Introduction
Breast cancer (BC) is the most common type of
malignant cancer in the female population globally, resulting in
the leading cause of cancer-associated mortality due to the
metastatic spread of the cancer to vital organs (1). Therapy for primary breast cancer usually
involves tumor resection, radiation therapy and chemotherapy.
Surgery usually extends the survival of patients; however, for
patients with the advanced stage of the disease, chemotherapy is
considered as the alternative option (2). Unfortunately, systemic chemotherapy is
usually ineffective due to the low sensitivity of cancer cells to
chemotherapeutic drugs (3).
Doxorubicin (DOX) is one type of antitumor antibiotic, which is
widely used in multiple cancer types, including BC (4). The mechanism by which DOX kills the
cancer cells is dependent on the apoptotic pathway, as DOX is able
to embed into the DNA, and inhibit the synthesis of RNA and DNA,
which is the initial signal of apoptosis (5). Despite the fact that DOX is widely used
for the treatment of BC, the resistance of BC cells to the
apoptotic pathway limits the clinical effectiveness of DOX
(6,7).
Therefore, there is a requirement to decrease DOX-resistance by
enhancing the sensitivity of BC cells to apoptosis.
MicroRNAs (miRNAs) are endogenous, small non-coding
RNAs with ~22 nucleotides in length. They are able to regulate
various genes by binding to the 3′-untranslated region (3′-UTR) of
their target mRNAs, leading to mRNA degradation or translational
inhibition. As >30% of protein-coding genes are regulated by
miRNAs, they are involved in various biological and pathological
processes, including cell proliferation, metastasis,
differentiation, and apoptosis (8–10). Studies
have demonstrated that numerous miRNAs are dysregulated in BC
(11,12). Therefore, the expression profiles of
miRNAs have been investigated for the development of diagnostic
biomarkers and therapeutic targets for the treatment of BC
(11,12). Recently, studies have indicated that
the dysregulation of miRNAs is associated with the sensitivity of
cancer cells to chemotherapy. For example, miR-221 and miR-27a is
usually dysregulated in breast cancer, which contributes to
cisplatin resistance (13,14). However, the role and the mechanism of
miRNAs in BC cell DOX-resistance remain unclear.
In the present study, the potential role of miR-125b
in BC was investigated. The expression of miR-125b in tumor tissue
samples from patients with BC were examined and it was demonstrated
that miR-125b was downregulated in BC. Notably, it was revealed
that the absence of miR-125b was involved in the development of
DOX-resistance. These findings may provide a novel strategy for
decreasing the resistance of chemotherapy in BC.
Materials and methods
Clinical specimens and cell lines
A total of 30 pairs of BC tumor and corresponding
adjacent non-tumor tissues were obtained from patients who
underwent tumor resection at Dongyang People's Hospital (Jinhua,
China) between August 2013 and December 2015. All of the patients
were female and the median age of the patients was 55 years (range,
32–78 years). The use of clinical tissues for the present study was
approved by the Ethical Committee of Dongyang People's Hospital and
all of the patients provided written informed consent. Human BC
cell lines MCF-7, MDA-MB-231, T-47D and the human normal breast
epithelial cell line MCF-10A were obtained from the Institute of
Biochemistry and Cell Biology, Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). Cells were cultured
at 37°C in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) in a humidified 5% CO2 incubator. To
investigate the chemoresistance of BC, a DOX-resistant MCF-7 cell
line (MCF-7/R) was established by stepwise exposure of MCF-7 cells
to increasing concentrations of DOX (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). Briefly, the MCF-7 cells were initially
treated with DOX at 0.2 µg/ml for 2 months and the DOX
concentration was increased every 3 weeks by 0.04 µg/ml up to a
final concentration of 0.6 µg/ml. To eliminate the influence of
residual DOX in culture medium, the MCF-7/R cells were cultured in
DOX-free DMEM for 3 weeks prior to the experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the tissues of patients
with BC patients or BC cell lines in vitro using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.).
For analysis of miR-125b expression, the total RNA was reverse
transcribed using stem-loop RT primers from the PrimeScript RT
reagent kit according to the manufacturer's protocol (Takara Bio,
Inc., Otsu, Japan). The miR-125b RT primer (Guangzhou RiboBio Co.,
Ltd., Guangzhou, China) had the following sequence:
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCACAGGT-3′. qPCR was
performed in triplicate using the SYBR Premix Ex Taq (Takara Bio,
Inc.) on an ABI PRISM 7900 Sequence Detection system (Applied
Biosystems). The relative expression of miR-125b was determined
using the 2−ΔΔCq analysis method (15), whereby U6 snRNA was used as an
internal reference. The PCR reaction mix consisted of 12.5 µl 2X
SYBR® Premix Ex Taq II (cat no. RR820A; Takara Bio,
Inc.), 1 µl forward primer (10 µM), 1 µl reverse primer (10 µM), 2
µl cDNA and 8.5 µl H2O. PCR was performed under the
following thermocycling conditions: 95°C for 30 sec, followed by 40
cycles of 95°C for 5 sec and 60°C for 30 sec, and one cycle of 95°C
for 15 sec, 60°C for 60 sec and 95°C for 15 sec for dissociation.
PCR primers were obtained from Guangzhou RiboBio Co., Ltd. and had
the following sequences: miR-125b forward,
5′-ACACTCCAGCTGGGTCCCTGAGACCCTTTAAC-3′ and reverse,
5′-TGGTGTCGTGGAGTCG-3′; and U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and
reverse, 5′-AACGCTTCACGAATTTGCGT-3′.
Plasmid and transfection
For enforced expression of hematopoietic
cell-specific protein 1-associated protein X-1 (HAX-1), the
eukaryotic expression vector (pcDNA3.1 plasmid containing HAX-1
open reading frame; Invitrogen; Thermo Fisher Scientific, Inc.) was
conducted. For transfection, 2 µg/ml HAX-1 vector, 50 pmol/ml
miR-125b mimics (5′-AGUGUUCAAUCCCAGAGUCCCU-3′), 50 pmol/ml negative
control oligonucleotide (miR-NC, 5′-AGUCAUCCGUACUCAGUGUCCA-3′), and
50 pmol/ml HAX-1 small interfering (si)RNA (forward
5′-GAGUGAUGCAAGAAGUGAAUU-3′, reverse 5′-UUCACUUCUUGCAUCACUCUU-3′)
were transfected into the MCF-7 and MCF-7/R cells using
Lipofectamine™ 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. All the RNA
oligonucleotides were purchased from Shanghai GenePharma Co., Ltd.
