Introduction
Liver cancer is the fifth most prevalent type of
cancer and the second most frequent cause of cancer-associated
mortality in males, with an estimated 782,500 new cases and 745,500
deaths worldwide in 2012 (1).
Hepatocellular carcinoma (HCC) is the most common type of liver
cancer (2). HCC is particularly
prevalent in East Asia, with an incidence rate twice that of Africa
and >4 times higher than North America. However, the incidence
rate of HCC has increased in Western countries (3). The dominant etiological cofactors that
contribute to the incidence rate of HCC vary according to ancestry
and region, including hepatitis B virus infection in East Asia and
Africa, hepatitis C virus infection in Japan, aflatoxin B1 exposure
in China and Africa and alcohol intake in Western countries
(4,5).
Mutational disruption of driver genes and pathways
occurs constantly in cancers, enabling tumor cells to proliferate
without constraints. A number of cancer genomics studies intend to
prioritize driver genes based on recurrent mutation status across a
cohort of tumor samples (6,7). For instance, Totoki et al
(8) applied MutSigCV to 503 pairs of
HCC and matched non-cancerous liver tissues or blood and identified
30 recurrently mutated driver genes in HCC, including telomerase
reverse transcriptase (TERT), catenin β1 (CTNNB1), tumor protein
p53 (TP53), AT-rich interaction domain 2 (ARID2) and axin 1
(AXIN1). However, little attention has been paid to driver genes
that are not frequently mutated, including genes that are enriched
for mutations with high functional impact (FI) and genes with
mutually exclusive mutation rates. Therefore, the development of
computational tools that are able to detect moderate or
low-frequency mutated driver genes is necessary to provide a more
complete understanding of cancer genomics. Methods including
Oncodrive-FM (9) aim to identify
genes with a bias toward the enrichment of variants with a high FI,
as measured by the Sorting Intolerant From Tolerant algorithm
(10), PolyPhen2 (11) and Mutation Assessor (12). Another method, Dendrix (13), was constructed to uncover sets of
genes which have at least one mutation in the majority of cancer
samples. These bioinformatics tools, which complement existing
methods, open novel avenues for the identification of potential
driver genes involved in the tumorigenesis and progression of
HCC.
In the present study, our group assessed the
moderate or low-frequency mutated driver genes predicted by
Oncodrive-FM and Dendrix in HCC. In addition to the previously
reported driver genes, our group identified novel cancer-driving
genes and pathways, including potential treatment targets and
prognostic biomarkers. Then, aberrant expression of these driver
genes, DNA methylation levels, copy number variations (CNVs) and
correlation with prognosis were assessed in patients with HCC. The
present study highlights the importance of analyzing cancer-driving
genes in an integrated fashion, and provides insights into the
carcinogenesis and progression of HCC.
Materials and methods
Classification of cancer
mutations
A total of 112,980 somatic mutations, comprised of
104,595 single-nucleotide variants (SNVs) and 8,385 small
insertions or deletions (indels), were generated by whole-exome
sequencing of 377 tumor tissues and paired normal tissues, and this
data was downloaded from The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/, accessed on January 15,
2016) (14). Somatic mutations were
classified into 11 categories based on their functional impacts in
the coding genome using Ensembl Variant Effect Predictor (VEP)
(15).
Prediction of driver genes and
pathways
Driver genes and pathways were determined using
OncodriveFM (https://www.intogen.org/analysis) and Dendrix
(http://compbio.cs.brown.edu/projects/dendrix/) with
default parameters following the criteria that genes and pathways
have q values smaller than 0.05. To clarify the functional
enrichment of driver genes, Gene Ontology (GO) terms (16,17) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) (18) pathway enrichment analyses were
conducted for all the driver genes on the home page of STRING
(http://string.embl.de/) (19). The GO terms and KEGG pathways were
considered to be significantly enriched for driver genes with the
cut-off of a false positive rate of P<0.05. Finally, a
protein-protein interaction network was constructed with STRING to
prioritize the pivotal driver genes in HCC. For each driver gene,
the overall STRING score that represents the degree to which the
driver gene is associated with other genes was computed by summing
up combined STRING scores of all its protein-protein
interactions.
Gene expression, DNA methylation and
principal component analyses
The present study used expression data from Gao
et al (20), which were
generated by RNA sequencing of 8 paired HCC and normal tissues, and
deposited in the Gene Expression Omnibus database (21) (accession no. GSE55758). DNA
methylation data from 340 HCC samples were obtained from TCGA
(14), and unavailable values were
replaced with the mean β value. Differentially expressed and
dysmethylated genes between HCC and normal tissues were determined
using the function of t-test in R with the cutoff value of
P<0.05. Next, principal component analysis (PCA) was conducted
using the princomp function in R to examine whether the
differentially expressed genes distinguished cancer tissues from
normal tissues.
