Introduction
Lung cancer remains the global leading cause of
malignancy-associated deaths, with the highest incidence and
mortality rate, causing 1.6 million deaths worldwide in 2012
(1). Non-small cell lung cancer
(NSCLC) accounts for >80% of the cases and is often diagnosed at
advanced stages of the disease. Platinum-based chemotherapy is the
standard first-line systemic treatment; however, it has limited
efficacy and significant toxicity (2–4). Over the
previous decades, epidermal growth factor receptor (EGFR) has
become a therapeutic target, as it triggers a signaling cascade
that facilitates cell proliferation, survival and invasion. In
particular, EGFR-tyrosine kinase inhibitors (TKIs) have emerged as
an alternative cancer treatment (2–4). Although
compared to chemotherapy the outcomes of studies involving random
populations are poor, TKIs elicit high response rates among NSCLC
patients with EGFR mutations (5,6). The
Iressa Pan-Asia Study reported that NSCLC patients with EGFR
mutations gained prolonged progression-free survival with gefitinib
treatment compared to chemotherapy (7). However, the prognosis in patients with
wild-type EGFR was improved with chemotherapy (7). EGFR can be activated with ErbB
receptors, either via autocrine or paracrine ligand binding. This
induces EGFR tyrosine kinase activity, which subsequently triggers
downstream signaling pathways (4,8).
Activating mutations in EGFR are able to activate tyrosine kinase
in the absence of ligands. Therefore, the downstream oncogenic
signaling pathways are intrinsically upregulated (9–11).
The two most commonly observed mutations are exon 19
deletions and L858R missense substitutions at position 858,
accounting for 60 and 35% of the total cases, respectively
(12,13). In the presence of these mutations, the
sensitivity of EGFR to EGFR-TKIs is increased and therefore these
mutations are beneficial. However, a variety of treatment responses
are observed and the sensitivity towards EGFR-TKIs diminished
following 8–12 months of treatment (7,10). This
effect is primarily due to the gaining of resistance to EGFR-TKIs,
either via primary resistance (de novo) or acquired
resistance, following exposure to targeted agents (14). In the majority of cases, acquired
resistance is due to secondary EGFR mutations (15). The most common mechanism is via
gaining EGFR T790M second-site mutation, which occurs following
EGFR-TKI treatment, and contributes to ~50% of cases (16). The second most common mechanism
involves MET amplification, which accounts for ~20% of cases
(17,18). The remaining cases are due to
mutations in phosphoinositide 3-kinase (PI3K) subunit α, Erb-B2
receptor tyrosine kinase 2 (ERBB2; HER2), BRAF, signal transducer
and activator of transcription 3, AXL receptor tyrosine kinase and
the amplification of CRK like proto-oncogene adaptor protein
(19–25).
Previous studies have reported that a common
Bcl2-like 11 (BIM) deletion polymorphism is associated with
EGFR-TKI resistance in NSCLC patients with EGFR mutations. The
polymorphism results in alternative splicing, which leads to
expression of BIM isoforms that lack a crucial pro-apoptotic
Bcl2-homology domain 3 (BH3) (26).
Failure to generate the functional pro-apoptotic isoform results in
a drug-resistant phenotype, whereby a reduced response is observed
following treatment with EGFR-TKI in patients with BMI
polymorphisms vs. patients without (27). It has also been reported that BIM
plays an essential role in EGFR-TKI-induced apoptosis, which may be
enhanced by BH3 mimetics (28–31).
Therefore, malfunction of BIM contributes considerably to the
development of drug resistance. Although the precise mechanism
remains to be elucidated, the MET oncogene is thought to be
involved in de novo and acquired resistance to EGFR-TKI in
NSCLC (32,33). Acquired resistance to EGFR-TKIs is a
major obstacle in the management of lung cancer; the present study
was initiated to investigate insights to tackle the issue.
Materials and methods
Cell lines and patient biopsies
HCC827 was obtained from the American Type Culture
Collection (Manassas, VA, USA) and four resistant cell lines
[gefitinib-cultured (GR) 1 and 2, erlotinib-cultured (ER) 1 and 2]
were successfully screened. The cells were screened via a gradual
increase in TKI dosage with a final concentration at 10 µM for 6
months. Formalin-fixed, paraffin-embedded NSCLC patient samples
were obtained from the Sun Yat-sen University Cancer Center between
January 2012 and December 2013 (State Key Laboratory of Oncology in
South China, Collaborative Innovation Center for Cancer Medicine,
Guangzhou, China). The age of the patients ranged from 43 to 71
years, with a median age of 56.5 years. The male to female sex
ratio was 3:7. Ethical approval and written informed consent was
obtained (Sun Yat-sen University Cancer Center Institutional Review
Board; approval no. YP2013-06-06). No personal information or
detailed clinical histories were disclosed.
Cytotoxicity assay
Cytotoxicity was assessed by a colorimetric assay
using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
dissolved in dimethyl sulfoxide. Cells were plated and treated with
gefitinib, erlotinib and sorafenib for 48 h. Cell proliferation
inhibition was expressed as the percentage of absorbance of control
cultures and measured at 570 nm with a microplate reader
(VICTOR3 Multilabel Reader; catalog no. 1420;
PerkinElmer, Inc., Waltham, MA, USA). The half maximal inhibitory
concentration (IC50) was calculated using GraphPad PRISM
software version 4.0 (GraphPad Software, Inc., La Jolla, CA,
USA).