(Shanghai, China).
Luciferase reporter assay
Bioinformatics analysis using TargetScan (http://www.targetscan.org/) indicated that HAX-1
represents a target gene of miR-125b. To verify this, the wild-type
of the putative miR-125b binding sites of HAX-1 3′-UTR were cloned
into the downstream of firefly luciferase gene in the pMIR-REPORT™
miRNA Expression Reporter Vector (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. To mutate the seed region
of the miR-125b-binding sites (CUCAGGG to CUCUAGG), the QuikChange
Site-Directed Mutagenesis kit (Agilent Technologies, Inc., Santa
Clara, CA, USA) was used based on the wild-type conducted
pMIR-REPORT™ vector following the manufacturer's protocol. To
perform the luciferase reporter assay, the MCF-7/R cells were
incubated in 48-well plates overnight at 37°C. The cells were then
co-transfected with the wild-type (or mutant-type) of pMIR-REPORT
vectors, Renilla luciferase pRL-TK vectors (Promega Corporation,
Madison, WI, USA), and the miR-125b mimics using Lipofectamine
2000. After 48 h of transfection, the cells were collected and
lysed using a lysis buffer provided by Promega Corporation (cat no.
E1910). Luciferase activity was then measured using a Dual
Luciferase Reporter Assay system according to the manufacturer's
protocol (cat no. E1910, Promega Corporation). The relative Firefly
luciferase activity was normalized to Renilla luciferase
activity.
Western blot analysis
BC cells were lysed in the RIPA lysis buffer (Cell
Signaling Technology, Inc., Danvers, MA, USA). A total of 50 µg
total protein extracted from the lysed cells was separated by 12.5%
SDS-PAGE and transferred to a polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). Non-specific binding was blocked
using 5% (w/v) skimmed milk in Tris-buffered saline with 1%
Tween-20 for 2 h at room temperature. The membranes were then
incubated with the primary antibodies mouse anti-HAX-1 monoclonal
antibody (mAb) (cat. no. sc-166845; dilution, 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), cleaved caspase-9 rabbit mAb
(cat. no. 7237; dilution, 1:1,000; Cell Signaling Technology,
Inc.), cleaved caspase-3 rabbit mAb (cat. no. 9661; dilution,
1:1,000; Cell Signaling Technology, Inc.), cleaved
poly(ADP-ribose)polymerase (PARP) rabbit mAb (cat. no. 5625;
dilution, 1:1,000; Cell Signaling Technology, Inc.) and β-actin
rabbit mAb (cat. no. 4970; dilution, 1:1,000; Cell Signaling
Technology, Inc.) overnight at 4°C. The membranes were then
incubated with horseradish peroxidase (HRP)-conjugated goat
anti-rabbit immunoglobulin G (IgG; cat no. 7074; dilution, 1:2,000;
Cell Signaling Technology, Inc.) for detection of cleaved
caspase-9, cleaved caspase-3, cleaved PARP and β-actin. Membranes
with HAX-1 protein were probed by mouse IgGκ light chain binding
protein conjugated with horseradish peroxidase (m-IgGκ BP-HRP, cat
no. sc-516102; dilution, 1:2,000; Santa Cruz Biotechnology, Inc.).
After 2 h incubation at 4°C, proteins were detected by using an
enhanced chemiluminescence detection kit (Pierce; Thermo Fisher
Scientific, Inc.).
Cell viability and the half maximal
inhibitory concentration (IC50)
BC cells were seeded in 96-well plates at a density
of 5×103 per well and transfected with RNAs and
plasmids. Then, 24 h after transfection, the cells were treated
with DOX for 48 h and the cell viability was evaluated using an MTT
assay, in which dimethyl sulfoxide was used to dissolve the purple
formazan (Sigma-Aldrich; Merck KGaA) (16). The absorbance was read at 570 nm using
a microplate reader. The IC50 was calculated according
to the cell viability curve.
Cell apoptosis detection
BC cells were collected after the 24 h transfection
followed by the 48 h DOX treatment. The cells were then stained
with Annexin V/propidium iodide (cat no. APOAF; Sigma-Aldrich;
Merck KGaA) for 15 min at room temperature according to the
manufacturer's protocols. Cell apoptosis was analyzed using a flow
cytometer and FlowJo 10 software (FlowJo LLC, Ashland, OR,
USA).
Measurement of mitochondrial membrane
potential (MMP; ΔΨm) and reactive oxygen species
(ROS)
BC cells were collected after the 24 h transfection
followed by the 48 h DOX treatment. The MMP was detected using JC-1
(Molecular Probes; Thermo Fisher Scientific, Inc.) as the indicator
(17) according to the manufacturer's
protocol. Generation of ROS was measured using dihydroethidium
(DHE; Molecular Probes; Thermo Fisher Scientific, Inc.) staining
(18) according to the manufacturer's
protocol. N-acetylcysteine (NAC; Sigma-Aldrich; Merck KGaA) was
used as a ROS scavenger (18). The
MMP and ROS were detected using a flow cytometer and analyzed using
FlowJo 10 software (FlowJo LLC).