Sources of CNV and survival
analyses
CNVs in 370 HCC samples, detected by single
nucleotide polymorphism (SNP) array, were acquired from the Broad
Institute TCGA GDAC Firehose (gdac.broadinstitute.org) (14). RNA sequencing (RNAseq) and clinical
outcome data were retrieved from TCGA to further evaluate whether
the expression of driver genes was associated with prognosis in
360/377 patients with HCC (14). For
each driver gene, the patients with HCC were divided into high and
low expression groups. The former refers to 25% (n=90) of patients
with the highest RNA expression levels of the driver gene, while
the latter refers to the 25% (n=90) of patients with the lowest
expression levels of driver gene. Kaplan-Meier survival analyses
were performed between the high and low expression groups using
oncolnc (http://www.oncolnc.org/) (22) and the log-rank test was utilized to
compare the difference of survival rates between different groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Catalog of somatic mutations
A total of 112,980 somatic mutations generated by
the whole-exome sequencing of 377 paired HCC and normal samples
were downloaded from TCGA. Among them, 104,595, 8,385 mutations
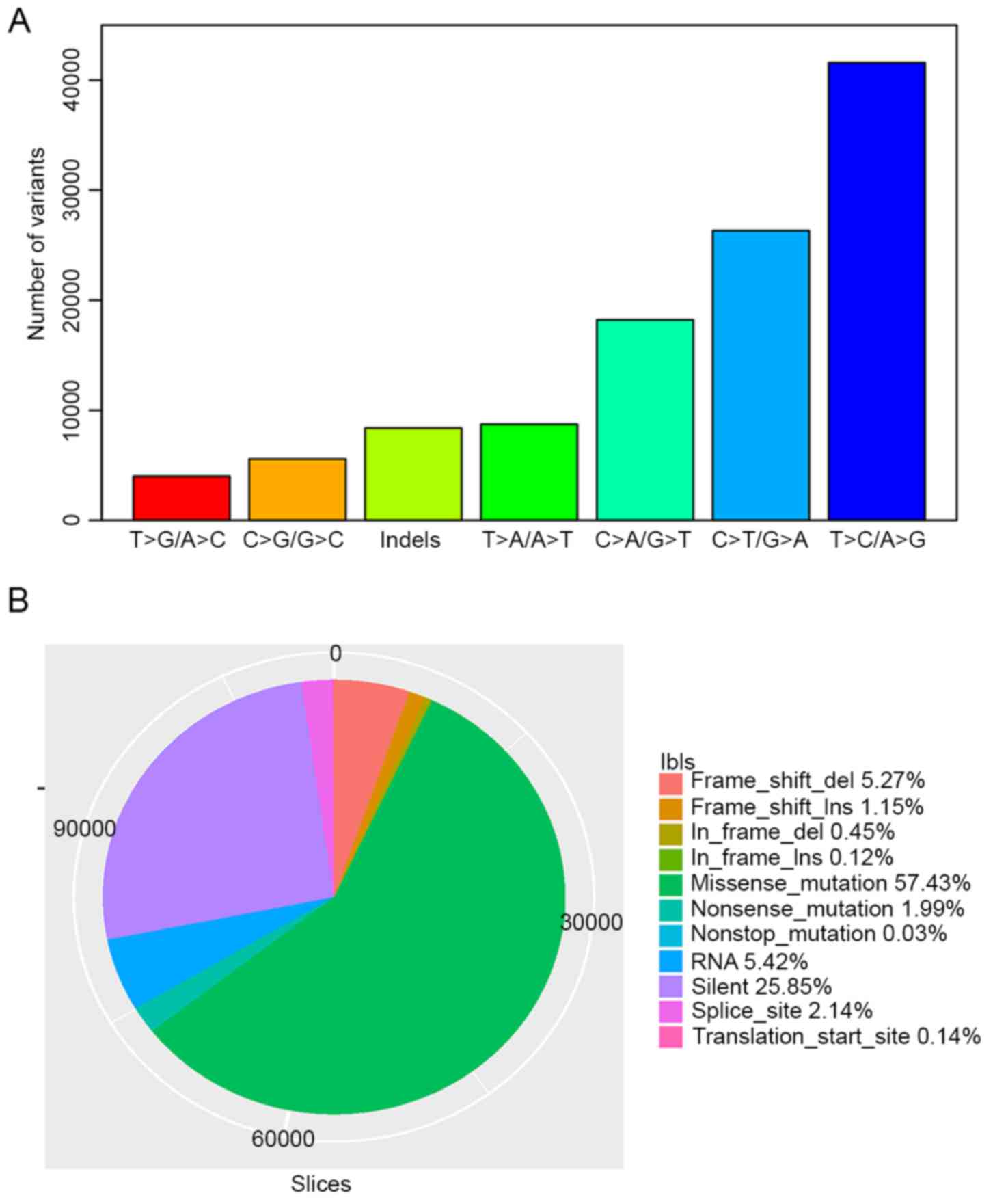
were SNVs and small indels, respectively. T>C/A>G,
C>T/G>A and C>A/G>T were the three most prevalent
transitions in HCC, with mutation rates of 36.82, 23.29 and 16.13%
in all variant types (Fig. 1A). There
were 64,890 missense mutations, 2,250 nonsense mutations, 34
nonstop mutations and 29,209 silent SNVs classified by VEP. In
total, 505 deletions and 135 insertions caused translational frame
shifts, while 5,957 deletions and 1,302 insertions were in frame
mutations. A total of 2,419 and 155 mutations occurred in splicing