Western blot analysis
To investigate the signaling properties of the cell
lines, western blotting was performed with antibodies against
various targets. Total protein lysate was collected with RIPA lysis
buffer (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing protease and phosphatase inhibitors (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) and quantified by BCA assay. Equal
amounts of protein (25–40 µg) were resolved on 10% SDS-PAGE gels
and subsequently transferred onto polyvinylidene difluoride (PVDF)
membrane. The PVDF membranes were blocked with 5% non-fat milk in
TBST for 30 min at room temperature and subsequently incubated
overnight at 4°C with primary antibodies of interest in 1:2,000
dilution as follows: ABCC4 (D2Q20), cat. no. 12705S; ABCG2, cat.
no. 4477S; Phospho-Akt (Ser473), cat. no. 9271; Phospho-Akt
(Thr308), cat. no. 9275S; Akt (pan) (11E7), cat. no. 4685; EGFR
E746-A750del, cat. no. 2085; EGFR, cat. no. 2232; GAPDH, cat. no.
2118; Phospho-MET (Tyr1234/1235) (3D7), cat. no. 3129 and pTEN
(138G6), cat. no. 9559 (Cell Signaling Technology, Inc., Danvers,
MA, USA); Bcl-2, cat. no. ab32124 (Abcam, Cambridge, UK), MET
(c-12), cat. no. sc-10 (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) and horseradish peroxidase-conjugated goat anti-rabbit (cat.
no. 166-2408) or goat anti-mouse (cat. no. 172-1011) secondary
antibodies in 1:5,000 dilution (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) for 2 h at room temperature. The blots were
developed with enhanced chemiluminescence substrate (GE Healthcare
Life Sciences, Chalfont, UK) and by autoradiography.
Immunohistochemistry
Tumor specimens were collected, processed and
sectioned. Pathological changes were observed by staining with
haematoxylin and eosin. For Bcl2 immunostaining, sections were
de-paraffinized and rehydrated through a gradient of ethanol. The
samples underwent antigen retrieval by incubating in 10 mM of
citrate buffer at 95°C for 20 minutes. Slides were subsequently
blocked with 3% bovine serum albumin in TBST and incubated with
monoclonal mouse anti-human Bcl2 (Clone 124) (cat. no. M0887;1:50;
DAKO; Agilent Technologies, Inc., Santa Clara, CA) for 2 hours.
After that, samples were rinsed with phosphate-buffered saline and
then incubated with DAKO REAL Envision HRP antibodies (cat. no.
K5007; DAKO; Agilent Technologies, Inc., Santa Clara, CA) for 30
mins. The stain was finally visualized in brown with
3,3-diaminobenzidine (DAB) as substrate following counterstained
with Mayer's hematoxylin. After mounting, images were captured
under the microscope Axio Observer Z1 (Carl Zeiss, Germany).
Immunofluorescence staining
Cells were plated on a sterilized cover glass and
fixed with 4% paraformaldehyde. The cells were permeabilized with
0.1% Triton X-100 and were subsequently incubated for 2 h at room
temperature with antibody against EGFR E746-A750del (cat. no. 2085;
Cell Signaling Technology, Inc.) at a dilution ratio of 1:100. EGFR
exon 19-deletion staining was visualized with appropriate
conjugated secondary antibodies (Alexa Fluor® 488;
Thermo Fisher Scientific, Inc.). Cell nuclei were visualized with
DAPI stain. Finally, the cover glasses were mounted on slides by
anti-fade prolonged gold media (Invitrogen; Thermo Fisher
Scientific, Inc.).
Quantitative (q)PCR
The RNA levels of Bcl2 in cells with resistance to
EGFR-TKI were validated by qPCR using a Bcl2 Taqman Gene Expression
Assay (cat. no. Hs00608023_m1; Thermo Fisher Scientific, Inc.). The
reverse transcription PCR reaction was performed using 7500
Software v2.0.6 and 7500 Real-time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) with the following thermocycler
protocol: 50°C for 2 min, 95°C for 10 min, and then a two-step
cycle of 95°C for 15 sec and 60°C for 60 sec, for 40 cycles. The
RNA expressions of Bcl2 were normalized to the parental HCC827 for
each sample by the 2−ΔΔCq method (34).