Statistical analysis
Data are represented as mean ± standard error
obtained from three independent experiments. For comparison
analysis, two-tailed Student's t-tests were used to estimate the
statistical differences between two groups. One-way analysis of
variance and Bonferroni's post hoc test were used to determine the
differences between three or more groups. Statistical analysis was
performed using SPSS 15.0 software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Downregulation of miR-125b is
associated with DOX-resistance in BC
The expression of miR-125b was detected in the tumor
tissues of 30 patients with BC and their adjacent normal tissues
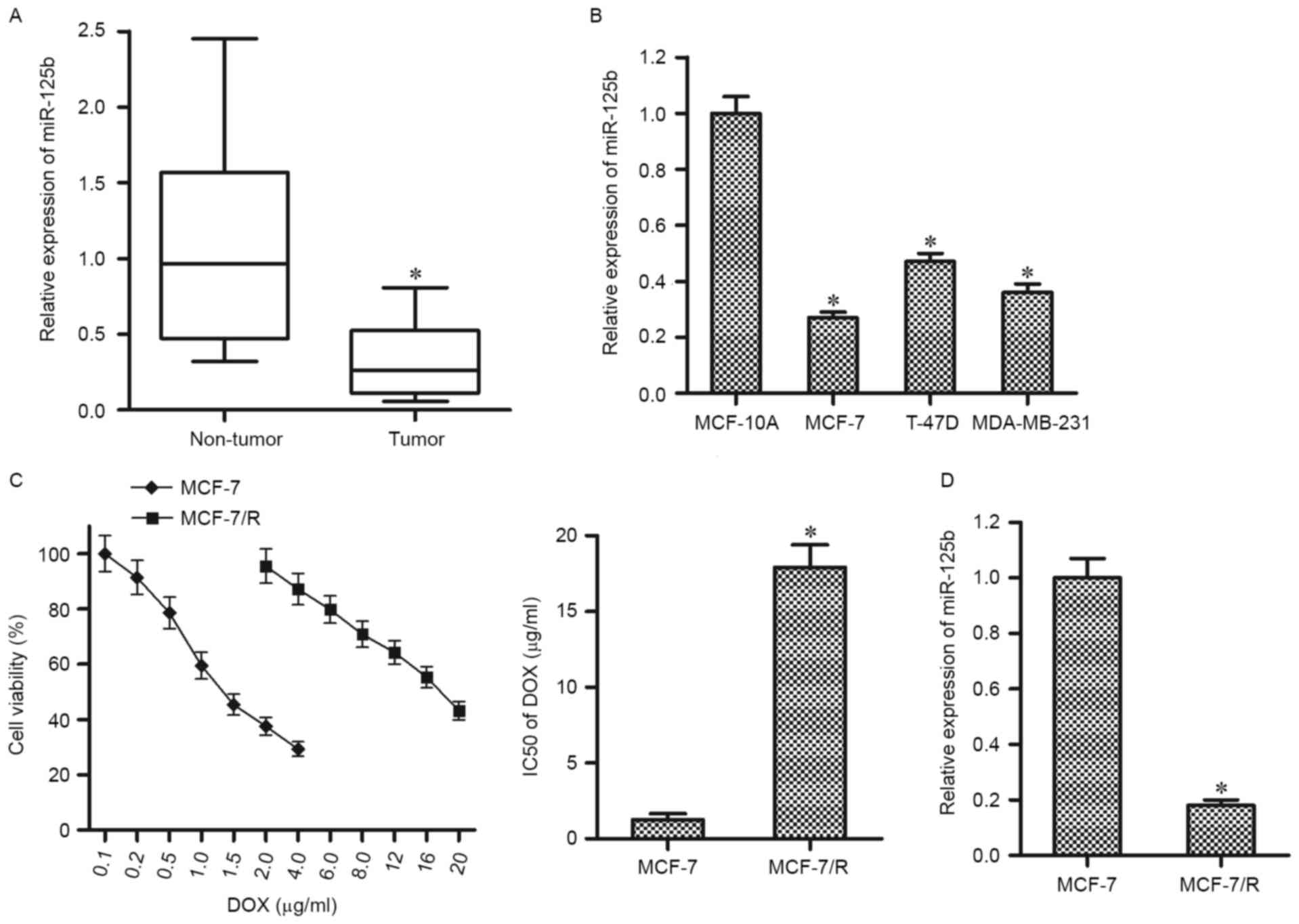
using RT-qPCR. As presented in Fig.
1A, the miR-125b levels were significantly downregulated in BC
tissues compared with the corresponding normal tissues.
Furthermore, the results in vitro demonstrated that the
expression levels of miR-125b in BC cell lines (MCF-7, MDA-MB-231,
and T-47D) were significantly higher compared with that in the
normal breast epithelial cell line MCF-10A (Fig. 1B). These results suggest that miR-125b
may function as a tumor suppressor in BC. To investigate
chemoresistance in BC, a DOX-resistant MCF-7 cell line (MCF-7/R)
was established. It was observed that the MCF-7/R cells were
significantly resistant to the DOX treatment compared with their
parental MCF-7 cells (Fig. 1C). To
investigate the association between miR-125b and DOX-resistance,
RT-qPCR analysis was performed in MCF-7 and MCF-7/R cells. Notably,
the expression of miR-125b was significantly lower in MCF-7/R cells
compared with that in the parental MCF-7 cells, suggesting the
dysregulation of miR-125b is an important factor to promote the
DOX-resistance in BC (Fig. 1D).
Enforced expression of miR-125b
resensitizes MCF-7/R cells to DOX treatment
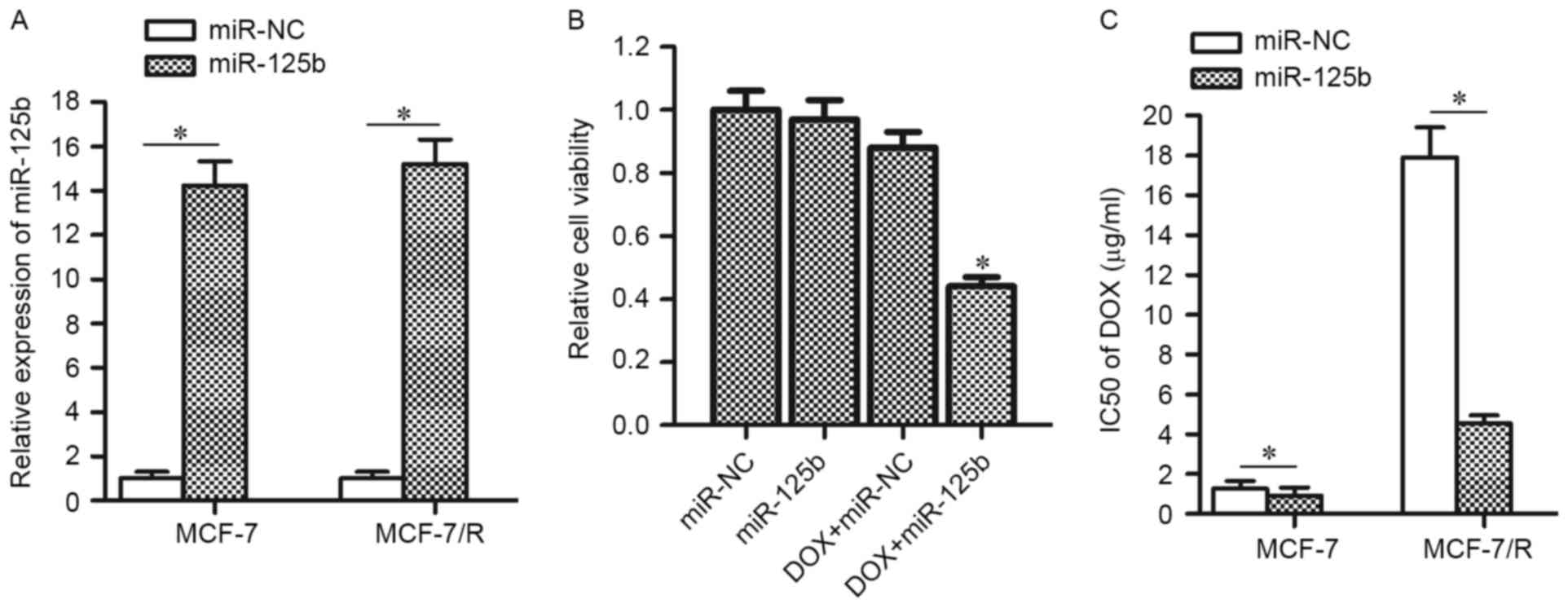
To confirm the hypothesis that the downregulation of
miR-125b promotes DOX-resistance, MCF-7/R cells as well their
parental MCF-7 cells were transfected with miR-125b mimics (the
transfection efficiency is presented in Fig. 2A). In addition, as treatment with 4
µg/ml DOX induced slight cell death in MCF-7/R (Fig. 1C), this concentration of DOX was
chosen for combination treatment with miR-125b mimics. As expected,
transfection of miR-125b mimics was observed to significantly
enhance the anti-tumor effect of DOX on MCF-7/R cells (Fig. 2B). Enforced expression of miR-125b
decreased the IC50 of DOX by 74.6% in MCF-7/R cells and
by 27.1% in MCF-7 cells (Fig. 2C).