sites and translation start sites, respectively. Over 60%
(72,149/112,980) of total variants were non-synonymous mutations
(Fig. 1B). HCC has a lower
nonsynonymous mutation density (2.03 nonsynonymous mutations per Mb
per sample, on average) in comparison with other types of cancer,
including melanoma and lung cancer (6).
Cancer driver genes and pathways in
HCC
OncodriveFM was used to identify driver genes and
pathways in HCC. In total, 108 driver genes were identified by
OncodriveFM. Among them, TP53, CTNNB1, ARID2, AXIN1 and TERT were
known driver genes, with mutation rates of 30.69, 27.11, 11.00,
7.16 and 2.05% across all HCC samples (8). However, the majority of driver genes
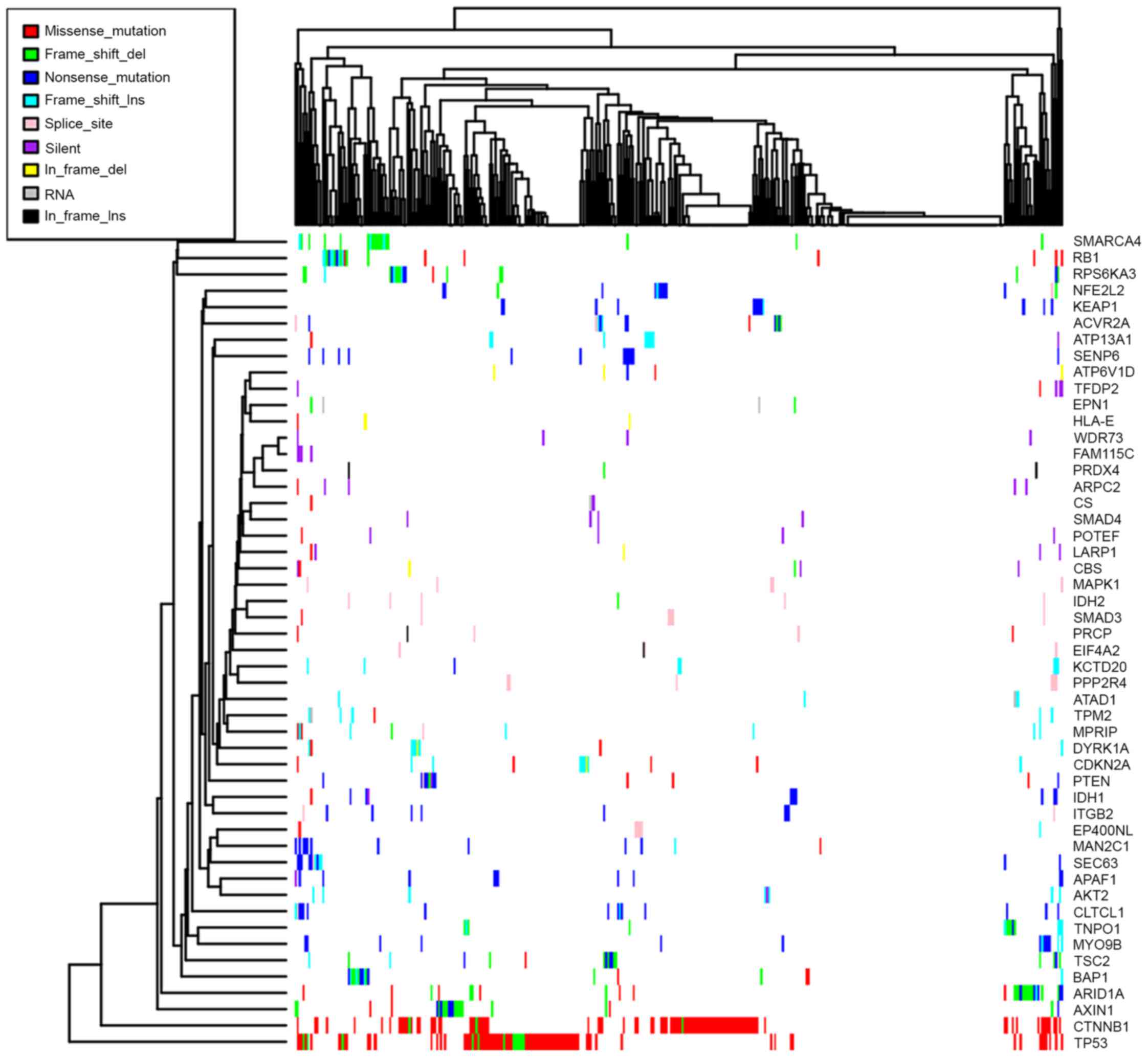
were middle or low-frequency mutated genes, Fig. 2 presents the mutation patterns of the
50 most frequently mutated driver genes, however data are not shown
for the remaining driver genes. Dendrix analysis was performed for
sets of size ranging from 2 to 4. When k=2, the pair TP53 and
CTNNB1 was sampled with a frequency of 24% (240/1,000). When k=3,
the triplet [BRCA associated protein 1 (BAP1), TP53 and CTNNB1] was
sampled with a frequency of 5.4% (54/1,000). For k=4, no gene set
had a sample frequency >1%. The pair (TP53 and CTNNB1) and
triplet (BAP1, TP53 and CTNNB1) were the most prevalent gene sets
in the mutual exclusivity test, further supporting the importance
of TP53 and CTNNB1 in the tumorigenesis of HCC.
The enrichment of GO terms was performed for 109
cancer genes on STRING, and 80 biological processes, 36 molecular
functions and 47 cellular components were reported with statistical
significance. The GO terms represented a wide variety of functional
processes, including ‘regulation of protein metabolic process’,
‘regulation of cellular process’, ‘regulation of gene expression’,
