Introduction
The receptor tyrosine-protein kinase erbB (ErbB)
family of receptor tyrosine kinases (RTKs) consists of four
members, ErbB1 [also known as epidermal growth factor receptor
(EGFR)/HER1], ErbB2 (also known as proto-oncogene Neu/HER2), ErbB3
(also termed HER3) and ErbB4 (also termed HER4) (1). Upon activation, ErbB family members form
homodimers or heterodimers that serve important roles in biological
processes associated with differentiation, proliferation and
apoptosis, primarily through mitogen-activated protein kinase and
the phosphoinositide-3 kinase/RAC-alpha serine/threonine-protein
kinase signaling pathways (2–6). ErbB mutations are frequently identified
in human cancer, leading to aberrant ErbB expression and subsequent
survival signals (7). Therefore,
drugs targeting the ErbB receptors have been a central focus of
cancer research (8,9).
RTKs are non-canonically regulated by
phosphorylation at their serine and threonine residues. For
example, extracellular signal-regulated kinase (ERK)-mediated
phosphorylation of a conserved threonine in the juxtamembrane
domain of EGFR (Thr-669) and ErbB2 (Thr-677) is a typical negative
feedback mechanism of ErbB dimers (10,11). Tumor
necrosis factor-α-induced and p38 mitogen-activated protein kinase
(p38)-mediated phosphorylation of EGFR (Ser-1046/1047) at the
C-terminal tail is associated endocytosis of the receptor. A
previous study demonstrated that cisplatin (CDDP), a well known
DNA-damaging agent, induces the non-canonical phosphorylation of
EGFR proteins harboring an activating mutation in lung cancer
cells, resulting in negative feedback inhibition and endocytosis
(12).
ErbB2, which has been demonstrated to be expressed
in 20–30% of breast cancer cases (13), is usually co-expressed with ErbB3, and
together they form a heterodimer that transmits survival signals
(14). A typical strategy for
treating ErbB2-overexpressing breast cancer is to block its
signaling using targeted therapies, including trastuzumab and
lapatinib (15); however, resistance
to these agents is emerging in clinical settings (15,16).
Chemotherapeutic DNA-damaging agents, including doxorubicin (DOX),
are frequently used to treat breast tumors (17); therefore, in the current study the
effects of CDDP and DOX on the non-canonical regulation of
ErbB2/ErbB3 activation were investigated in breast cancer
cells.
Materials and methods
Antibodies and reagents
The following antibodies were used; phosphorylation
(phospho)-specific antibodies against ERK (Thr-202 and Tyr-204,
cat. no. 9101, 1:2,000 dilution), p38 (Thr-180 and Tyr-182, cat.
no. 9211, 1:2,000 dilution), ErbB2 (Tyr-1196, cat. no. 6942,
1:2,000 dilution) and ErbB3 (Tyr-1289, cat. no. 4791, 1:2,000
dilution) (Cell Signaling Technology, Inc., Danvers, MA, USA). In
addition, antibodies against ErbB2 (cat. no. sc-284, 1:2,000
dilution), ErbB3 (cat. no. sc-285, 1:2,000 dilution) and actin
(cat. no. sc-1615, 1:2,000 dilution) were obtained from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA. A monoclonal recombinant
antibody against phospho-ErbB2 Thr-677 (clone no. 18-4, 1:1,000
dilution) was generated using the rabbit immunospot array assay on
a chip, as described previously (11,18,19).
Trametinib (AdooQ BioScience LLC, Irvine, CA, USA) and SB203580
(Merck KGaA, Darmstadt, Germany), CDDP, DOX and G418 (Wako Pure
Chemical Industries, Ltd., Osaka, Japan) were also used. All
chemical agents were dissolved in dimethylsulphoxide (DMSO), and
the final concentration of DMSO was <0.1%.
Cell culture and treatment
Human BT474, MDA-MB-453 and 293 cells were
maintained in Dulbecco's modified Eagle's medium (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
calf serum (FCS), 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin. Human MKN45 gastric cancer cells were maintained in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FCS, 2 mM glutamine, 100 U/ml penicillin and
100 µg/ml streptomycin. Cells were incubated at 37°C with 5%
CO2. 293 cells were cultured to maintain the
transfection stably expressing ErbB2/ErbB3 maintained in media
containing 0.5 mg/ml G418 (11).