These results indicated that the DOX-resistant MCF-7 cells are more
sensitive to miR-125b compared with the routine MCF-7 cells and
enforced expression of miR-125b is able to resensitize the MCF-7/R
cells to DOX treatment.
HAX-1 is the target of miR-125b in
MCF-7/R
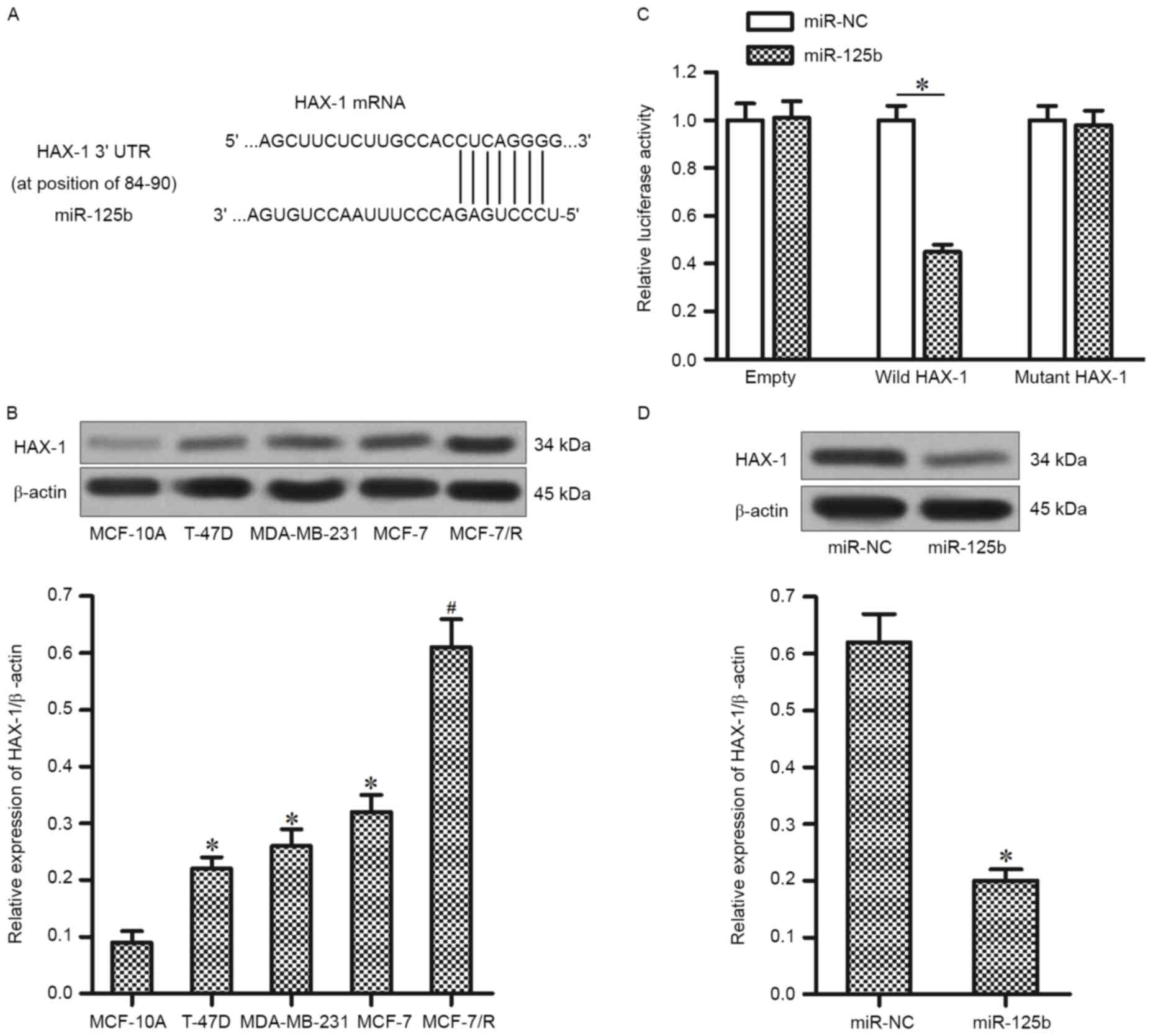
To investigate the target of miR-125b, which is
associated with the DOX-resistance in MCF-7/R, the public database
of TargetScan was used. It was demonstrated that the HAX-1 gene was
a putative target of miR-125b (Fig.
3A). In accordance with this, the expression of HAX-1 was
significantly higher in T-47D, MDA-MB-231 and MCF-7 BC cell lines
compared with the MCF-10A cells. Furthermore, the expression level
of HAX-1 was upregulated in MCF-7/R cells compared with that in the
parental MCF-7 cells (Fig. 3B).
