
(95°C for 15 sec, 60°C for 60 sec, and 95°C for 15 sec). Relative
expression levels of target genes of interest were calculated using
the 2−ΔΔCq method, and GAPDH was used as an internal
control (47). Each sample was
investigated in triplicate and mean values were used for
quantitation.
Cell proliferation and cytotoxicity
assay
Cell viability was measured using Cell Counting
Kit-8 (CCK-8; 5 mg/ml; Beyotime Institute of Biotechnology) for 3 h
according to the manufacturer's protocol. The absorbance was then
measured at a wavelength of 492 nm using a microplate reader.
Autophagosome formation
The LC3-green fluorescent protein (GFP) lentivirus
expression vector was produced as previously described (48). H9C2 cells were infected with LC3-GFP
lentivirus in the presence of polybrene (6 µg/ml).
Lentivirus-infected cells were selected with blasticidin (5 µg/ml)
and maintained for 10 days in medium containing blasticidin.
Stably-infected, blasticidin-resistant H9C2 cells were cloned and
expanded as previously described (26). Following different treatments, H9C2
cells were washed three times in PBS and fixed with 4%
paraformaldehyde in PBS (Guangzhou Chemical Reagent Factory,
Guangzhou, China) for 10 min at room temperature. H9C2 cells were
observed at ×650 magnification laser scanning confocal microscopy
(IX81 + FV10-MCPSU + IX2-UCB + U-RFL-T; Olympus Corporation, Tokyo,
Japan). GFP-LC3 puncta represented the autophagosome. Autophagy was
analyzed by quantifying the mean number of GFP-LC3 puncta per cell
in all cells in the population by using Photoshop CS5 (Adobe
Systems, Inc., San Jose, CA, USA).
Immunostaining
Cells transfected with LC3-GFP vectors were seeded
on coverslips at a density of 1×104 cells/ml), and fixed
with PBS solution containing 4% paraformaldehyde for 10 min at room
temperature (25°C). The coverslips were blocked with 10% normal
goat serum (Vector Labs, Burlingame, CA, USA) at room temperature
(25°C) for 60 min. The coverslips were then incubated with LAMP1
antibody (cat. no. ab24170, dilution, 1:200; Abcam) at 4°C
overnight, and the following day with Alexa Fluor-conjugated
secondary antibody (ab150077, dilution 1:1,000; Abcam, Shanghai,
China) at 37°C for 30 min. Finally, fluorescent mounting media
containing DAPI was added. The results were observed at ×100
magnification laser scanning confocal microscopy (IX81 + FV10-MCPSU
+ IX2-UCB + U-RFL-T; Olympus Corporation).
Hematoxylin and eosin (H&E)
staining
Following drug treatment, H9C2 cells were incubated
in 4% paraformaldehyde for 20 min at room temperature (25°C), and
washed with PBS followed by hematoxylin solution incubation for 10
min at room temperature (25°C). Cells were next washed in running
tap water for 10 min, immersed in distilled water briefly and in
95% alcohol for 5 sec. The cells were then counterstained in eosin
solution for 30 sec to 2 min at room temperature (25°C). Cell
morphology was examined under a ×40 light microscope.
Statistical analysis
Data were analyzed by SPSS 19.0 statistical software
(IBM SPSS, Armonk, NY, USA). Data are presented as the mean ±
standard deviation. Statistical significance was estimated by
one-way analysis of variance with least significant difference
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
TPO increases viability of DOX-treated
cardiomyocytes
The CCK-8 assays demonstrated that DOX treatment (5
µg/ml, 24 h) significantly impaired H9C2 cell viability (reduced by
27.1±1.1%, n=3; P<0.001; Fig. 1A)
when compared with the control, while TPO pretreatment prior to DOX
significantly improved cell viability (increased by 7.8±0.3%, n=3;
P<0.001; Fig. 1A) when compared
with the DOX treatment group. These results indicated that TPO can
protect cardiomyocytes from DOX-induced growth inhibition.