Statistical analysis
Analyses were performed using PRISM software version
4.0 (GraphPad Software, Inc., La Jolla, CA). Unpaired t-test with
Welch Correction was used unless specified. The significance of the
Bcl2 RNA levels between resistant cell lines and parental HCC827
was determined by one-way ANOVA followed by Tukey's Honest
Significant difference post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Sensitivity of the resistant cell
lines to TKIs
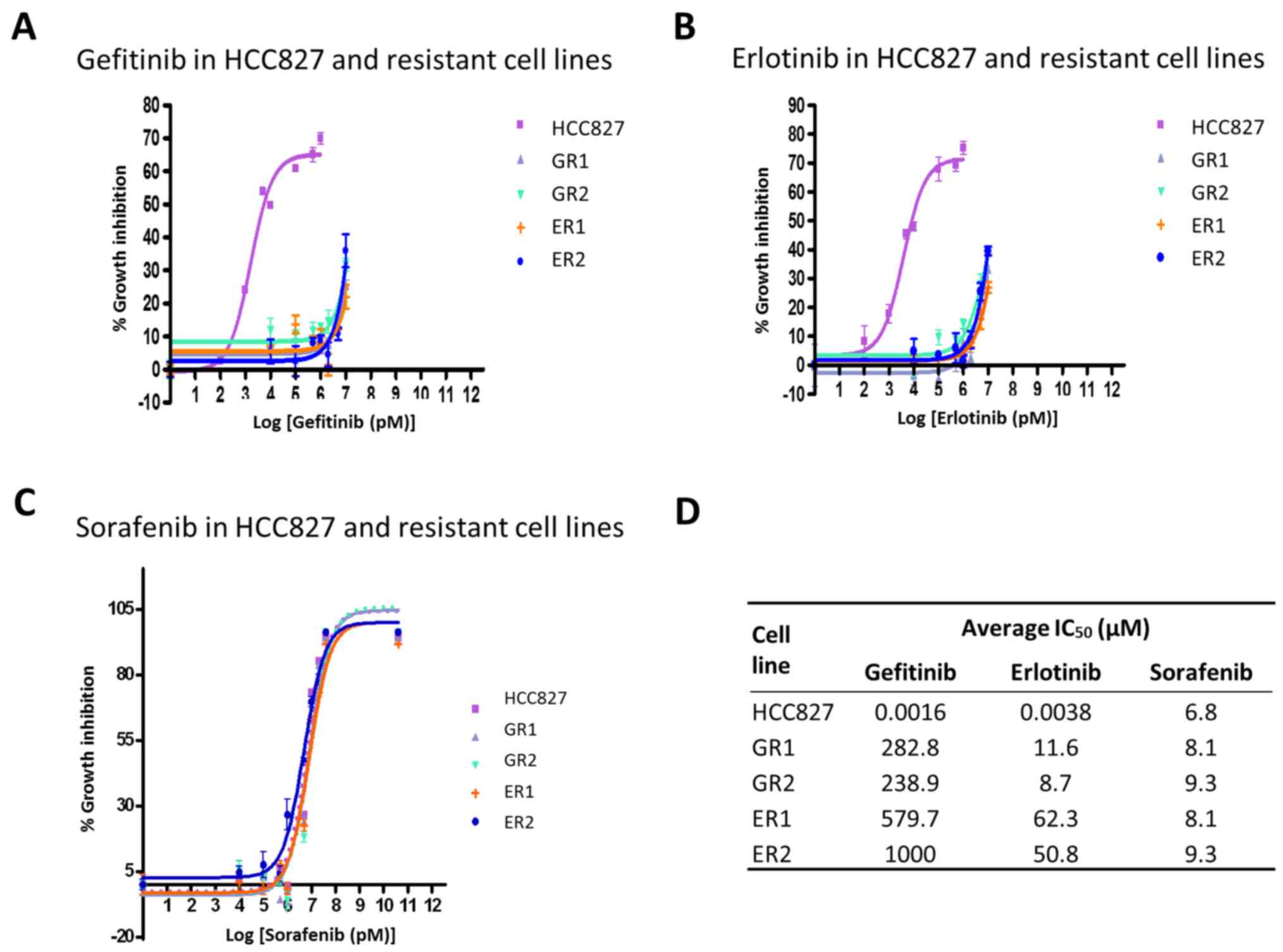
The EGFR-mutant HCC827 is the only NSCLC cell line
sensitive to EGFR-TKIs, with IC50 values of ~5 nM. The
cells were treated with gefitinib, erlotinib and sorafenib for 48
h. The IC50 values of the resistant cell lines, treated
with gefitinib and erlotinib, increased 1,000-fold from 1.6 nM-1.0
mM (Fig. 1A and B). The
IC50 values of the cell lines treated with gefitinib
were 1.6 nM, 282.8, 238.9 and 579.7 µM and 1.0 mM for HCC827, GR1,
GR2, ER1 and ER2, respectively. The IC50 values of the
cell lines treated with erlotinib were 3.8 nM, 11.6, 8.7, 62.3 and
50.8 µM for HCC827, GR1, GR2, ER1 and ER2, respectively. The
sensitivity of the cell lines towards sorafenib, a specific
multikinase inhibitor for vascular endothelial growth factor
receptors and platelet-derived growth factor receptors, was also
investigated. No statistically significant differences were
observed between the parental HCC827 cell line and the resistant
clones, with IC50 values ~5–10 µM (Fig. 1C). The origin of the resistant clones
was verified by the presence of exon 19 deletion (E746-750) in the
resistant clones via immunofluorescence. This confirmed that the
screened resistant clones (GR1, GR2, ER1 and ER2) originated from
the parental HCC827 cell line. The specificity of the antibody was
verified by parallel testing with H358 (EGFR wild type) by western
blotting (Fig. 2).