Therefore, it was hypothesized that the HAX-1 is the target of
miR-125b, its overexpression of which is responsible for inducing
the DOX-resistance in MCF-7/R. To validate this, a luciferase
reporter assay was performed to directly investigate whether HAX-1
was targeted by miR-125b. It was observed that the miR-125b mimics
significantly decreased the luciferase activity of wild-HAX-1
reporter, but not the mutant or empty reporter (Fig. 3C). Furthermore, transfection of
miR-125b significantly downregulated the protein level of HAX-1 in
MCF-7/R cells (Fig. 3D). Taken
together, these results suggest that HAX-1 is the target of
miR-125b in DOX-resistant MCF-7 cells.
Enforced expression of miR-125b
resensitizes MCF-7/R cells to DOX via downregulation of HAX-1
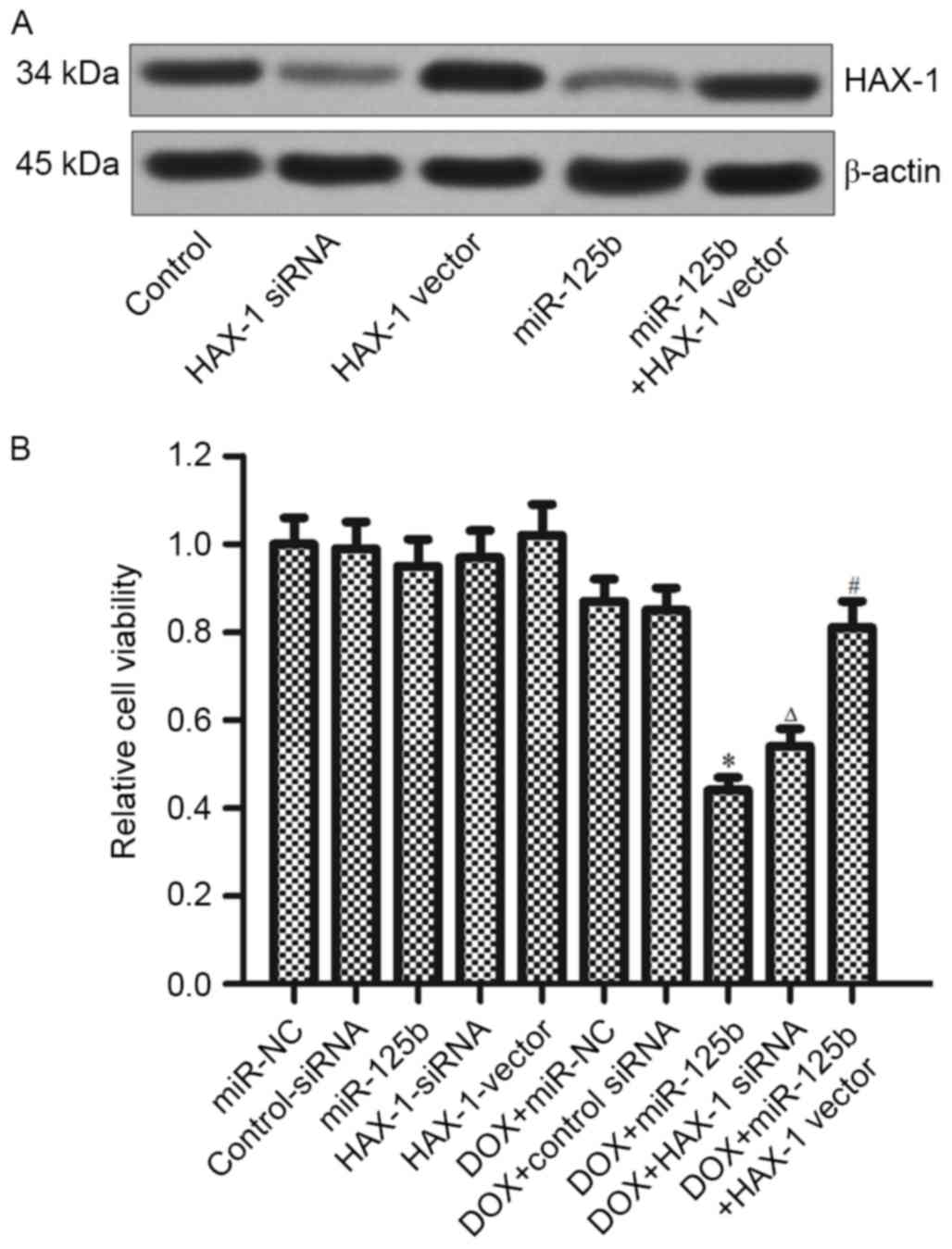
To confirm whether miR-125b regulates the
DOX-resistance via targeting the HAX-1, MCF-7/R cells were
transfected with HAX-1 siRNA, HAX-1 vector and miR-125b mimics (the
transfection efficiency is presented in Fig. 4A). It was demonstrated that HAX-1
siRNA serves a similar role with the miR-125b in promoting
DOX-induced cell death. However, overexpression of HAX-1
significantly decreased the cytotoxicity in the combination
treatment of DOX+miR-125b (Fig. 4B).
These results indicated that the miR-125b/HAX-1 signaling pathway
regulates the DOX-resistance in BC.