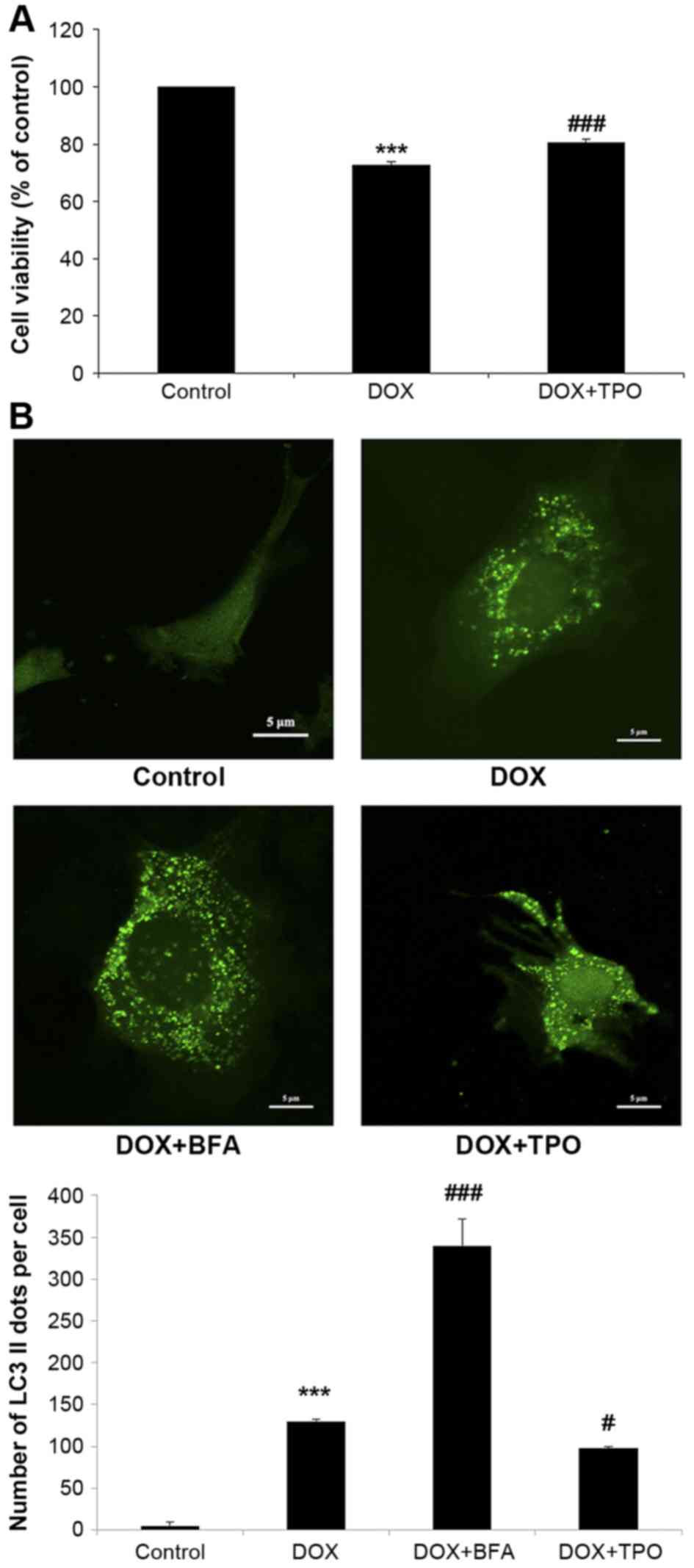 | Figure 1.TPO increased the viability of H9C2
cells after DOX treatment and reduced DOX-induced autophagy. H9C2
cells were divided into four groups. Control group was untreated.
DOX groups were all incubated in the presence of DOX (5 µg/ml, 24
h); besides, DOX + BFA group was subsequently treated with BFA (10
nM, 6 h), while DOX + TPO group pretreated with TPO (10 ng/ml, 36
h). (A) The viability was estimated by Cell Counting Kit-8 assay.
(B) H9C2 cells were transduced with LC3-GFP lentivirus.
Representative fluorescent images were shown. Scale bar, 5 µm.
Autophagosomes were calculated as the mean number of GFP-LC3 puncta
per cell for all cells in the population. Results are presented as
the mean ± standard deviation and were estimated by one-way
analysis of variance with least significant difference post-hoc
test (n=3). ***P<0.001 vs. control group; #P<0.05
and ###P<0.001 vs. DOX treated alone group. TPO,
thrombopoietin; DOX, doxorubicin; BFA, bafilomycin A1; GFP, green
fluorescent protein; LC3, microtubule-associated protein
1A/1B-light chain 3. |
TPO reduces DOX-induced autophagy in
cardiomyocytes
The mean number of GFP-LC3 puncta per cell was
counted in all cells in each field (Fig.
1B). There were negligible GFP-LC3 puncta in the control cells,
while DOX markedly increased the GFP-LC3 puncta, indicating that
DOX was able to induce cardiomyocyte autophagy (n=3; control 5±5.0
vs. DOX 130±14.0; P<0.001). H9C2 cells were treated with the
lysosomal inhibitor BFA to block autolysosome formation, and the
autophagosome formation was significantly increased compared with
the DOX alone treated cells (n=3; DOX 130±14.0 vs. DOX+BFA
340±32.2; P<0.001). However, the mean number of GFP-LC3 puncta
per cell was significantly decreased by TPO pretreatment compared
with DOX alone, indicating the ability of TPO to inhibit
DOX-induced autophagy (n=3; DOX 130±14.0 vs. DOX + TPO 98±9.6;
P<0.05).
TPO regulates autophagy-associated
proteins
The mRNA and protein levels of LC3-II and Beclin 1
were all upregulated by DOX treatment (n=3; all P<0.001;
Fig. 2), and were further increased
by lysosomal inhibitor BFA (P<0.01 or P<0.001). However, TPO
pretreatment prior to DOX treatment reduced the mRNA level of LC3
and Beclin 1 compared with the DOX-treated alone group, which was
consistent with the protein expression (P<0.01 or P<0.001).
DOX treatment significantly decreased ATG14L and Rubicon mRNA
levels compared with the control (n=3; both P<0.001; Fig. 2). BFA post-treatment and TPO
pretreatment lead to a significant increase in ATG14L mRNA upon DOX
treatment (P<0.05 or P<0.01; Fig.
2A).