‘cell cycle’ and ‘cellular component biogenesis and organization’,
indicating that driver gene candidates are actively involved in
cancer-associated processes in HCC (data not shown). The 109 driver
genes were significantly enriched in 58 KEGG pathways, including
‘cell cycle’, ‘p53 signaling pathway’, ‘mTOR signaling pathway’ and
‘PI3K-Akt signaling pathway’ (data not shown). In addition,
Oncodrive-FM also identified 119 pathways with statistical FI bias
in HCC, including ‘colorectal cancer’, ‘endometrial cancer’, ‘basal
cell carcinoma’, ‘thyroid cancer’, ‘prostate cancer’, ‘glioma’,
‘wnt signaling pathway’, ‘pathways in cancer’, ‘p53 signaling
pathway’, ‘pancreatic cancer’, ‘non-small cell lung cancer’ and
‘melanoma’ (data not shown). A total of 71.19% (42/59) of driver
gene-enriched pathways were overlapped with driver pathways in HCC.
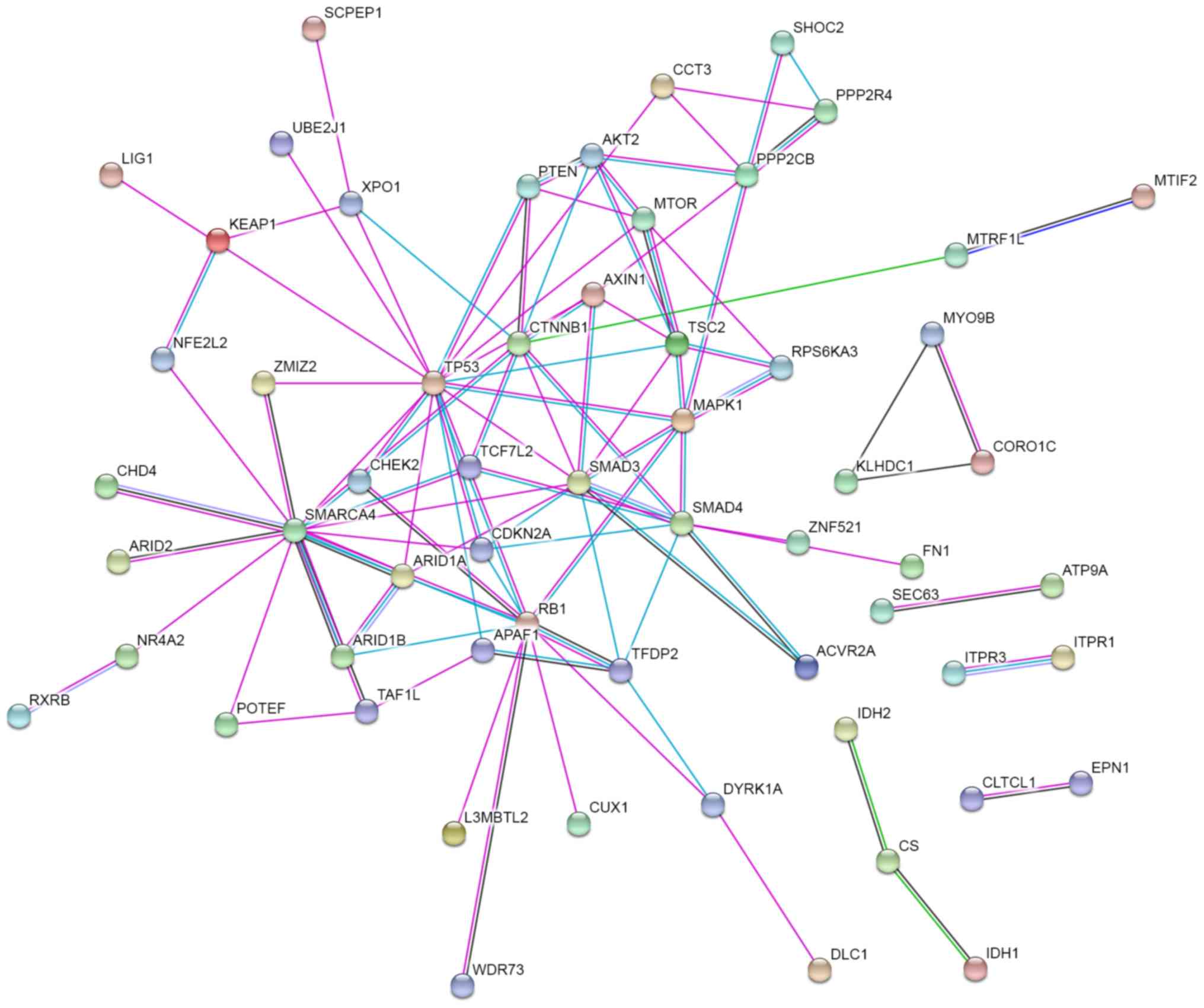
Finally, a protein-protein interaction network was constructed with
STRING to prioritize the pivotal driver genes. As presented in
Fig. 3, TP53, SWI/SNF related, matrix
associated, actin dependent regulator of chromatin, subfamily a,
member 4 (SMARCA4), smad family member 3, RB transcriptional
corepressor 1 (RB1), CTNNB1, smad family member 4 (SMAD4),
mitogen-activated protein kinase 1 (MAPK1) and TSC complex subunit
2 are at the core of the protein-protein interaction network. This
suggests that they may serve key functions in the network, because
they possessed the strongest protein-protein interactions (overall
STRING score >5; data not shown).
Expression profiling and DNA
methylation analyses in HCC
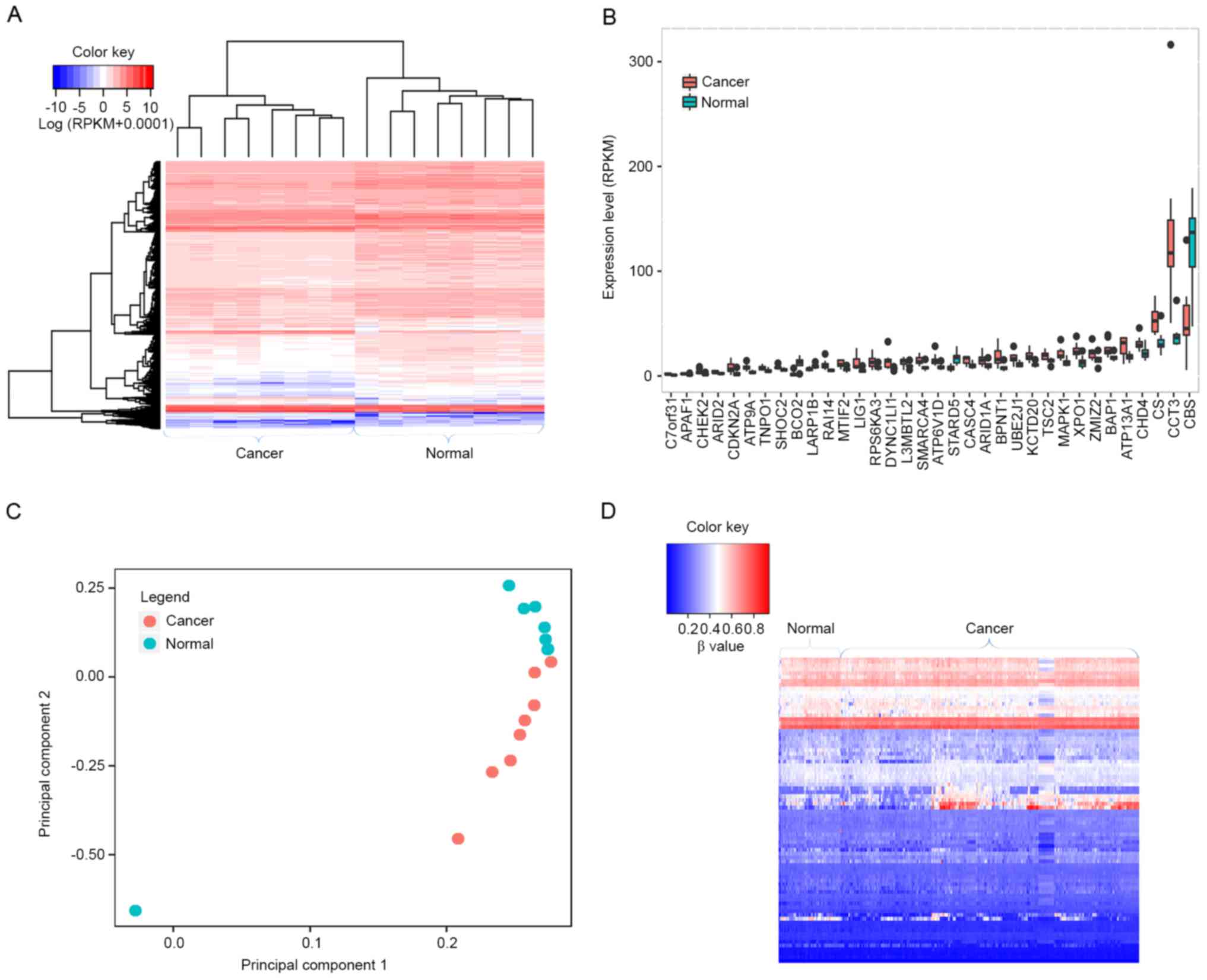
To analyze the gene expression profile in HCC,
RNA-seq data from 8 paired HCC and normal tissues were obtained.