miR-125b promotes DOX-induced cell
death through caspase-dependent apoptosis in MCF-7/R cells
As the aforementioned results demonstrated that the
downregulation of HAX-1 is essential for reversing DOX-resistance
in MCF-7/R cells, the effect of the combination with DOX and
miR-125b on the cell death pathway was next investigated. As
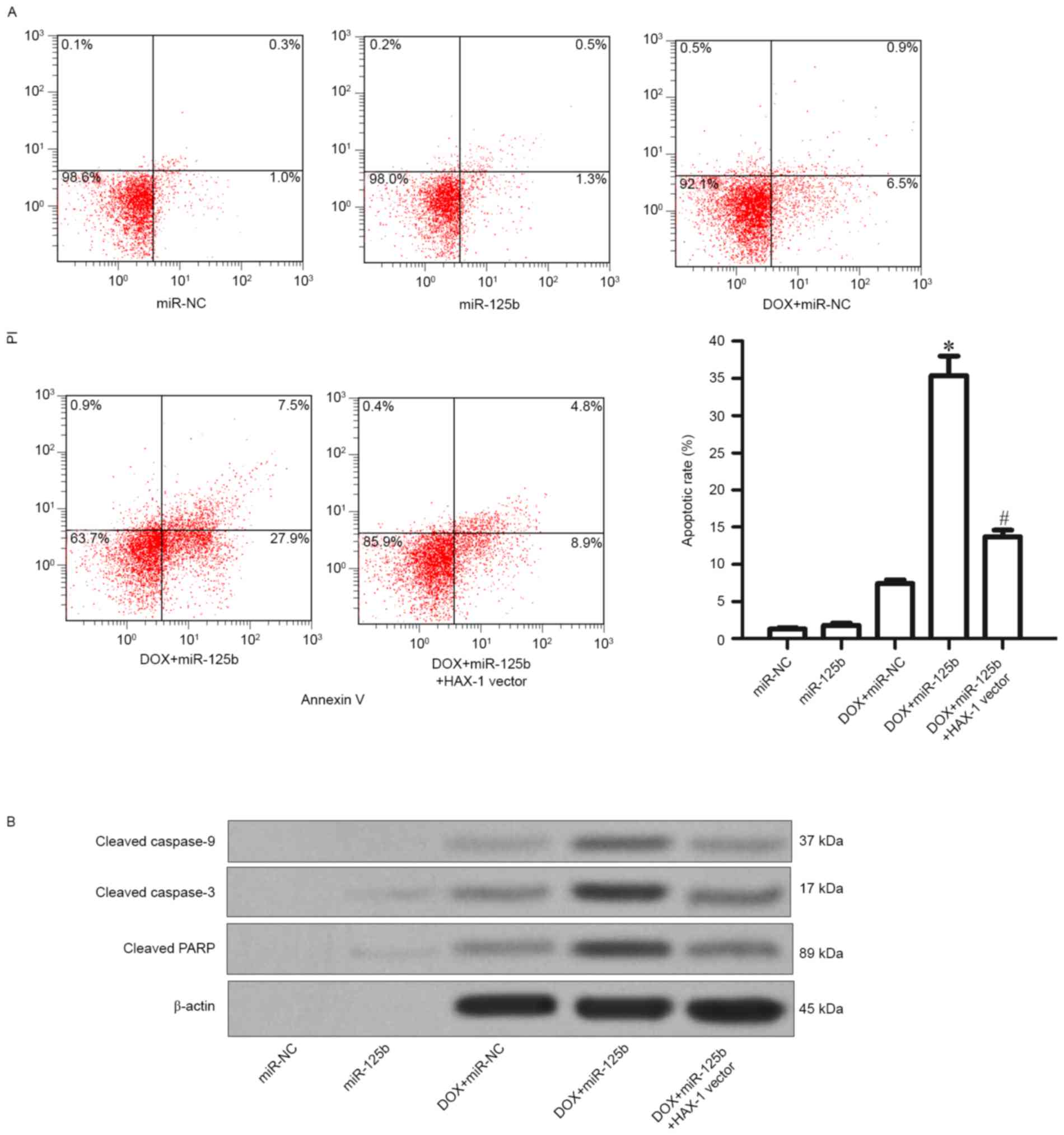
presented in Fig. 5A, it was observed
that although the DOX alone treatment induced apoptosis slightly in
MCF-7/R cells, the combination with miR-125b promoted the
DOX-induced apoptosis significantly (Fig.
5A). Furthermore, it was revealed that the combination of DOX
and miR-125b resulted in significant activation of caspase-9,
caspase-3 and their substrate PARP, in addition, the activation of
caspase activity was significantly impaired by the HAX-1 vector
(Fig. 5B). Thus, it was demonstrated
that miR-125b promotes DOX-induced apoptosis through the
caspase-dependent signaling pathway by targeting HAX-1.
miR-125b-promoted apoptosis is
dependent on the HAX-1-MMP-ROS signaling pathway
Previous reports have indicated that HAX-1 is an
important regulator for the mitochondrial pathway of apoptosis
(19). Therefore, the role of the
mitochondrial pathway in miR-125b-promoted apoptosis was
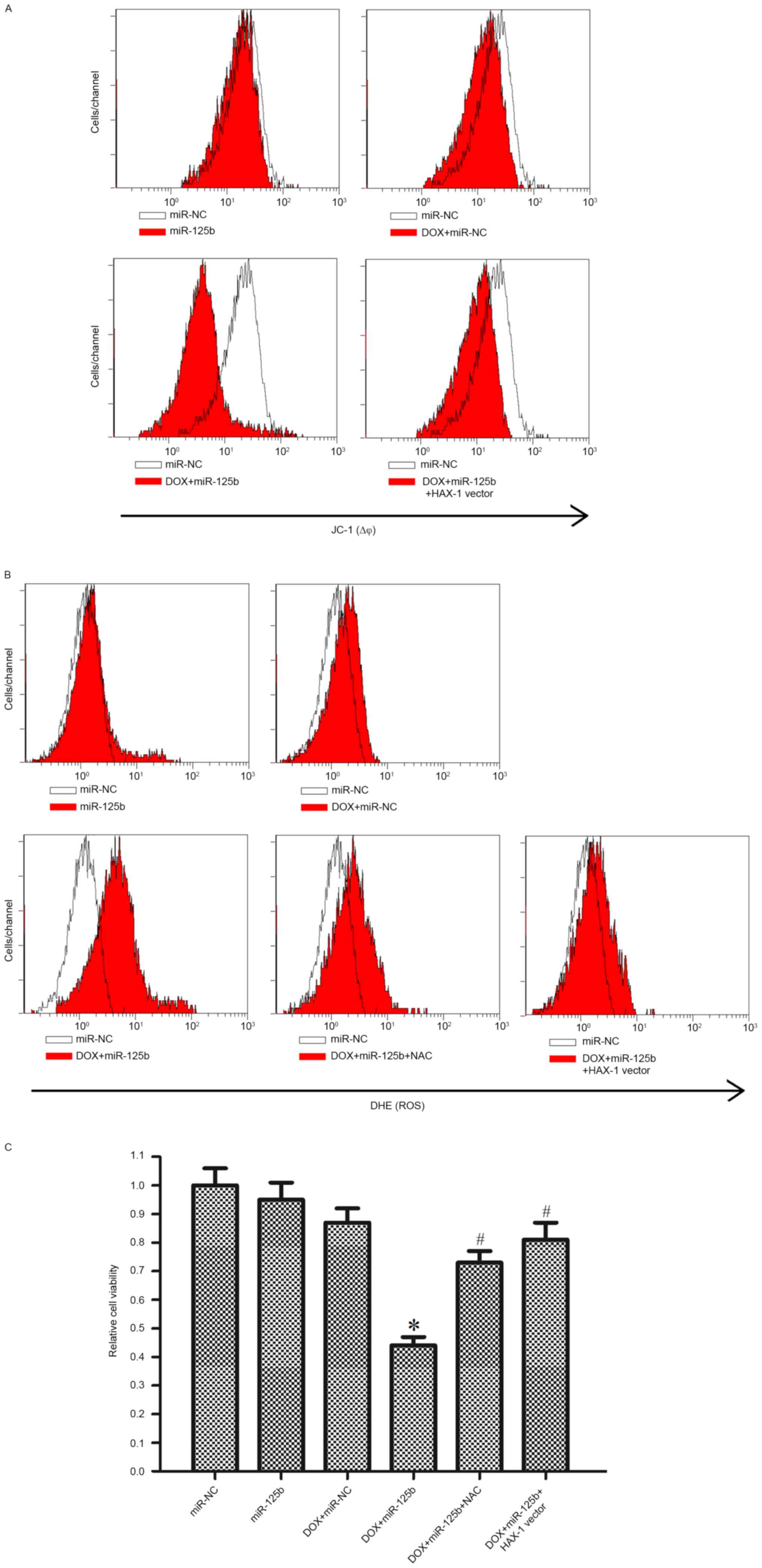
investigated. As presented in Fig.