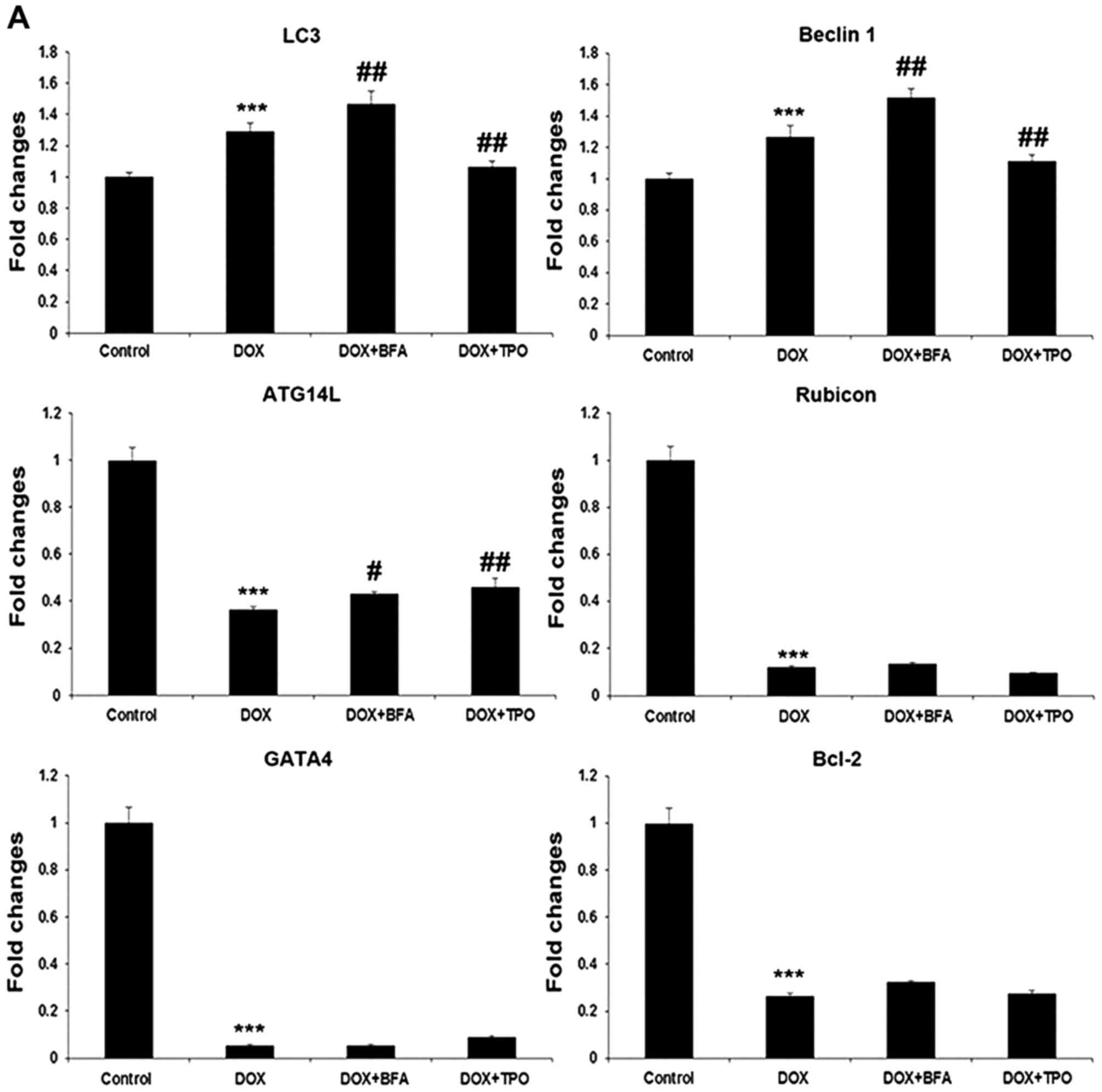 | Figure 2.TPO pretreatment reduced
autophagy-promoting factors while increased autophagy-inhibitory
factors. Grouping was set the same as described in Fig. 1. (A) Reverse
transcription-quantitative polymerase chain reaction for the genes
encoding LC3, Beclin 1, ATG14L, Rubicon, GATA-4 and Bcl-2. (B)
Western blots: Representative images and quantification histograms
for LC3-I/II, Beclin 1, p62, GATA-4 and Bcl-2. Results are
presented as the mean ± standard deviation and were estimated by
one-way analysis of variance with least significant difference
post-hoc test (n=3). *P<0.05 and ***P<0.001, compared with
control group; #P<0.05, ##P<0.01 and
###P<0.001 vs. DOX treated alone group. LC3,
microtubule-associated protein 1A/1B-light chain 3; GATA-4,
GATA-binding protein-4; Bcl-2, B cell lymphoma 2; p62, nucleoporin
62; DOX, doxorubicin; ATG14L, autophagy related 14; TPO,
thrombopoietin; BFA, bafilomycin A1. |
The protein amount of p62 was significantly
decreased in DOX-treated cells compared with the control cells
(P<0.05), and increased following BFA addition compared with the
DOX single treatment; TPO-pretreated H9C2 cells compared with
non-TPO-pretreated H9C2 cells also showed increased p62 levels
following exposure to DOX (P<0.001; Fig. 2B).
The expression of anti-autophagic proteins was also
detected. GATA-4 mRNA was decreased 18-fold following DOX treatment
(n=3; P<0.001; Fig. 2A); neither
TPO nor BFA could upregulate GATA4 expression upon DOX treatment.