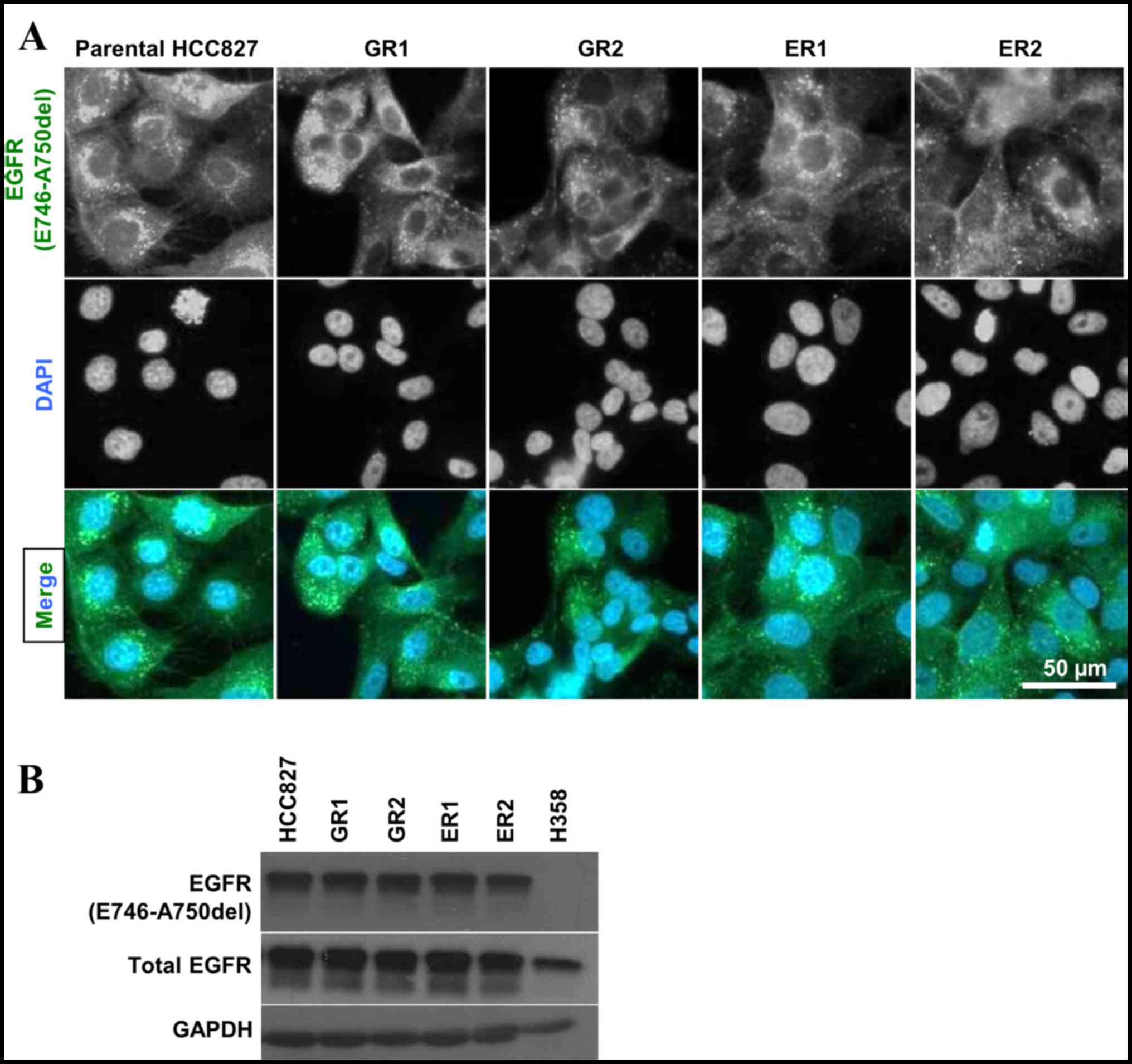 | Figure 2.(A) Immunofluorescence staining of
EGFR in HCC827 and four EGFR-TKI resistant cell lines (GR1, GR2,
ER1 and ER2) with antibody against EGFR exon 19 deletion
(E746-A750del; green) and DAPI (blue). The presence of EGFR
(E746-A750del) was detected in all samples. (B) EGFR exon 19
deletion (E746-A750del) was detected by immunoblotting in HCC827,
all four resistant cell lines (GR1, GR2, ER1 and ER2) and H358.
Specificity of the antibody against E746-A750del was validated with
H358, which served as a negative control. EGFR-TKI, epidermal
growth factor receptor-tyrosine kinase inhibitor; EGFR, epidermal
growth factor receptor; ER, erlotinib-cultured; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; GR, gefitinib-cultured.
Scale bar, 50 µm. |
Upregulation of Bcl2 in EGFR-TKI
resistant NSCLC cell line and associated signaling pathway
Strong expression of Bcl2 was observed in resistant
clones, with the exception of ER2. Bcl2 was not detected in the
parental cell line HCC827. The associated signaling pathways were
investigated via western blotting. PTEN, phosphorylated (p)-MET and
MET were downregulated in the resistant clones, with the exception
of ER2. ER2 had similar levels of expression of PTEN,
phosphorylated (p)-MET and MET proteins compared with HCC827.
Phosphorylated and un-phosphorylated forms of Akt were more
strongly expressed in the resistant clones than HCC827, as a
consequence of the reduced PTEN expression in the resistant clones.
Furthermore, there were no differences in expression of the drug
resistant pumps, ATP-binding cassette transporter G2 (ABCG2) and
ATP-binding cassette subfamily C member 4 (ABCC4), in the resistant
clones compared with HCC827 (Fig.
3A).