6A, it was demonstrated that although miR-125b alone did not
influence the MMP of MCF-7/R cells, miR-125b significantly promoted
DOX-induced mitochondrial damage. Furthermore, the effect of
miR-125b on mitochondria was inhibited by the HAX-1 vector.
Previous studies have indicated that the generation of ROS may be
induced by the mitochondrial dysfunction. Thus, ROS generation was
assessed by DHE staining using a previously descried method
(18,20). It was observed that miR-125b
significantly promoted the generation of ROS induced by DOX
treatment. Notably, the HAX-1 vector and NAC, a scavenger of ROS
(18), inhibited ROS generation,
indicating the important role of HAX-1 in the mitochondrial pathway
of DOX-based treatment in MCF-7/R cells (Fig. 6B). Furthermore, the HAX-1 vector and
NAC significantly inhibited DOX- and miR-125b-induced cell death in
MCF-7/R cells (Fig. 6C). Taken
together, this data suggested that miR-125b promoted DOX-induced
apoptosis through the HAX-1-MMP-ROS pathway.
Discussion
An increasing number of studies have demonstrated
that miR-125b acts as a tumor suppressor in multiple cancer types.
Zhao et al (21) demonstrated
that the expression of miR-125b in tumor tissues is lower compared
with that in adjacent normal tissues. Furthermore, the
overexpression of miR-125b inhibited cell proliferation, migration
and caused cell cycle arrest in G1 phase in bladder cancer
(21). In ovarian cancer cells,
miR-125b was reported to suppress tumor invasion and migration by
targeting eukaryotic translation initiation factor 4E binding
protein 1 (22). The present study
demonstrated that miR-125b was significantly downregulated in the
tumor tissues of patients with BC and BC cell lines, when compared
with adjacent non-tumor, and normal MCF-10A cells, respectively.
Furthermore, it was revealed that the MCF-7 cells exhibited a more
extensive decline in miR-125b expression when they became
DOX-resistance. Therefore, we hypothesized that miR-125b is a tumor
suppressor in BC.
Previously, studies have confirmed that the
dysregulation of miRNAs is associated with the curative effect of
chemotherapy in cancer. For example, miR-193b which is
downregulated in HCC has been demonstrated to be essential for
cisplatin-induced apoptosis (23).
The knockdown of miR-155 is able to reverse DOX-resistance in lung
cancer (24). Overexpression of
miR-125b in chondrosarcoma cells inhibits cell growth rates and
increases the sensitivity of cells to DOX (25). To investigate chemoresistance in BC, a
DOX-resistant MCF-7 cell line was established. Several reports have
demonstrated that P-glycoprotein, which leads to drug efflux, is
overexpressed and mediates drug-resistance in chemoresistant BC
cells (26). However, in the current
study, it was revealed that the miRNA dysregulation also mediated
chemoresistance. To investigate whether or not downregulated
miR-125b was associated the chemoresistance, the MCF-7/R cells were
treated with DOX in the presence or absence of miR-125b mimics. It
was observed that the combination with miR-125b significantly
decreased the IC50 of DOX in MCF-7/R cells. Therefore,
the role of miR-125b in reversing DOX-resistance was
demonstrated.
HAX-1 is an important anti-apoptotic protein, which
is located on the mitochondrial outer membrane (27). Previous studies have reported that the
overexpression of HAX-1 impedes apoptosis of cancer cells,
resulting from the protection of mitochondria from damage induced
by drugs and environmental factors (28). Therefore, numerous malignancies
exhibit a high level of HAX-1, including BC (29). In the present study, HAX-1 was
identified as a target of miR-125b and the promotion of miR-125b
mimics on DOX-induced cell death is dependent on the downregulation
of HAX-1 in MCF-7/R cells.
As HAX-1 is an apoptosis-associated protein, the
results of the current study revealed that the overexpression of
miR-125b reversed DOX-resistance and promoted DOX-induced apoptosis
through the HAX-1 signaling pathway. To further explore the
underlying mechanisms, the MMP was measured, which is the
downstream of HAX-1 signaling. The results indicated that
miR-125b-induced inhibition of HAX-1 promoted the DOX to damage the
mitochondria. In combination with miR-125b and DOX, HAX-1 induced a
decrease in the MMP, which was responsible for the release of
mitochondrial components into the cytoplasm (30). Among these components, ROS is a key
inducer of mitochondrial apoptosis (20). In the current study, it was observed
that the elimination of ROS by NAC inhibited cell death induced by
the combination of miR-125b and DOX in MCF-7/R cells. This suggests
that the overexpression of miR-125b resensitizes the MCF-7/R cells
to DOX through the mitochondrial-ROS pathway.