Cells were treated with CDDP (100 µM) for 3–12 h or DOX (10 µM) for
12–24 h with or without pretreatment with Trametinib (0.03 µM) or
SB203580 (10 µM) for 30 min.
Transfection
293 cells were transfected with plasmid DNA
(11), encoding human ErbB2 and ErbB3
complementary DNA sequences using Lipofectamine® 2000
according to manufacturer's protocol (Invitrogen; Thermo Fisher
Scientific, Inc.). The KOD FX neo kit (Toyobo, Co., Ltd., Osaka,
Japan) was used for substitution of Thr-677 to Ala (T677A) in
ErbB2, as previously described (11).
Western blotting
BT474, MDA-MB-453, 293, and MKN45 whole cell
lysates, prepared as described previously (20,21), were
resolved using SDS-PAGE (8 or 10% gel; 15–20 µg protein loaded per
lane) and transferred to an Immobilon-P nylon membrane (EMD
Millipore, Billerica, MA, USA). The membrane was treated with 100%
BlockAce (Dainippon Sumitomo Pharma Co., Ltd., Osaka, Japan) and
probed with primary antibodies aforementioned for 2–3 h at room
temperature. Antibodies were detected using horseradish
peroxidase-conjugated anti-rabbit (cat. no. P0448; 1:2,000) and
anti-goat immunoglobulin G (cat. no. P0449, 1:2,000) (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA) and incubated for 1 h at
room temperature. Proteins were visualized using the
Pierce™ enhanced chemiluminescence western blotting
substrate (Thermo Fisher Scientific, Inc.). Actin was used as a
loading control.
Results
CDDP induces feedback inhibition of
ErbB2 and ErbB3 phosphorylation at Thr-677
A previous study demonstrated that
12-otetradecanoylphorbol-13-acetate (TPA) induces ERK-mediated
Thr-677 phosphorylation of ErbB2 (11). Given the role of CDDP in the
ERK-mediated phosphorylation of EGFR at Thr-669 in lung cancer
cells (22), in the present study the
role of CDDP-induced activation of ERK on ErbB2 and ErbB3
phosphorylation in breast cancer cells was investigated. CDDP
caused a time-dependent induction of ERK activation and subsequent
Thr-677 phosphorylation in BT474 cells (Fig. 1A). Concomitantly, tyrosine
phosphorylation of ErbB2 and ErbB3 was markedly reduced (Fig. 1A). Similarly, DOX stimulated ERK
activation and Thr-677 phosphorylation, which in turn led to
suppression of ErbB2 and ErbB3 tyrosine phosphorylation (Fig. 1B). Similar phosphorylation patterns
were observed for ErbB2 and ErbB3 in 293 stably transfected cells
(Fig. 1C) and in MDA-MB-453, another
ErbB2/ErbB3-overexpressing breast cancer cell line (Fig. 1D). Since CDDP is commonly used for
treating gastric cancers (23), the
effect of CDDP on ErbB2/3 phosphorylation in the ErbB2-expressing
MKN45 gastric cancer cells was examined. As predicted, CDDP induced
feedback control of ErbB2 and ErbB3 tyrosine phosphorylation
(Fig. 1E). Notably, total ErbB2 and
ErbB3 expression was downregulated by either CDDP or DOX treatments
in all cell lines (Fig. 1).