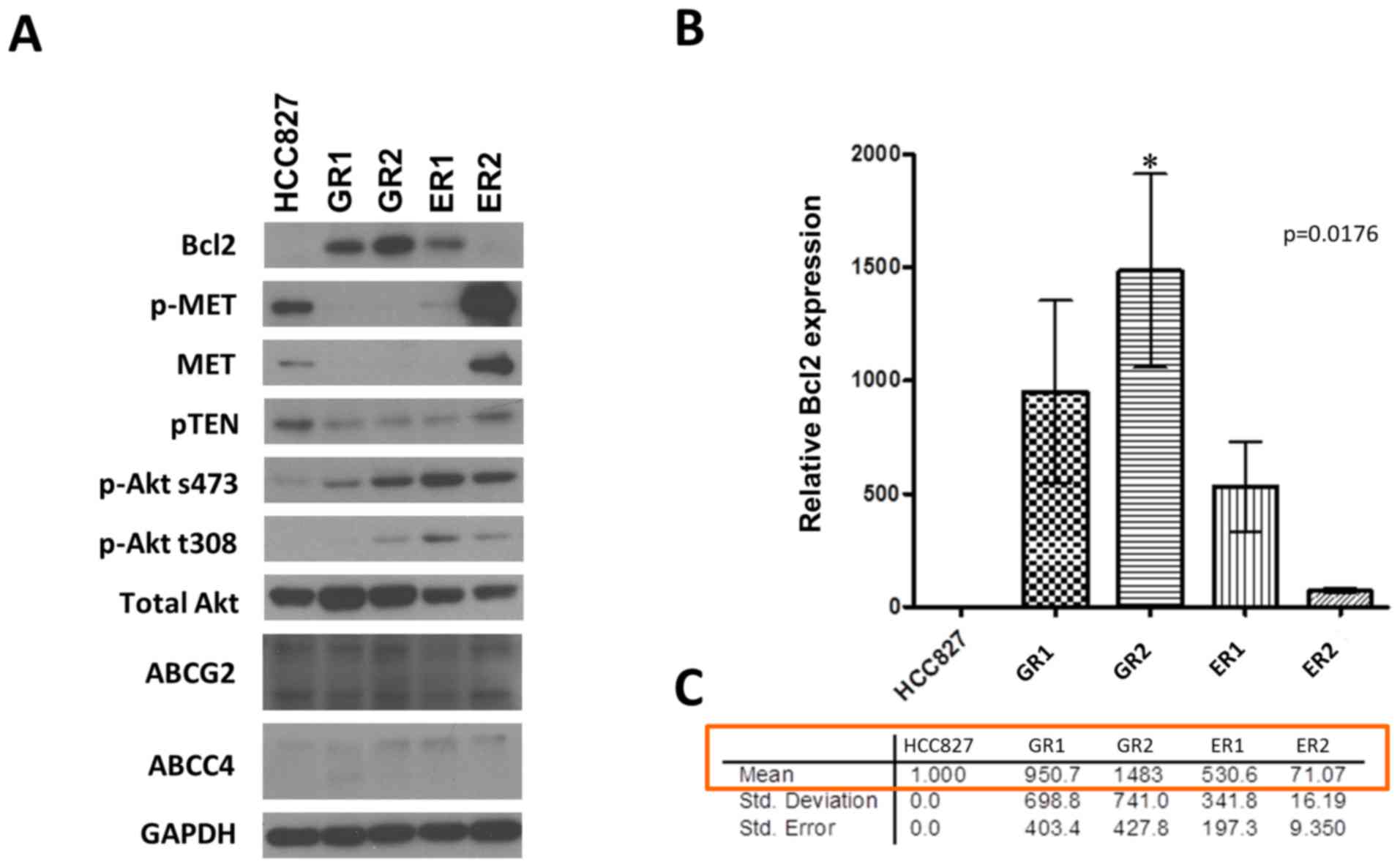 | Figure 3.(A) Detection of protein expression
by immunoblotting. A marked Bcl2 upregulation was observed in
EGFR-TKI resistant cell lines (GR1, GR2, ER1 and ER2). (B) Bcl2 RNA
expression level was quantified by qPCR and normalized to GAPDH
expression with the error bars representing standard deviation. The
significance of the Bcl2 RNA levels between resistant cell lines
and parental HCC827 was determined by one-way ANOVA with P=0.0176,
followed by Tukey's HSD post-hoc test. *P<0.05, HCC827 vs. GR2.
RNA levels of Bcl2 in resistant cell lines were 70–1,000-fold
higher vs. HCC827. (C) Using parental HCC827 as the standard, the
Bcl2 RNA levels were upregulated by 951-, 1,483-, 531- and 71-folds
for GR1, GR2, ER1 and ER2, respectively. The Bcl2 level in ER2
increased by a relatively less extent compared with the other
resistant clones. ABCC4, ATP-binding cassette subfamily C member 4;
ABCG2, ATP-binding cassette transporter G2; EGFR-TKI, epidermal
growth factor receptor-tyrosine kinase inhibitor; ER,
erlotinib-cultured; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase; GR, gefitinib-cultured; p, phosphorylated; PTEN,
phosphatase and tensin homolog; qPCR, quantitative PCR. |
The level of Bcl2 RNA expression was examined to
validate the upregulation of Bcl2 in resistant clones (Fig. 3B). It was observed that the levels of
RNA expression of Bcl2 in resistant clones were 70–1,500-fold
higher compared to the parental HCC827 cell line. The highest level
of expression (1,483 fold) was observed in GR2, whilst the lowest
level (71 fold) was observed in ER2 (Fig.
3B). This is a potential explanation for the observation that
ER2 did not resemble the other three clones as demonstrated by the
western blots (Fig. 3A).