However, at protein level, both the BFA post-treatment and TPO
pretreatment showed significantly increased GATA-4 compared to DOX
alone treated cells (n=3; P<0.01 or P<0.001; Fig. 2B). Furthermore, although BFA and TPO
had no effect on Bcl-2 mRNA level upon DOX treatment, it
significantly increased the protein level (n=3; P<0.05 or
P<0.001; Fig. 2B).
Taken together, these results confirmed that TPO
reduced DOX-induced autophagy in H9C2 cells.
TPO has no significant effect on
autophagosome maturation
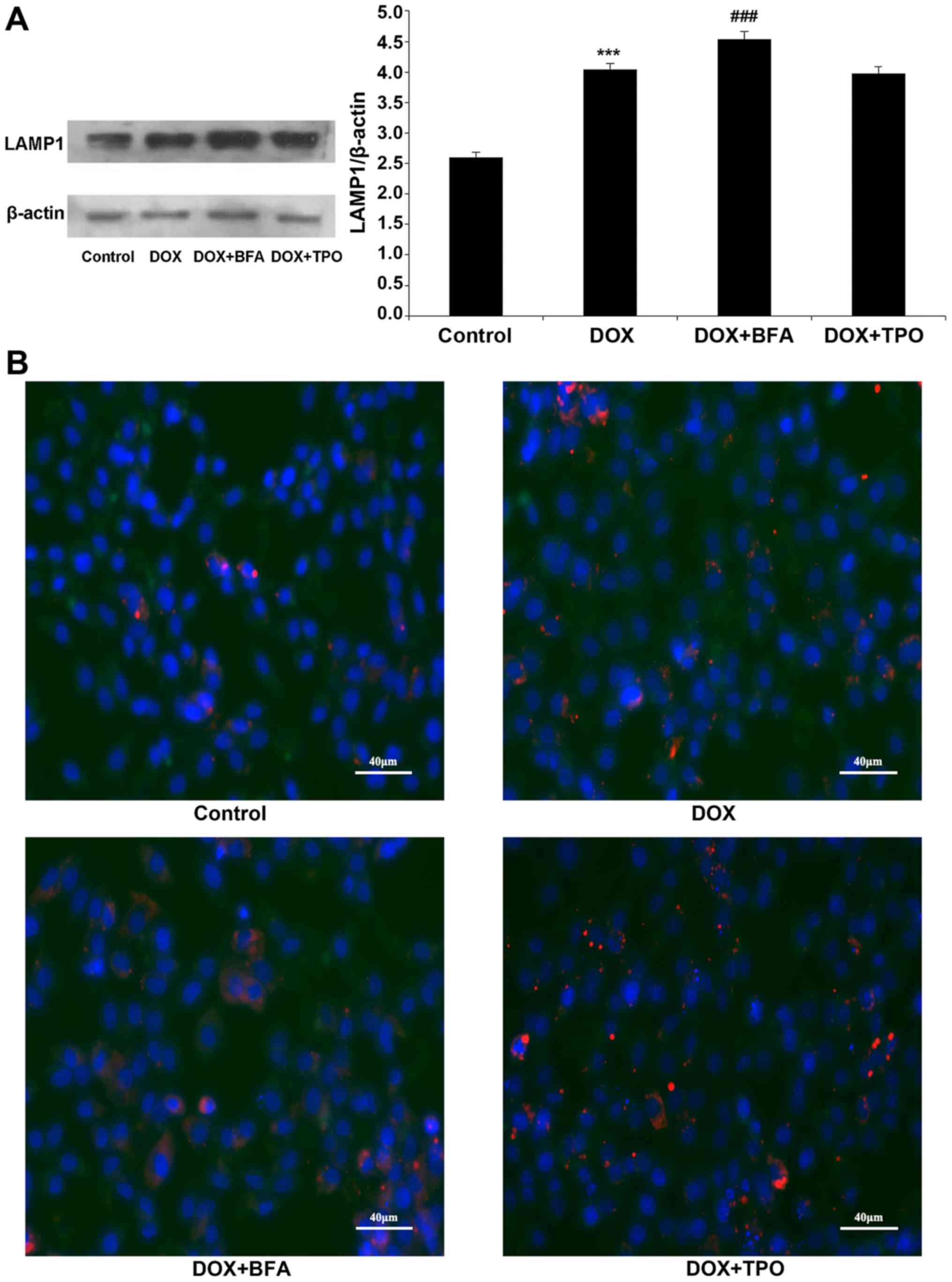
LAMP1 protein level significantly increased
following DOX treatment compared with the control, indicating that
DOX may regulate lysosome function (n=3; P<0.001; Fig. 3A). However, TPO had no effect on LAMP
expression compared with DOX treatment alone. In order to assess
the effect of TPO on autophagosome maturation, the co-localization
of GFP-LC3-labelled autophagosomes with LAMP1-stained lysosomes was
measured. The number of LC3+/LAMP1+
co-stained autophagosomes was higher in each of the DOX-treated
groups compared with that in the control group. BFA addition
reduced LC3 and LAMP1 co-localization, indicating that BFA blocked
DOX-induced autophagosome maturation. However, the numbers of
LC3+/LAMP1+ co-stained autophagosomes in the DOX + TPO groups were
not statistically significant different from the DOX group,
indicating that TPO did not block DOX-induced autophagosome
maturation (Fig. 3B).