Overall, 4,665 differentially expressed genes were identified
between the 8 paired HCC and normal samples (Fig. 4A), of which 34 were driver gene
candidates, including transportin 1 (TNPO1), apoptotic peptidase
activating factor 1 (APAF1), AT-rich interaction domain 1A (ARID1A)
and BAP1 (Fig. 4B). PCA was applied
to examine whether differentially expressed genes were able to
discriminate cancer tissues from normal tissues. As presented in
Fig. 4C, normal tissues were
aggregated to the upper side of the graph, whereas cancer tissues
were clustered to the lower side, suggesting that differentially
expressed genes were able to separate the tissue samples into two
distinct groups.
DNA methylation of cytosine within CpG dinucleotides
maintains the proper regulation of gene expression and stable gene
silencing; therefore, DNA methylation alterations make a
substantial contribution to tumorigenesis (23). In order to characterize the DNA
methylation status of driver gene candidates, our group downloaded
DNA methylation data from TCGA and analyzed the DNA methylation
levels of the identified driver genes in HCC. Overall, 83 driver
gene candidates had dysmethylation at their promoters, including 45
hypomethylated and 38 hypermethylated driver genes (Fig. 4D). These results suggest that the
majority of driver genes may be implicated in HCC through
modulating methylation status. 13 driver genes are overexpressed
and hypomethylated, including chaperonin containing TCP1 subunit 3
(CCT3), checkpoint kinase 2 (CHEK2), SMARCA4, MAPK1, TNPO1 and
mitochondrial translational initiation factor 2, suggesting they
may have oncogenic functions in HCC. In contrast, β-carotene
oxygenase 2 (BCO2) and cystathionine-β-synthase (CBS) exhibited
hypermethylation and low expression, and may therefore function as
tumor suppressors in HCC.
CNVs in HCC
CNVs of 370 HCC samples detected by SNParray were
also obtained from the Broad Institute. Significant focal gains and
deletions (q<0.25) were identified in 360 samples (360/370,
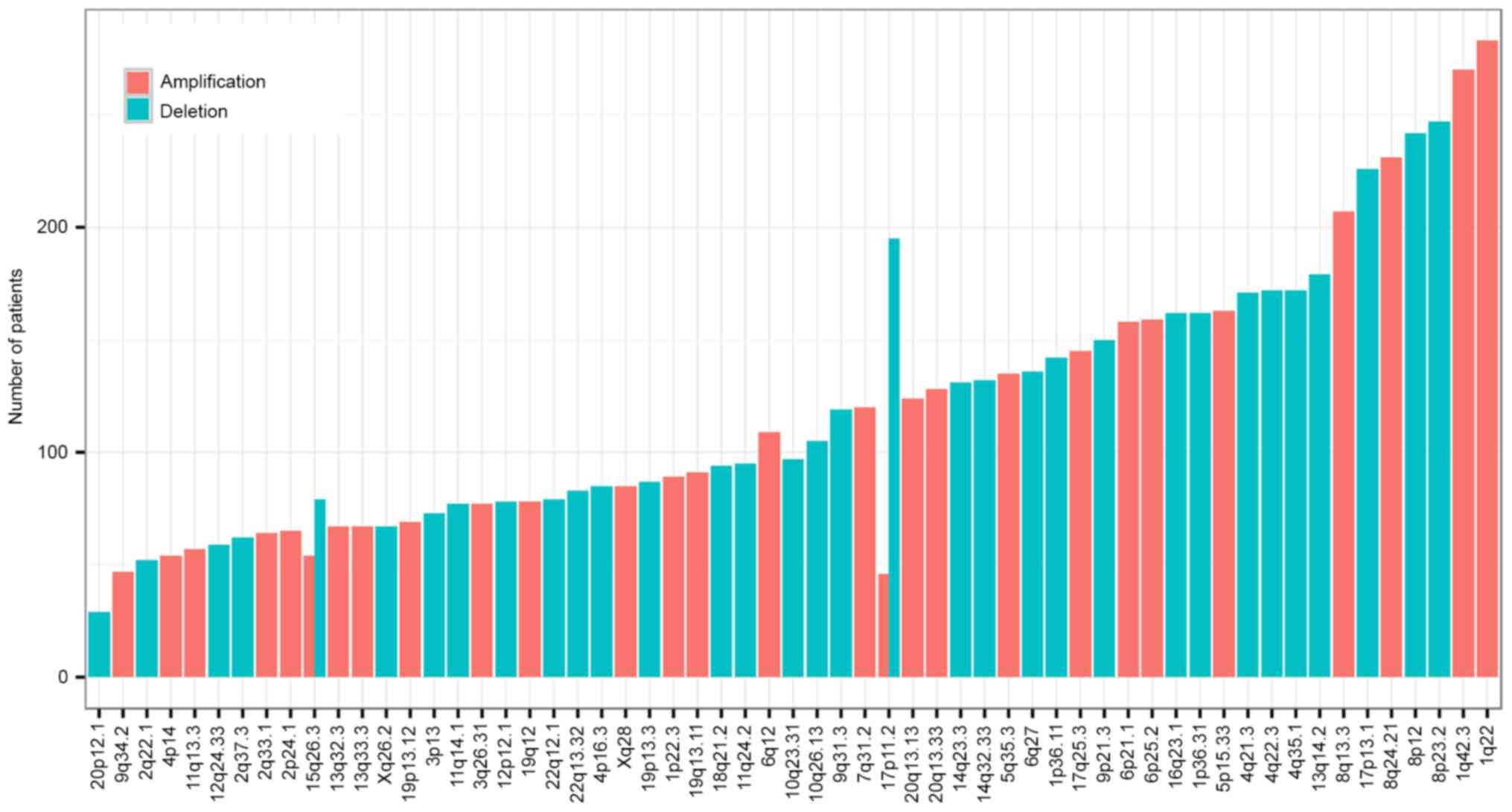
97.30%) at 61 loci (34 amplifications and 27 deletions). Among
these, amplifications at 1q22 and 1q42.3 and deletions at 8p23.2
and 8p12 were the most frequent CNVs in HCC, with occurrence rates
of 76.49% (90/370), 72.97% (89/370), 66.76% (89/370) and 65.41%
(90/370), respectively (Fig. 5). A
total of 17 cancer driver genes were involved in CNVs, including
known tumor suppressors and oncogenes, for example TP53 (deletion,
17p13.1), phosphatase and tensin homolog (deletion, 10q23.31) and
RB1 (deletion 13q14.2). Multiple driver candidates were also
implicated in the CNVs, including SHOC2, leucine rich repeat
scaffold protein (deletion, 10q25.2), transcription factor 7 like 2
(TCF7L2; deletion, 10q25.2), SMAD4 (deletion, 18q21.2), TRPM8
channel associated factor 2 (deletion, 7q35), isocitrate
dehydrogenase [NADP (+2)] mitochondrial (IDH2) and DNA polymerase
γ, catalytic subunit (deletion, 15q26.1), solute carrier family 2
member 6 (deletion, 9q34.2), outer dense fiber of sperm tails 2 and
protein phosphatase 2 phosphatase activator (deletion, 9q34.11),
ARID1A (deletion, 1p36.11), cut like homeobox 1 (deletion, 7q22.1),
cyclin dependent kinase inhibitor 2A (CDKN2A; deletion, 9p21.3),
BCO2 (deletion, 11q23.1) and EP400 N-terminal like (deletion,
12q24.33).