Upregulation of Bcl2 in patient
biopsies in relation to TKI resistance
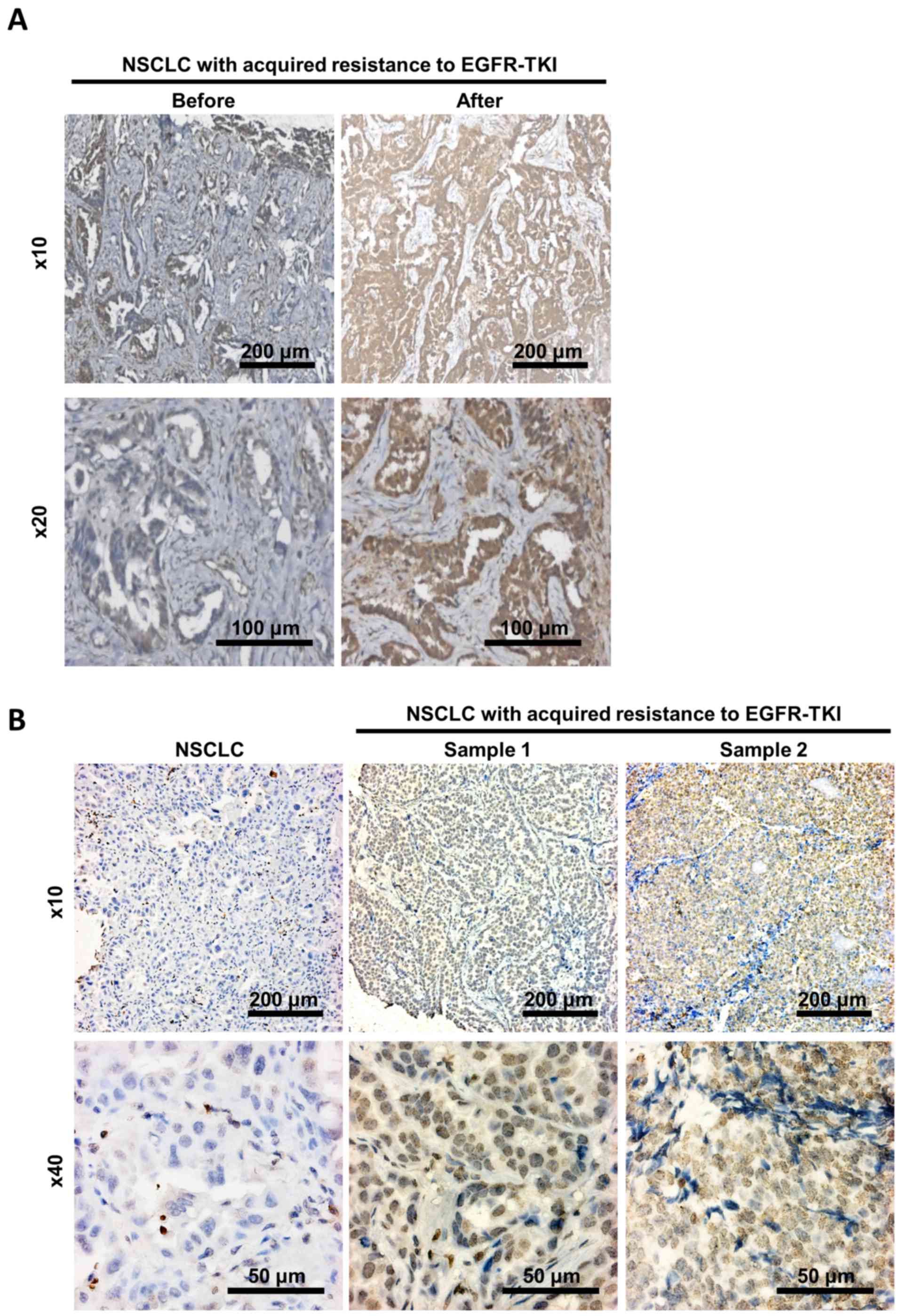
Small scale IHC staining was conducted on the NSCLC
patient samples. Low levels of Bcl2 expression were detected in
samples collected prior to resistance development, whereas strong
levels were detected subsequent to acquiring resistance (Fig. 4A). The staining of independent samples
indicated that Bcl2 was weakly expressed in non-resistant samples,
but that it was markedly detected in resistant NSCLC samples
(Fig. 4B). Together, these results
provided preliminary clinical evidence to the in vitro
observation that upregulation of Bcl2 was identified to be
associated with TKI resistance.
Discussion
Lung carcinoma is an important human health issue,
with respect to its high incidence and mortality rate (1). Platinum-based chemotherapy is the
conventional standard treatment regime, but it has several
drawbacks including limited efficacy and significant toxicity. The
emergence of EGFR-TKIs signifies a remarkable breakthrough in the
development of NSCLC therapeutics, particularly for patients with
EGFR mutations. However, patients who initially respond to
EGFR-TKIs develop resistance following varying periods of time on
account of heterogeneous responses to TKIs (14,15).
Secondary mutations are closely associated with the development of
drug resistance. Great interest has been generated in overcoming
this impediment in the management of lung cancer (35,36).
BIM deletion polymorphisms were reported to be one
of the molecular mechanisms contributing to intrinsic EGFR-TKI
resistance (26). BIM is a
proapoptotic member of the Bcl2 family, encoding gene products with
a BH3 domain, including BIMEL, BIML and
BIMS, which are essential elements for inducing
apoptosis, activating proapoptotic and antagonizing antiapoptotic
proteins (37–39). The deletion polymorphism switches BIM
splicing from exon 4 to exon 3, which generates BIM isoforms that
lack the BH3 domain, resulting in a diminished ability to trigger
cell death (27).
In the present study, four HCC827 resistant cell
lines were screened successfully by inducing mild selection
pressure; GR1 and GR2 were screened with gefitinib, while ER1 and
ER2 were screened with erlotinib. The resistant cell lines were
shown to be highly insensitive to EGFR-TKIs, with the
IC50 values against EGFR-TKIs 1,000-fold higher than the
parental HCC827 cell line. The results indicated a cross-resistance
across the same TKI family, as GR and ER cell lines demonstrated
high tolerance to erlotinib. Furthermore, the gaining of acquired
resistance did not alter the sensitivity of cells against TKIs of
other families e.g., sorafenib.
The resistance mechanism of the parental and
resistant cell lines was investigated by examining their signaling
pathways. The results illustrated a marked upregulation of Bcl2 in
resistant clones, which has not been previously reported in
EGFR-TKI acquired resistance. RNA expression of the resistant cell
lines further confirmed that Bcl2 was markedly upregulated.