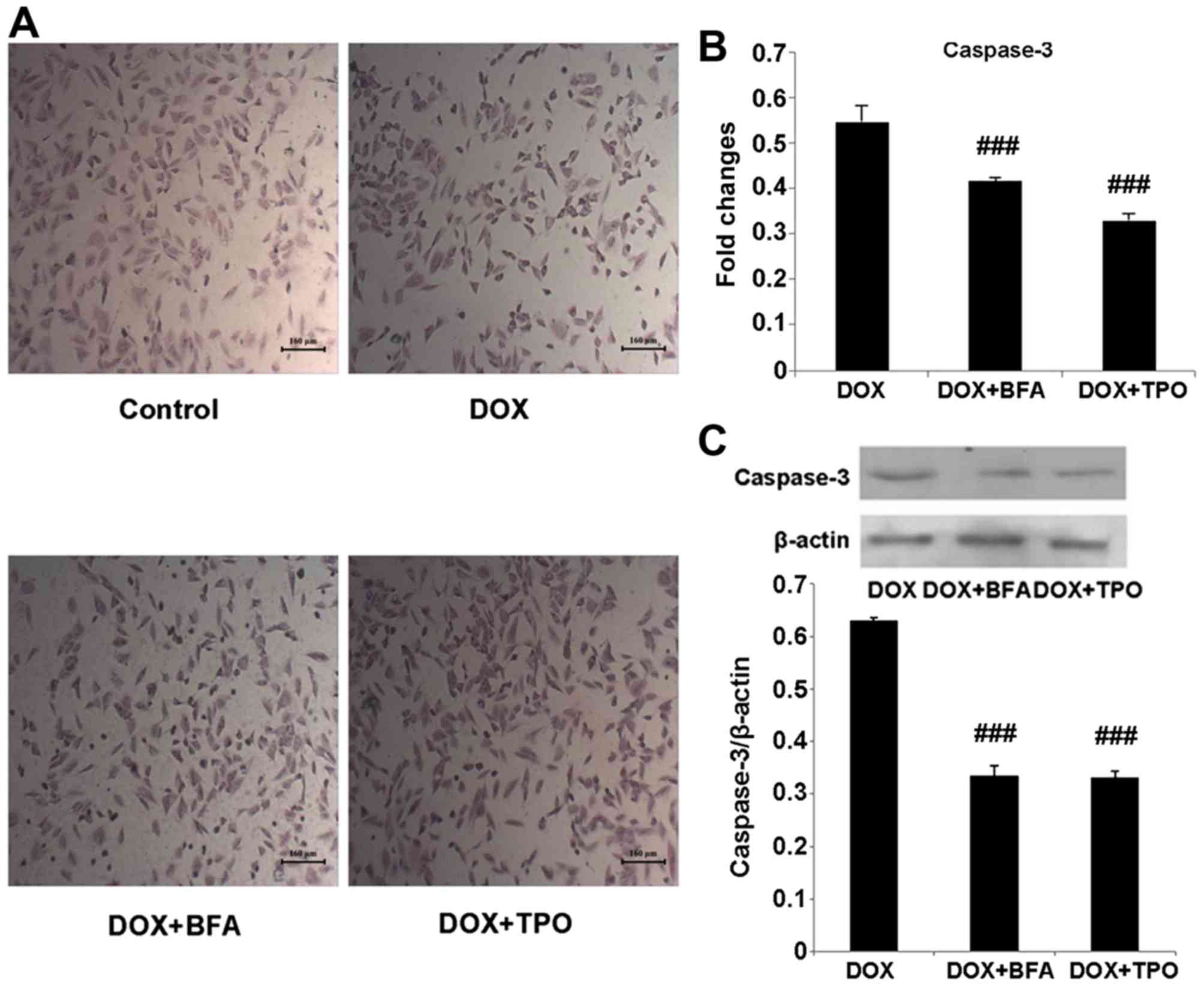
TPO treatment reduces apoptosis in
H9C2 cells
In the H&E stained specimens of H9C2 cells, the
morphology of control cells was not altered, whereas cells in the
DOX-treated group decreased in size and altered to a round shape.
With TPO or BFA treatment, a number of cells were restored to their
normal morphology (Fig. 4A). The mRNA
and protein levels of caspase-3 in DOX + TPO-treated groups were
significantly reduced compared with that in the DOX-treated alone
group (Fig. 4B and C; both
P<0.001). The DOX + BFA-treated groups showed the same change
(Fig. 4B and C; both P<0.001).
Taken together, these results demonstrated that in DOX-treated H9C2
cells, the inhibition of autophagy reduced apoptosis.
Discussion
In the present study, it was revealed that TPO could
protect H9C2 cells from DOX-induced excessive autophagy and
apoptosis. These results explored a novel mechanism of the
cardioprotective function of TPO on doxorubicin-induced myocardial
damage. To the best of our knowledge, this is the first study to
demonstrate that apart from apoptosis, TPO may significantly reduce
DOX-induced autophagy.