Survival analyses
TCGA RNAseq and clinical outcome data were acquired
from TCGA to evaluate whether the expression of 109 driver genes is
associated with survival and prognosis in patients with HCC.
Kaplan-Meier survival analysis showed that the expression of 24
driver genes was significantly associated with clinical outcomes in
patients with HCC. High expression of 14 driver genes indicated
worse survival rates in patients with HCC, including AXIN1, serine
carboxypeptidase 1, APAF1, TRPM8 channel associated factor 2, actin
related protein 2/3 complex subunit 2, autophagy related 9A,
exportin 1 (XPO1), Basic Leucine Zipper and W2 domains 2, CCT3,
CDKN2A, chromodomain helicase DNA binding protein 4, coronin 1C,
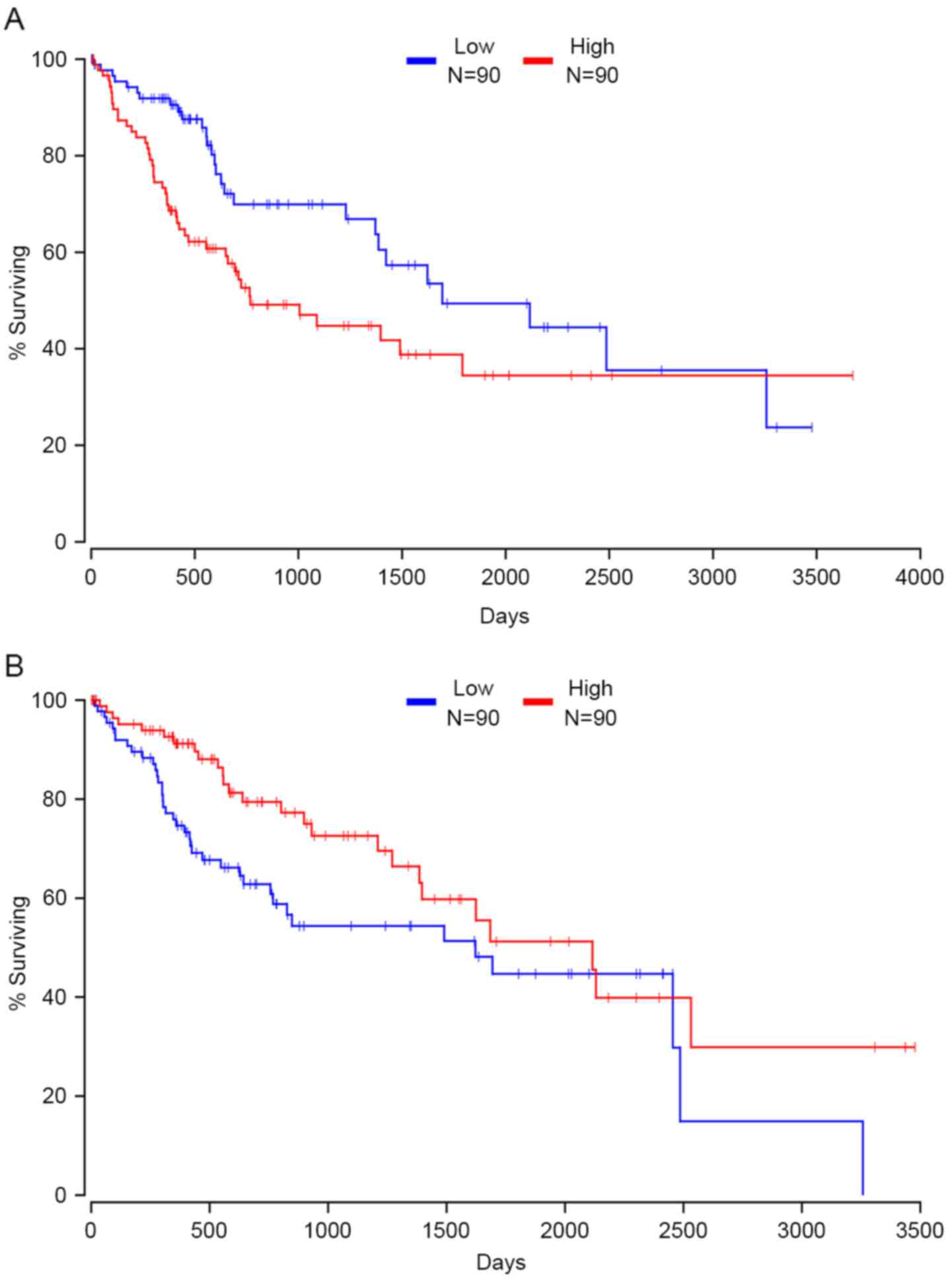
RAB32, member RAS oncogene family and TNPO1. Fig. 6A presents the correlation between the
expression of XPO1 and patients' clinical outcome; data are not
shown for the remaining driver genes (Fig. 6A). In contrast, patients with high
expression of 10 other driver genes had a relatively favorable
prognosis, including kinesin 2, BCO2, cancer susceptibility 4,
GTF2I repeat domain containing 2B, major histocompatibility
complex, class I, E, IDH2, La ribonucleoprotein domain family
member 1B, mannosidase alpha class 2C member 1, zinc finger protein
521 and StAR related lipid transfer domain containing 5 (data not
shown). TNPO1 and CCT3 were hypomethylated, overexpressed and
associated with a poor prognosis in HCC. BCO2 was hypermethylated,
under-expressed and associated with a favorable prognosis in HCC.
These three driver genes may be potential targets for treatment and
prognostic biomarkers for patients with HCC in the future.