Furthermore, the resistance was not due to ATP-binding cassette
(ABC) multidrug transporters, as the western blots showed no
effects on ABCG2 and ABCC4 proteins. These proteins were detected
in patients undergoing chemotherapy, and shown to confer resistance
against cytotoxic compounds applied in cancer therapy by blocking
the drugs from reaching intracellular targets (40–42). This
further confirmed the role of aberrant BIM function on acquired
resistance to EGFR-TKI.
MET amplification also contributes to the
development of acquired resistance to EGFR-TKI by activating
PI3K/Akt signaling through ERBB3 (32). It was reported that MET, EGFR, ERBB2
and ERBB3 were all phosphorylated in parental cell lines and the
proteins were reduced upon gefitinib treatment. Downregulation of
the ERBB3/PI3K/Akt signaling axis leads to the induction of
apoptosis. By contrast, high levels of phosphorylation remained in
the resistant cells in the presence of gefitinib, which caused
little or no apoptotic effect (32,43). Based
on the results, phosphorylation of MET was reduced in GR1, GR2 and
ER1, whilst phosphorylation of Akt was increased. Meanwhile, PTEN
expression was decreased in accordance with the level of Akt
phosphorylation. The hypothesis that Bcl-2 is a contributing factor
to the EGFR-TKI-acquired resistant was further supported by
immunohistochemical staining of Bcl2 on patient biopsies. This
implied that MET amplification may not be a potential mechanism of
acquired resistance, PTEN and Bcl2 are more likely to be
contributing factors of acquired resistance, meaning that the
resistance mechanism is potentially pathway-specific.
In conclusion, EGFR-TKI acquired resistance is a
major obstacle in lung cancer therapy. The results of the present
study provide evidence that upregulation of Bcl2 may be a latent
driver for resistance exploitation. This warrants further
investigation in targeting Bcl2 as a strategy in treating NSCLC
with acquired resistance.
Acknowledgements
The authors would like to thank the Charlie Lee
Charitable Foundation (Hong Kong, Hong Kong, SAR, P.R. China) for
their support.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hotta K, Fujiwara Y, Matsuo K, Suzuki T,
Kiura K, Tabata M, Takigawa N, Ueoka H and Tanimoto M: Recent
improvement in the survival of patients with advanced nonsmall cell
lung cancer enrolled in phase III trials of first-line, systemic
chemotherapy. Cancer. 109:939–948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peters S, Adjei AA, Gridelli C, Reck M,
Kerr K and Felip E; ESMO Guidelines Working Group, : Metastatic
non-small-cell lung cancer (NSCLC): ESMO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 23
Suppl 7:vii56–vii64. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arteaga CL: The epidermal growth factor
receptor: From mutant oncogene in nonhuman cancers to therapeutic
target in human neoplasia. J Clin Oncol. 19 18 Suppl:32S–40S.
2001.PubMed/NCBI
|
|
5
|
Gatzemeier U, Pluzanska A, Szczesna A,
Kaukel E, Roubec J, De Rosa F, Milanowski J, Karnicka-Mlodkowski H,
Pesek M, Serwatowski P, et al: Phase III study of erlotinib in
combination with cisplatin and gemcitabine in advanced
non-small-cell lung cancer: The Tarceva Lung Cancer Investigation
Trial. J Clin Oncol. 25:1545–1552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herbst RS, Prager D, Hermann R,
Fehrenbacher L, Johnson BE, Sandler A, Kris MG, Tran HT, Klein P,
Li X, et al: TRIBUTE: A phase III trial of erlotinib hydrochloride
(OSI-774) combined with carboplatin and paclitaxel chemotherapy in
advanced non-small-cell lung cancer. J Clin Oncol. 23:5892–5899.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wilson KJ, Gilmore JL, Foley J, Lemmon MA
and Riese DJ II: Functional selectivity of EGF family peptide
growth factors: Implications for cancer. Pharmacol Ther. 122:1–8.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Laurent-Puig P, Lievre A and Blons H:
Mutations and response to epidermal growth factor receptor
inhibitors. Clin Cancer Res. 15:1133–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kancha RK, von Bubnoff N, Peschel C and
Duyster J: Functional analysis of epidermal growth factor receptor
(EGFR) mutations and potential implications for EGFR targeted
therapy. Clin Cancer Res. 15:460–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jackman DM, Yeap BY, Sequist LV, Lindeman
N, Holmes AJ, Joshi VA, Bell DW, Huberman MS, Halmos B, Rabin MS,
et al: Exon 19 deletion mutations of epidermal growth factor
receptor are associated with prolonged survival in non-small cell
lung cancer patients treated with gefitinib or erlotinib. Clin
Cancer Res. 12:3908–3914. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rosell R, Moran T, Queralt C, Porta R,
Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M,
et al: Screening for epidermal growth factor receptor mutations in
lung cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jackman D, Pao W, Riely GJ, Engelman JA,
Kris MG, Jänne PA, Lynch T, Johnson BE and Miller VA: Clinical
definition of acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small-cell lung cancer.
J Clin Oncol. 28:357–360. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun JM, Ahn MJ, Choi YL, Ahn JS and Park
K: Clinical implications of T790M mutation in patients with
acquired resistance to EGFR tyrosine kinase inhibitors. Lung
Cancer. 82:294–298. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ayoola A, Barochia A, Belani K and Belani
CP: Primary and acquired resistance to epidermal growth factor
receptor tyrosine kinase inhibitors in non-small cell lung cancer:
An update. Cancer Invest. 30:433–446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bean J, Brennan C, Shih JY, Riely G, Viale
A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:pp. 20932–20937. 2007; View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ohashi K, Sequist LV, Arcila ME, Moran T,
Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, et
al: Lung cancers with acquired resistance to EGFR inhibitors
occasionally harbor BRAF gene mutations but lack mutations in KRAS,
NRAS, or MEK1. Proc Natl Acad Sci USA. 109:pp. E2127–E2133. 2012;
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Serizawa M, Takahashi T, Yamamoto N and
Koh Y: Genomic aberrations associated with erlotinib resistance in
non-small cell lung cancer cells. Anticancer Res. 33:5223–5233.