DOX-induced cardiotoxicity has been a major
limitation on its usage in cancer therapy (1). TPO has been approved to exert
cardioprotective effects when used with DOX in cancer therapy
(35). Although mechanisms of its
cardioprotective effects remain unclear, previous studies have
indicated that TPO regulates the expression of numerous genes,
including genes involved in apoptosis, Akt and extracellular
signal-regulated kinase pathways, cell division regulators, blood
vessel remodeling and matrix, channel regulators, muscle and
contractile proteins (37). The
present study firstly demonstrated that TPO also regulates
DOX-induced autophagy in H9C2 cells. The present data confirmed
that autophagy and apoptosis are upregulated in H9C2 cells
following DOX treatment, whereas TPO inhibits DOX-induced autophagy
and apoptosis.
Autophagy is a conserved cellular process involving
the degradation of a cell's own components, which may be either
protective or detrimental in DOX-treated cells depending on the
stress level (21). A number of
studies have revealed that DOX treatment can increase autophagy in
cardiac cells, which mediates DOX-induced cardiotoxicity (12,13). In
the present study, DOX increased H9C2 autophagy was indicated by
the accumulation of GFP-LC3 puncta and increased expression of
Beclin 1, which is in agreement with other studies (5,49).
Notably, it was demonstrated that upon DOX treatment, TPO inhibits
H9C2 cell autophagy as shown by the decreased expression of GFP-LC3
puncta, LC3II, Beclin 1 and increased amount of p62 compared with
the DOX alone-treated groups.
In the present study, the regulatory effect of BFA
upon DOX-induced autophagy was not completely consistent with that
of TPO. BFA and TPO promoted the accumulation of p62 upon DOX
treatment, indicating that the ultimate outcome of their regulation
on autophagy was inhibitory. However, BFA significantly increased
GFP-LC3 puncta compared with DOX treatment alone, which was in
accordance with the well-accepted conclusion that BFA suppresses
autophagic degradation through inhibiting the fusion between
autophagosomes and lysosomes, which then leads to an increase in
autophagosomes (12,13); while TPO has no such effect on
autophagosomes. It was confirmed that BFA blocked the DOX-induced
autophagosome maturation by detecting co-localization of LC3 and
LAMP (26). LAMP1 is an important
lysosomal membrane protein, which is expressed at high levels in a
number of normal tissue cells and is responsible in part for
maintaining lysosomal integrity (50). It was revealed that TPO did not affect
the appearance of LC3+/LAMP1+ autophagosomes,
which indicated that the inhibitory effect of TPO on DOX-induced
autophagy was irrelevant to autophagosome maturation arrest.
Furthermore, it was revealed that TPO downregulated
the autophagy-promoting factor Beclin 1, while BFA brought about an
opposite result of Beclin 1 regulation, which may be associated
with feedback mechanisms for compensating the blocked autophagy
flux. Taken together, these results indicated that TPO may
negatively regulate autophagy at the early stage, which is
different from the autophagy-inhibitory functions of BFA primarily
in autolysosome formation.
It has been demonstrated that autophagy can promote
survival by inhibiting apoptosis or facilitating apoptosis
(51). The present study indicated
that DOX induces autophagy and facilitates apoptosis in
cardiomyocytes. GATA-4 is a zinc finger transcription factor, which
protects cardiomyocytes from DOX-induced toxicity by inhibiting
autophagy as well as apoptosis through regulating expression of
Bcl-2 and Beclin1 (5). GATA-4
performs important roles in the development and hypertrophy of
adult cardiac myocytes (33). Its
downregulation is critical in DOX-induced cardiotoxicity (33). In the present study, the mRNA and
protein level of GATA-4 and Bcl-2 were all significantly reduced in
cardiomyocytes exposed to DOX, which was consistent with a previous
study (5). Notably, TPO reversed the
protein amount decrease. Additional studies are necessary to reveal
the comprehensive roles of TPO in the cross talk between autophagy
and apoptosis at the molecular level, particularly to investigate
whether TPO inhibits autophagy and apoptosis of cardiomyocytes
through regulating GATA-4 and/or Bcl-2 expression.
In conclusion, the present study demonstrated that
DOX induces autophagy in cardiomyocytes, which contributes to
DOX-induced cardiomyocyte death. TPO is able to attenuate
DOX-induced cardiomyocyte autophagy and apoptosis. GATA-4 and Bcl-2
may be involved in the regulation of TPO on autophagy. This study
investigated the new mechanism that the cardioprotective role of
TPO on doxorubicin-induced myocardial damage and could provide a
theoretical basis for TPO used as a cardioprotective agent in
clinical.
Acknowledgements
The present study was supported by the Hainan
Provincial Natural Science Foundation of China (grant no. 20158307)
and the Hainan Province Association of Science and Technology for
Youth Science and Technology Talents Innovation project (grant no.
HAST201632).
References
|
1
|
Zhang YW, Shi J, Li YJ and Wei L:
Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch
Immunol Ther Exp (Warsz). 57:435–445. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: from molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Simůnek T, Stérba M, Popelová O, Adamcová
M, Hrdina R and Gersl V: Anthracycline-induced cardiotoxicity:
Overview of studies examining the roles of oxidative stress and
free cellular iron. Pharmacol Rep. 61:154–171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kumar D, Kirshenbaum LA, Li T, Danelisen I
and Singal PK: Apoptosis in adriamycin cardiomyopathy and its
modulation by probucol. Antioxid Redox Signal. 3:135–145. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi S, Volden P, Timm D, Mao K, Xu X
and Liang Q: Transcription factor GATA4 inhibits
doxorubicin-induced au-tophagy and cardiomyocyte death. J Biol
Chem. 285:793–804. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nitobe J, Yamaguchi S, Okuyama M, Nozaki
N, Sata M, Miyamoto T, Takeishi Y, Kubota I and Tomoike H: Reactive
oxygen species regulate FLICE inhibitory protein (FLIP) and
susceptibility to Fas-mediated apoptosis in cardiac myocytes.