Discussion
In the present study, our group conducted an
integrated investigation of 109 cancer-driving genes and 119
pathways determined with Oncodrive-FM and Dendrix. Only a small
fraction of these driver genes are repeatedly mutated in HCC
samples, including TP53, CTNNB1, ARID2, AXIN1 and TERT (8). The P53-retinoblastoma (RB) signaling
pathway was consistently mutated in cancer samples, including TP53
and CHEK2 of the P53 pathway, and RB1 and CDKN2A of the RB pathway,
reflecting the inactivation of the P53-RB pathway in HCC. In
addition to those identified in the P53-RB pathway, inactivating
mutations were frequently observed in WNT pathway genes, including
AXIN1 and CTNNB1. Multiple cancer driver genes initially identified
in other types of cancer were first identified as drivers in HCC,
including BAP1 in renal cell carcinoma (24), IDH2 in angioimmunoblastic T-cell
lymphoma (25), CDKN2A in melanoma
(26), TCF7L2 in colorectal cancer
(27) and SMAD4 in colorectal and
pancreatic cancer (28). In addition,
multiple novel driver candidates were identified by our group; for
instance, LARP1 and APAF1. LARP1, as a conserved RNA-binding
protein, interacts with poly-A-binding protein and modulates
5′-terminal oligopyrimidine tract mRNA translation. Enhanced
expression of LARP1 increases cell migration, invasion, growth and
tumorigenicity in vivo through post-transcriptionally
altering gene expression, including mechanistic target of
rapamycin, in Hela cells (29). APAF1
is an important factor that regulates the mitochondrial apoptotic
pathway, and loss of APAF1 causes cellular resistance against
apoptotic signals. Reduced expression of APAF1 has been observed in
colorectal adenocarcinoma and is associated with deeper tumor
invasion, frequent lymph node metastasis, higher stage and poorer
differentiation (30). In addition,
APAF1 is frequently methylated in bladder and kidney cancer, and
demethylation of APAF1 increased APAF1 expression and inhibited the
proliferation of bladder and kidney cancer cell lines (31).
One notable strength of OncodriveFM and Dendrix
analysis lies in the identification of cancer-associated genes and
pathways which have a high FI bias towards accumulating high FI
variants or mutational exclusivity, independent of cancer mutation
frequency. Implementing these tools allows for a comprehensive
exploration of cancer-driving genes and pathways. In addition, our
group revealed 35 differentially expressed genes, 83 dysmethylated
driver genes and 17 cancer driver genes involved in CNVs,
suggesting these genes may contribute to the development and
progression of HCC in various ways.
Of the 109 driver gene candidates, 24 genes whose
expression levels were associated with HCC patient prognosis were
identified, including XPO1 and IDH2. XPO1, also known as CRM1,
encodes a protein which serves a critical function in the
trafficking of over 230 proteins, including tumor suppressors (for
example, p53, p73 and forkhead box O1), growth
regulator/pro-inflammatory (for example, IκB, Rb, p21, p27, BRCA1,
DNA repair associated and APC, WNT signaling pathway regulator),
and anti-apoptotic proteins (for example, nucleophosmin 1 and AP-1)
(32). XPO1 is an oncogenic,
anti-apoptotic protein in transformed cells and is overexpressed in
a number of types of cancer, including mantle cell lymphoma
(33), lung adenocarcinoma (34) and gastric cancer (35). Similar to the results of the present
study, high expression of XPO1 is an independent poor prognostic
factor in gastric carcinoma (35),
acute myeloid leukemia (36),
pancreatic cancer (37) and lung
adenocarcinoma (34). Another gene,
IDH2, is frequently mutated in multiple types of cancer, including
T-cell lymphoma (38), glioma
(39) and acute myeloid leukemia
(40). In line with the results of
the present study, Liu et al (41) reported that decreased expression of
IDH2 was associated with lower overall survival rates in HCC.
Furthermore, mutated IDH2 was an independent prognostic factor for
improved overall survival in acute myeloid leukemia (40) and glioma (42). Therefore, these genes may be ideal
candidates for the development of HCC prognostic indicators in
future studies.
In summary, our group have successfully compiled a
list of cancer driver genes and pathways in HCC, enhancing our
understanding of pathogenesis and progression of HCC. Furthermore,
the novel driver genes and pathways identified by the present study
pave the way for the development of therapies targeting driver
genes and prognostic biomarkers in HCC.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Massarweh NN and El-Serag HB: Epidemiology
of hepatocellular carcinoma and intrahepatic cholangiocarcinoma.
Cancer Control. 24:10732748177292452017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Center MM and Jemal A: International
trends in liver cancer incidence rates. Cancer Epidemiol Biomarkers
Prev. 20:2362–2368. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu J, Shen J, Sun TT, Zhang X and Wong N:
Obesity, insulin resistance, NASH and hepatocellular carcinoma.
Semin Cancer Biol. 23:483–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Augustine MM and Fong Y: Epidemiology and
risk factors of biliary tract and primary liver tumors. Surg Oncol
Clin N Am. 23:177–188. 2014. View Article : Google Scholar
|
|
6
|
Lawrence MS, Stojanov P, Polak P, Kryukov
GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH,
Roberts SA, et al: Mutational heterogeneity in cancer and the
search for new cancer-associated genes. Nature. 499:214–218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dees ND, Zhang Q, Kandoth C, Wendl MC,
Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis
ER, et al: MuSiC: Identifying mutational significance in cancer
genomes. Genome Res. 22:1589–1598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Totoki Y, Tatsuno K, Covington KR, Ueda H,
Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H,
et al: Trans-ancestry mutational landscape of hepatocellular
carcinoma genomes. Nat Genet. 46:1267–1273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gonzalez-Perez A and Lopez-Bigas N:
Functional impact bias reveals cancer drivers. Nucleic Acids Res.
40:e1692012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40(Web Server
Issue): W452–W457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reva B, Antipin Y and Sander C: Predicting
the functional impact of protein mutations: Application to cancer
genomics. Nucleic Acids Res. 39:e1182011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vandin F, Upfal E and Raphael BJ: De novo
discovery of mutated driver pathways in cancer. Genome Res.
22:375–385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cancer Genome Atlas Research Network, .
Electronic address. simplewheeler@bcm.eduCancer
Genome Atlas Research Network: Comprehensive and integrative
genomic characterization of hepatocellular carcinoma. Cell.
169:1327–1341.e23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McLaren W, Pritchard B, Rios D, Chen Y,
Flicek P and Cunningham F: Deriving the consequences of genomic
variants with the ensembl API and SNP effect predictor.