2013.PubMed/NCBI
|
|
22
|
Takezawa K, Pirazzoli V, Arcila ME, Nebhan
CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ,
Melnick MA, et al: HER2 amplification: A potential mechanism of
acquired resistance to EGFR inhibition in EGFR mutant lung cancers
that lack the second-site EGFRT790M mutation. Cancer Discov.
2:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu K, Chang Q, Lu Y, Qiu P, Chen B, Thakur
C, Sun J, Li L, Kowluru A and Chen F: Gefitinib resistance resulted
from STAT3-mediated Akt activation in lung cancer cells.
Oncotarget. 4:2430–2438. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR TKI therapy in 155 patients with EGFR mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nature Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko
TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, et al: A
common BIM deletion polymorphism mediates intrinsic resistance and
inferior responses to tyrosine kinase inhibitors in cancer. Nat
Med. 18:521–528. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakagawa T, Takeuchi S, Yamada T, Ebi H,
Sano T, Nanjo S, Ishikawa D, Sato M, Hasegawa Y, Sekido Y and Yano
S: EGFR-TKI resistance due to BIM polymorphism can be circumvented
in combination with HDAC inhibition. Cancer Res. 73:2428–2434.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Costa DB, Halmos B, Kumar A, Schumer ST,
Huberman MS, Boggon TJ, Tenen DG and Kobayashi S: BIM mediates EGFR
tyrosine kinase inhibitor-induced apoptosis in lung cancers with
oncogenic EGFR mutations. PLoS Med. 4:1669–1680. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cragg MS, Kuroda J, Puthalakath H, Huang
DC and Strasser A: Gefitinib-induced killing of NSCLC cell lines
expressing mutant EGFR requires BIM and can be enhanced by BH3
mimetics. PLoS Med. 4:1681–1690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Faber AC, Corcoran RB, Ebi H, Sequist LV,
Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, et
al: BIM expression in treatment-naive cancers predicts
responsiveness to kinase inhibitors. Cancer Discov. 1:352–365.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gong Y, Somwar R, Politi K, Balak M,
Chmielecki J, Jiang X and Pao W: Induction of BIM is essential for
apoptosis triggered by EGFR kinase inhibitors in mutant
EGFR-dependent lung adenocarcinomas. PLoS Med. 4:e2942007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benedettini E, Sholl LM, Peyton M, Reilly
J, Ware C, Davis L, Vena N, Bailey D, Yeap BY, Fiorentino M, et al:
Met activation in non-small cell lung cancer is associated with de
novo resistance to EGFR inhibitors and the development of brain
metastasis. Am J Pathol. 177:415–423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Balak MN, Gong Y, Riely GJ, Somwar R, Li
AR, Zakowski MF, Chiang A, Yang G, Ouerfelli O, Kris MG, et al:
Novel D761Y and common secondary T790M mutations in epidermal
growth factor receptor-mutant lung adenocarcinomas with acquired
resistance to kinase inhibitors. Clin Cancer Res. 12:6494–6501.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kosaka T, Yatabe Y, Endoh H, Yoshida K,
Hida T, Tsuboi M, Tada H, Kuwano H and Mitsudomi T: Analysis of
epidermal growth factor receptor gene mutation in patients with
non-small cell lung cancer and acquired resistance to gefitinib.
Clin Cancer Res. 12:5764–5769. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen L, Willis SN, Wei A, Smith BJ,
Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC:
Differential targeting of prosurvival Bcl-2 proteins by their
BH3-only ligands allows complementary apoptotic function. Mol Cell.
17:393–403. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
O'Connor L, Strasser A, O'Reilly LA,
Hausmann G, Adams JM, Cory S and Huang DC: Bim: A novel member of
the Bcl-2 family that promotes apoptosis. EMBO J. 17:384–395. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Özvegy-Laczka C, Cserepes J, Elkind NB and
Sarkadi B: Tyrosine kinase inhibitor resistance in cancer: Role of
ABC multidrug transporters. Drug Resist Updat. 8:15–26. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jonker JW, Buitelaar M, Wagenaar E, van
der Valk MA, Scheffer GL, Scheper RJ, Plosch T, Kuipers F, Elferink
RP, Rosing H, et al: The breast cancer resistance protein protects
against a major chlorophyll-derived dietary phototoxin and
protoporphyria. Proc Natl Acad Sci USA. 99:pp. 15649–15654. 2002;
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Allen JD and Schinkel AH: Multidrug
resistance and pharmacological protection mediated by the breast
cancer resistance protein (BCRP/ABCG2). Mol Cancer Ther. 1:427–434.
2002.PubMed/NCBI
|
|
43
|
Engelman JA, Jänne PA, Mermel C, Pearlberg
J, Mukohara T, Fleet C, Cichowski K, Johnson BE and Cantley LC:
ErbB-3 mediates phosphoinositide 3-kinase activity in
gefitinib-sensitive non-small cell lung cancer cell lines. Proc
Natl Acad Sci USA. 102:pp. 3788–3793. 2005; View Article : Google Scholar : PubMed/NCBI
|