Cardiovasc Res. 57:119–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grethe S, Coltella N, Di Renzo MF and
Pörn-Ares MI: p38 MAPK downregulates phosphorylation of Bad in
doxorubicin-induced endothelial apoptosis. Biochem Biophys Res
Commun. 347:781–790. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arai M, Yoguchi A, Takizawa T, Yokoyama T,
Kanda T, Kurabayashi M and Nagai R: Mechanism of
doxorubicin-induced inhibition of sarcoplasmic reticulum
Ca(2+)-ATPase gene transcription. Circ Res. 86:8–14. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang B, Ye D and Wang Y: Caspase-3 as a
therapeutic target for heart failure. Expert Opin Ther Targets.
17:255–263. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dirks-Naylor AJ: The role of autophagy in
doxorubicin-induced cardiotoxicity. Life Sci. 93:913–916. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sciarretta S, Hariharan N, Monden Y,
Zablocki D and Sadoshima J: Is autophagy in response to ischemia
and reperfusion protective or detrimental for the heart? Pediatr
Cardiol. 32:275–281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen K, Xu X, Kobayashi S, Timm D,
Jepperson T and Liang Q: Caloric restriction mimetic 2-deoxyglucose
antagonizes doxorubicin-induced cardiomyocyte death by multiple
mechanisms. J Biol Chem. 286:21993–22006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu X, Chen K, Kobayashi S, Timm D and
Liang Q: Resveratrol attenuates doxorubicin-induced cardiomyocyte
death via inhibition of p70 S6 kinase 1-mediated autophagy. J
Pharmacol Exp Ther. 341:183–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gottlieb RA and Mentzer RM Jr:
Cardioprotection through autophagy: Ready for clinical trial?
Autophagy. 7:434–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu H, Tannous P, Johnstone JL, Kong Y,
Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill
JA: Cardiac autophagy is a maladaptive response to hemodynamic
stress. J Clin Invest. 117:1782–1793. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hein S, Arnon E, Kostin S, Schönburg M,
Elsässer A, Polyakova V, Bauer EP, Klövekorn WP and Schaper J:
Progression from compensated hypertrophy to failure in the
pressure-overloaded human heart: Structural deterioration and
compensatory mechanisms. Circulation. 107:984–991. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kostin S, Pool L, Elsässer A, Hein S,
Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klövekorn
WP and Schaper J: Myocytes die by multiple mechanisms in failing
human hearts. Circ Res. 92:715–724. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen HH, Mekkaoui C, Cho H, Ngoy S,
Marinelli B, Waterman P, Nahrendorf M, Liao R, Josephson L and
Sosnovik DE: Fluorescence tomography of rapamycin-induced autophagy
and cardioprotection in vivo. Circ Cardiovasc Imaging. 6:441–447.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang C, Yitzhaki S, Perry CN, Liu W,
Giricz Z, Mentzer RM Jr and Gottlieb RA: Autophagy induced by
ischemic preconditioning is essential for cardioprotection. J
Cardiovasc Transl Res. 3:365–373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haar L, Ren X, Liu Y, Koch SE, Goines J,
Tranter M, Engevik MA, Nieman M, Rubinstein J and Jones WK: Acute
consumption of a high-fat diet prior to ischemia-reperfusion
results in cardioprotection through NF-κB-dependent regulation of
autophagic pathways. Am J Physiol Heart Circ Physiol.
307:H1705–H1713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsui Y, Takagi H, Qu X, Abdellatif M,
Sakoda H, Asano T, Levine B and Sadoshima J: Distinct roles of
autophagy in the heart during ischemia and reperfusion: Roles of
AMP-activated protein kinase and Beclin 1 in mediating autophagy.
Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsunaga K, Saitoh T, Tabata K, Omori H,
Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe
T, et al: Two Beclin 1-binding proteins, Atg14L and Rubicon,
reciprocally regulate autophagy at different stages. Nat Cell Biol.
11:385–396. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhong Y, Wang QJ and Yue Z: Atg14L and
Rubicon: yin and yang of Beclin 1-mediated autophagy control.
Autophagy. 5:890–891. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM,
Chait BT, Heintz N and Yue Z: Distinct regulation of autophagic
activity by Atg14L and Rubicon associated with Beclin
1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 11:468–476.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kimura S, Fujita N, Noda T and Yoshimori
T: Monitoring autophagy in mammalian cultured cells through the
dynamics of LC3. Methods Enzymol. 452:1–12. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang C, Lee JS, Inn KS, Gack MU, Li Q,
Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C and Jung JU:
Beclin1-binding UVRAG targets the class C Vps complex to coordinate
autophagosome maturation and endocytic trafficking. Nat Cell Biol.
10:776–787. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
BenYounès A, Tajeddine N, Tailler M, Malik
SA, Shen S, Métivier D, Kepp O, Vitale I, Maiuri MC and Kroemer G:
A fluorescence-microscopic and cytofluorometric system for
monitoring the turnover of the autophagic substrate p62/SQSTM1.