Bioinformatics. 26:2069–2070. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium. Nat Genet. 25:25–29. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
The Gene Ontology Consortium, . Expansion
of the gene ontology knowledgebase and resources. Nucleic Acids
Res. 45:D331–D338. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanehisa M, Furumichi M, Tanabe M, Sato Y
and Morishima K: KEGG: New perspectives on genomes, pathways,
diseases and drugs. Nucleic Acids Res. 45:D353–D361. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Szklarczyk D, Morris JH, Cook H, Kuhn M,
Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al:
The STRING database in 2017: Quality-controlled protein-protein
association networks, made broadly accessible. Nucleic Acids Res.
45:D362–D368. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao F, Liang H, Lu H, Wang J, Xia M, Yuan
Z, Yao Y, Wang T, Tan X, Laurence A, et al: Global analysis of DNA
methylation in hepatocellular carcinoma by a liquid hybridization
capture-based bisulfite sequencing approach. Clin Epigenetics.
7:862015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-Update. Nucleic Acids Res. 41(Database Issue):
D991–D995. 2013.PubMed/NCBI
|
|
22
|
Anaya J: OncoLnc: Linking TCGA survival
data to mRNAs, miRNAs and lncRNAs. PeerJ Comput Sci. 2:e672016.
View Article : Google Scholar
|
|
23
|
Kulis M and Esteller M: 2-DNA methylation
and cancerEpigenetics and Cancer, Part A. 70. Herceg Z and Ushijima
TBT-A G: Academic Press; pp. 27–56. 2010, View Article : Google Scholar
|
|
24
|
Sato Y, Yoshizato T, Shiraishi Y, Maekawa
S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki
H, et al: Integrated molecular analysis of clear-cell renal cell
carcinoma. Nat Genet. 45:860–867. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang C, Mckeithan TW, Gong Q, Zhang W,
Bouska A, Rosenwald A, Gascoyne RD, Wu X, Wang J, Muhammad Z, et
al: IDH2R172 mutations define a unique subgroup of patients with
angioimmunoblastic T-cell lymphoma. Blood. 126:1741–1752. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Betti M, Aspesi A, Biasi A, Casalone E,
Ferrante D, Ogliara P, Gironi LC, Giorgione R, Farinelli P, Grosso
F, et al: CDKN2A and BAP1 germline mutations predispose to melanoma
and mesothelioma. Cancer Lett. 378:120–130. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nome T, Hoff AM, Bakken AC, Rognum TO,
Nesbakken A and Skotheim RI: High frequency of fusion transcripts
involving TCF7L2 in colorectal cancer: Novel fusion partner and
splice. PLoS One. 9:e912642014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Inamoto S, Itatani Y, Yamamoto T,
Minamiguchi S, Hirai H, Iwamoto M, Hasegawa S, Taketo MM, Sakai Y
and Kawada K: Loss of SMAD4 promotes colorectal cancer progression
by accumulation of myeloid-derived suppressor cells through the
CCL15-CCR1 chemokine axis. Clin Cancer Res. 22:492–501. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mura M, Hopkins TG, Michael T, Abd-Latip
N, Weir J, Aboagye E, Mauri F, Jameson C, Sturge J, Gabra H, et al:
LARP1 post-transcriptionally regulates mTOR and contributes to
cancer progression. Oncogene. 34:5025–5036. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Paik SS, Jang KS, Song YS, Jang SH, Min
KW, Han HX, Na W, Lee KH, Choi D and Jang SJ: Reduced expression of
Apaf-1 in colorectal adenocarcinoma correlates with tumor
progression and aggressive phenotype. Ann Surg Oncol. 14:3453–3459.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Christoph F, Kempkensteffen C, Weikert S,
Köllermann J, Krause H, Miller K, Schostak M and Schrader M:
Methylation of tumour suppressor genes APAF-1 and DAPK-1 and in
vitro effects of demethylating agents in bladder and kidney cancer.
Br J Cancer. 95:1701–1707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ishizawa J, Kojima K, Hail N Jr, Tabe Y
and Andreeff M: Expression, function, and targeting of the nuclear
exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol
Ther. 153:25–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yoshimura M, Ishizawa J, Ruvolo V, Dilip
A, Quintás-Cardama A, McDonnell TJ, Neelapu SS, Kwak LW, Shacham S,
Kauffman M, et al: Induction of p53-mediated transcription and
apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma.
Cancer Sci. 105:795–801. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gao W, Lu C, Chen L and Keohavong P:
Overexpression of CRM1: A characteristic feature in a transformed
phenotype of lung carcinogenesis and a molecular target for lung
cancer adjuvant therapy. J Thorac Oncol. 10:815–825. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhou F, Qiu W, Yao R, Xiang J, Sun X, Liu
S, Lv J and Yue L: CRM1 is a novel independent prognostic factor
for the poor prognosis of gastric carcinomas. Med Oncol.
30:7262013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kojima K, Kornblau SM, Ruvolo V, Dilip A,
Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N, et al:
Prognostic impact and targeting of CRM1 in acute myeloid leukemia.
Blood. 121:4166–4174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH
and Sun YJ: Prognostic value of CRM1 in pancreas cancer. Clin
Invest Med. 32:E3152009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cairns RA, Iqbal J, Lemonnier F, Kucuk C,
de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, et
al: IDH2 mutations are frequent in angioimmunoblastic T-cell
lymphoma. Blood. 119:1901–1903. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koh J, Cho H, Kim H, Kim SI, Yun S, Park
CK, Lee SH, Choi SH and Park SH: IDH2 mutation in gliomas including
novel mutation. Neuropathology. 35:236–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Green CL, Evans CM, Zhao L, Hills RK,
Burnett AK, Linch DC and Gale RE: The prognostic significance of
IDH2 mutations in AML depends on the location of the mutation.
Blood. 118:409–412. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu WR, Tian MX, Jin L, Yang LX, Ding ZB,
Shen YH, Peng YF, Zhou J, Qiu SJ, Dai Z, et al: High expression of
5-hydroxymethylcytosine and isocitrate dehydrogenase 2 is
associated with favorable prognosis after curative resection of
hepatocellular carcinoma. J Exp Clin Cancer Res. 33:322014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang XW, Ciccarino P, Rossetto M,
Boisselier B, Marie Y, Desestret V, Gleize V, Mokhtari K, Sanson M
and Labussière M: IDH mutations: Genotype-phenotype correlation and
prognostic impact. Biomed Res Int. 2014:5402362014.PubMed/NCBI
|