In summary, the present study demonstrates that
miR-125b acts as a tumor suppressor in BC. Additionally, the
absence of miR-125b may be essential for developing DOX-resistance
in BC. miR-125b/HAX-1-targeted therapy may be considered as a novel
strategy for reversing the resistance of cancer cells to DOX
treatment.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Du XL, Key CR, Osborne C, Mahnken JD and
Goodwin JS: Discrepancy between consensus recommendations and
actual community use of adjuvant chemotherapy in women with breast
cancer. Ann Intern Med. 138:90–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen YY, Li ZZ, Ye YY, Xu F, Niu RJ, Zhang
HC, Zhang YJ, Liu YB and Han BS: Knockdown of SALL4 inhibits the
proliferation and reverses the resistance of MCF-7/ADR cells to
doxorubicin hydrochloride. BMC Mol Biol. 17:62016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H, Yu Y, Jiang Z, Cao WM, Wang Z, Dou
J, Zhao Y, Cui Y and Zhang H: Next-generation proteasome inhibitor
MLN9708 sensitizes breast cancer cells to doxorubicin-induced
apoptosis. Sci Rep. 6:264562016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen NT, Wu CY, Chung CY, Hwu Y, Cheng SH,
Mou CY and Lo LW: Probing the dynamics of doxorubicin-DNA
intercalation during the initial activation of apoptosis by
fluorescence lifetime imaging microscopy (FLIM). PLoS One.
7:e449472012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zheng Y, Lv X, Wang X, Wang B, Shao X,
Huang Y, Shi L, Chen Z, Huang J and Huang P: MiR-181b promotes
chemoresistance in breast cancer by regulating Bim expression.
Oncol Rep. 35:683–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chakravarty G, Mathur A, Mallade P,
Gerlach S, Willis J, Datta A, Srivastav S, Abdel-Mageed AB and
Mondal D: Nelfinavir targets multiple drug resistance mechanisms to
increase the efficacy of doxorubicin in MCF-7/Dox breast cancer
cells. Biochimie. 124:53–64. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ambros V: MicroRNA pathways in flies and
worms: Growth, death, fat, stress, and timing. Cell. 113:673–676.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan YY, Xu XY, Wang JF, Zhang CW and Zhang
SC: MiR-654-5p attenuates breast cancer progression by targeting
EPSTI1. Am J Cancer Res. 6:522–532. 2016.PubMed/NCBI
|
|
12
|
Gu X, Xue JQ, Han SJ, Qian SY and Zhang
WH: Circulating microRNA-451 as a predictor of resistance to
neoadjuvant chemotherapy in breast cancer. Cancer Biomark.
16:395–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye Z, Hao R, Cai Y, Wang X and Huang G:
Knockdown of miR-221 promotes the cisplatin-inducing apoptosis by
targeting the BIM-Bax/Bak axis in breast cancer. Tumour Biol.
37:4509–4515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou S, Huang Q, Zheng S, Lin K, You J and
Zhang X: miR-27a regulates the sensitivity of breast cancer cells
to cisplatin treatment via BAK-SMAC/DIABLO-XIAP axis. Tumour Biol.
37:6837–6845. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang J, Tian X, Han R, Zhang X, Wang X,
Shen H, Xue L, Liu Y, Yan X, Shen J, et al: Downregulation of
miR-486-5p contributes to tumor progression and metastasis by
targeting protumorigenic ARHGAP5 in lung cancer. Oncogene.
33:1181–1189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Prathapan A, Vineetha VP and Raghu KG:
Protective effect of Boerhaavia diffusa L. against mitochondrial
dysfunction in angiotensin II induced hypertrophy in H9c2
cardiomyoblast cells. PLoS One. 9:e962202014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Duan W, Jin X, Li Q, Tashiro S, Onodera S
and Ikejima T: Silibinin induced autophagic and apoptotic cell
death in HT1080 cells through a reactive oxygen species pathway. J
Pharmacol Sci. 113:48–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Radhika V, Onesime D, Ha JH and
Dhanasekaran N: Galpha13 stimulates cell migration through
cortactin-interacting protein Hax-1. J Biol Chem. 279:49406–49413.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao X, He W, Li J, Huang S, Wan X, Luo H
and Wu D: MiRNA-125b inhibits proliferation and migration by
targeting SphK1 in bladder cancer. Am J Transl Res. 7:2346–2354.
2015.PubMed/NCBI
|
|
22
|
Lee M, Kim EJ and Jeon MJ: MicroRNAs 125a
and 125b inhibit ovarian cancer cells through post-transcriptional
inactivation of EIF4EBP1. Oncotarget. 7:8726–8742. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin W, Nie Y, Zhang Z, Xie L and He X:
miR-193b acts as a doxorubicin sensitizer via the
caspase-3-dependent pathway in HCC chemotherapy. Oncol Rep.
34:368–3674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lv L, An X, Li H and Ma L: Effect of
miR-155 knockdown on the reversal of doxorubicin resistance in
human lung cancer A549/dox cells. Oncol Lett. 11:1161–1166.
2016.PubMed/NCBI
|
|
25
|
Tang XY, Zheng W, Ding M, Guo KJ, Yuan F,
Feng H, Deng B, Sun W, Hou Y and Gao L: miR-125b acts as a tumor
suppressor in chondrosarcoma cells by the sensitization to
doxorubicin through direct targeting the ErbB2-regulated glucose
metabolism. Drug Des Devel Ther. 10:571–583. 2016.PubMed/NCBI
|
|
26
|
Clarke R, Leonessa F and Trock B:
Multidrug resistance/P-glycoprotein and breast cancer: Review and
meta-analysis. Semin Oncol. 32(6 Supp 7): S9–S15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suzuki Y, Demoliere C, Kitamura D,
Takeshita H, Deuschle U and Watanabe T: HAX-1, a novel
intracellular protein, localized on mitochondria, directly
associates with HS1, a substrate of Src family tyrosine kinases. J
Immunol. 158:2736–2744. 1997.PubMed/NCBI
|
|
28
|
Trebinska A, Högstrand K, Grandien A,
Grzybowska EA and Fadeel B: Exploring the anti-apoptotic role of
HAX-1 versus BCL-XL in cytokine-dependent bone marrow-derived cells
from mice. FEBS Lett. 588:2921–2927. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Trebinska A, Rembiszewska A, Ciosek K,
Ptaszynski K, Rowinski S, Kupryjanczyk J, Siedlecki JA and
Grzybowska EA: HAX-1 overexpression, splicing and cellular
localization in tumors. BMC Cancer. 10:762010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Halestrap AP, Clarke SJ and Javadov SA:
Mitochondrial permeability transition pore opening during
myocardial reperfusion-a target for cardioprotection. Cardiovasc
Res. 61:372–385. 2004. View Article : Google Scholar : PubMed/NCBI
|