Autophagy. 7:883–891. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rubinstein AD and Kimchi A: Life in the
balance-a mechanistic view of the crosstalk between autophagy and
apoptosis. J Cell Sci. 125:5259–5268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI,
Woo HN, Cho DH, Choi B, Lee H, Kim JH, et al: Essential roles of
Atg5 and FADD in autophagic cell death: Dissection of autophagic
Cell death into vacuole formation and cell death. J Biol Chem.
280:20722–20729. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yousefi S, Perozzo R, Schmid I, Ziemiecki
A, Schaffner T, Scapozza L, Brunner T and Simon HU:
Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis.
Nat Cell Biol. 8:1124–1132. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aries A, Paradis P, Lefebvre C, Schwartz
RJ and Nemer M: Essential role of GATA-4 in cell survival and
drug-induced cardiotoxicity. Proc Natl Acad Sci USA. 101:pp.
6975–6980. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: Apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li K, Sung RY, Huang WZ, Yang M, Pong NH,
Lee SM, Chan WY, Zhao H, To MY, Fok TF, et al: Thrombopoietin
protects against in vitro and in vivo cardiotoxicity induced by
doxorubicin. Circulation. 113:2211–2220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kuter DJ and Begley CG: Recombinant human
thrombopoietin: Basic biology and evaluation of clinical studies.
Blood. 100:3457–3469. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Majka M, Ratajczak J, Villaire G, Kubiczek
K, Marquez LA, Janowska-Wieczorek A and Ratajczak MZ:
Thrombopoietin, but not cytokines binding to gp130 protein-coupled
receptors, activates MAPKp42/44, AKT, and STAT proteins in normal
human CD34+ cells, megakaryocytes, and platelets. Exp Hematol.
30:751–760. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chan KY, Xiang P, Zhou L, Li K, Ng PC,
Wang CC, Zhang L, Deng HY, Pong NH, Zhao H, et al: Thrombopoietin
protects against doxorubicin-induced cardiomyopathy, improves
cardiac function, and reversely alters specific signalling
networks. Eur J Heart Fail. 13:366–376. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baker JE, Su J, Hsu A, Shi Y, Zhao M,
Strande JL, Fu X, Xu H, Eis A, Komorowski R, et al: Human
thrombopoietin reduces myocardial infarct size, apoptosis, and
stunning following ischaemia/reperfusion in rats. Cardiovasc Res.
77:44–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu Y, Shiou SR, Guo Y, Lu L, Westerhoff M,
Sun J, Petrof EO and Claud EC: Erythropoietin protects epithelial
cells from excessive autophagy and apoptosis in experimental
neonatal necrotizing enterocolitis. PLoS One. 8:e696202013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
de Sauvage FJ, Hass PE, Spencer SD, Malloy
BE, Gurney AL, Spencer SA, Darbonne WC, Henzel WJ, Wong SC, Kuang
WJ, et al: Stimulation of megakaryocytopoiesis and thrombopoiesis
by the c-Mpl ligand. Nature. 369:533–538. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tramontano AF, Muniyappa R, Black AD,
Blendea MC, Cohen I, Deng L, Sowers JR, Cutaia MV and El-Sherif N:
Erythropoietin protects cardiac myocytes from hypoxia-induced
apoptosis through an Akt-dependent pathway. Biochem Biophys Res
Commun. 308:990–994. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Parsa CJ, Matsumoto A, Kim J, Riel RU,
Pascal LS, Walton GB, Thompson RB, Petrofski JA, Annex BH, Stamler
JS and Koch WJ: A novel protective effect of erythropoietin in the
infarcted heart. J Clin Invest. 112:999–1007. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peter AK, Bjerke MA and Leinwand LA:
Biology of the cardiac myocyte in heart disease. Mol Biol Cell.
27:2149–2160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yoshimori T, Yamamoto A, Moriyama Y, Futai
M and Tashiro Y: Bafilomycin A1, a specific inhibitor of
vacuolar-type H(+)-ATPase, inhibits acidification and protein
degradation in lysosomes of cultured cells. J Biol Chem.
266:17707–17712. 1991.PubMed/NCBI
|
|
46
|
Hescheler J, Meyer R, Plant S, Krautwurst
D, Rosenthal W and Schultz G: Morphological, biochemical, and
electrophysiological characterization of a clonal cell (H9c2) line
from rat heart. Circ Res. 69:1476–1486. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hannan NR, Jamshidi P, Pera MF and
Wolvetang EJ: BMP-11 and myostatin support undifferentiated growth
of human embryonic stem cells in feeder-free cultures. Cloning Stem
Cells. 11:427–435. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhang YY, Meng C, Zhang XM, Yuan CH, Wen
MD, Chen Z, Dong DC, Gao YH, Liu C and Zhang Z: Ophiopogonin D
attenuates doxorubicin-induced autophagic cell death by relieving
mitochondrial damage in vitro and in vivo. J Pharmacol Exp Ther.
352:166–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Eskelinen EL: Roles of LAMP-1 and LAMP-2
in lysosome biogenesis and autophagy. Mol Aspects Med. 27:495–502.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sishi BJ, Loos B, van Rooyen J and
Engelbrecht AM: Autophagy upregulation promotes survival and
attenuates doxorubicin-induced cardiotoxicity. Biochem Pharmacol.
85:124–134. 2013. View Article : Google Scholar : PubMed/NCBI
|