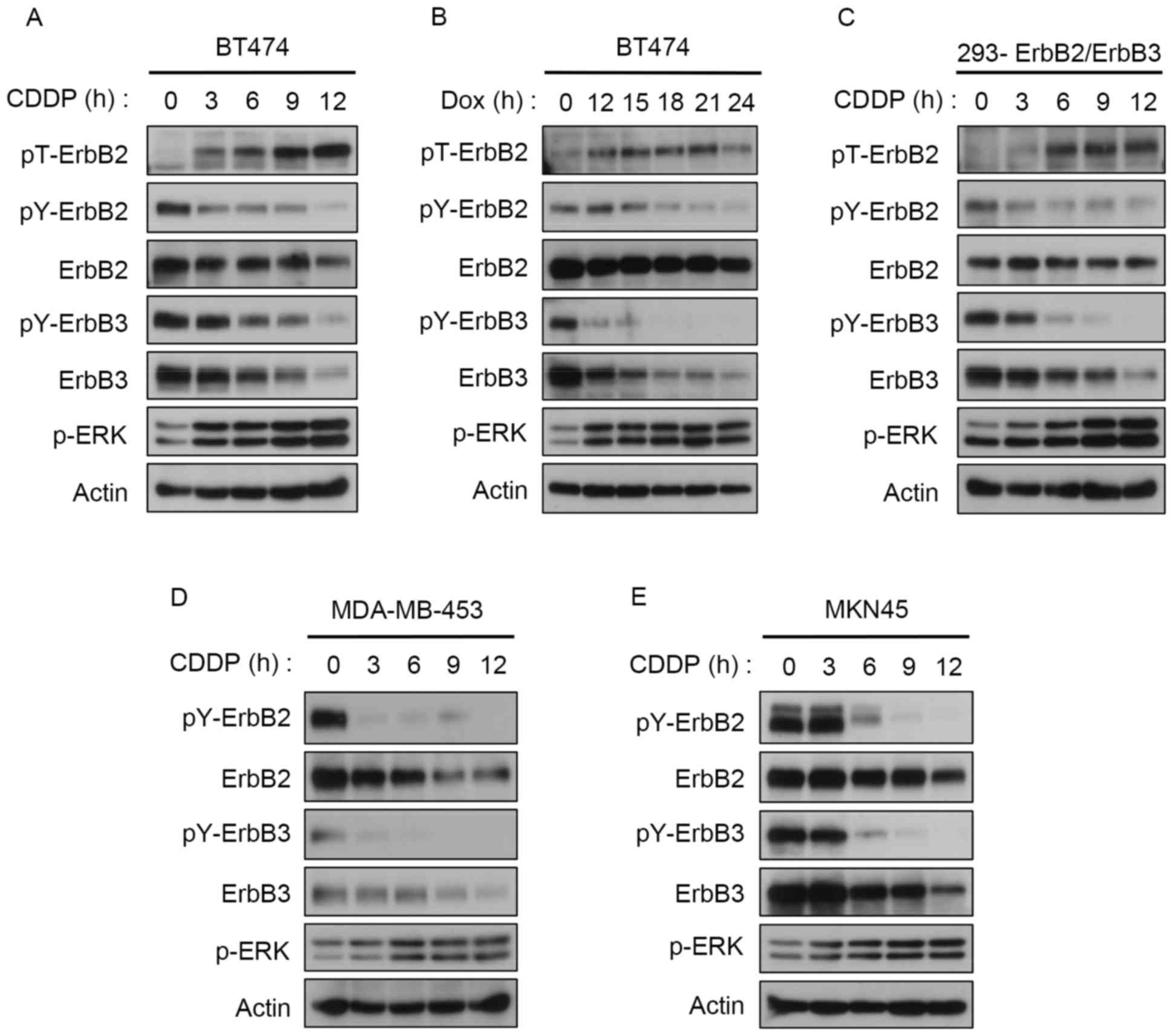 | Figure 1.DNA-damaging agents induce feedback
inhibition of ErbB2 and ErbB3. Western blotting analysis of (A)
BT474 cells treated with 100 µM CDDP, (B) BT474 cells treated with
10 mM DOX, (C) 293 cells stably expressing ErbB2 and ErbB3 treated
with 100 µM CDDP, (D) MDA-MB-453 cells treated with 100 µM CDDP and
(E) MKN45 cells treated with 100 µM CDDP for the indicated time
points. ErbB, receptor tyrosine-protein kinase erbB; CDDP,
cisplatin; DOX, doxorubicin; ERK, extracellular signal-regulated
kinase; p, phosphorylated; Y, tyrosine; T, threonine. |
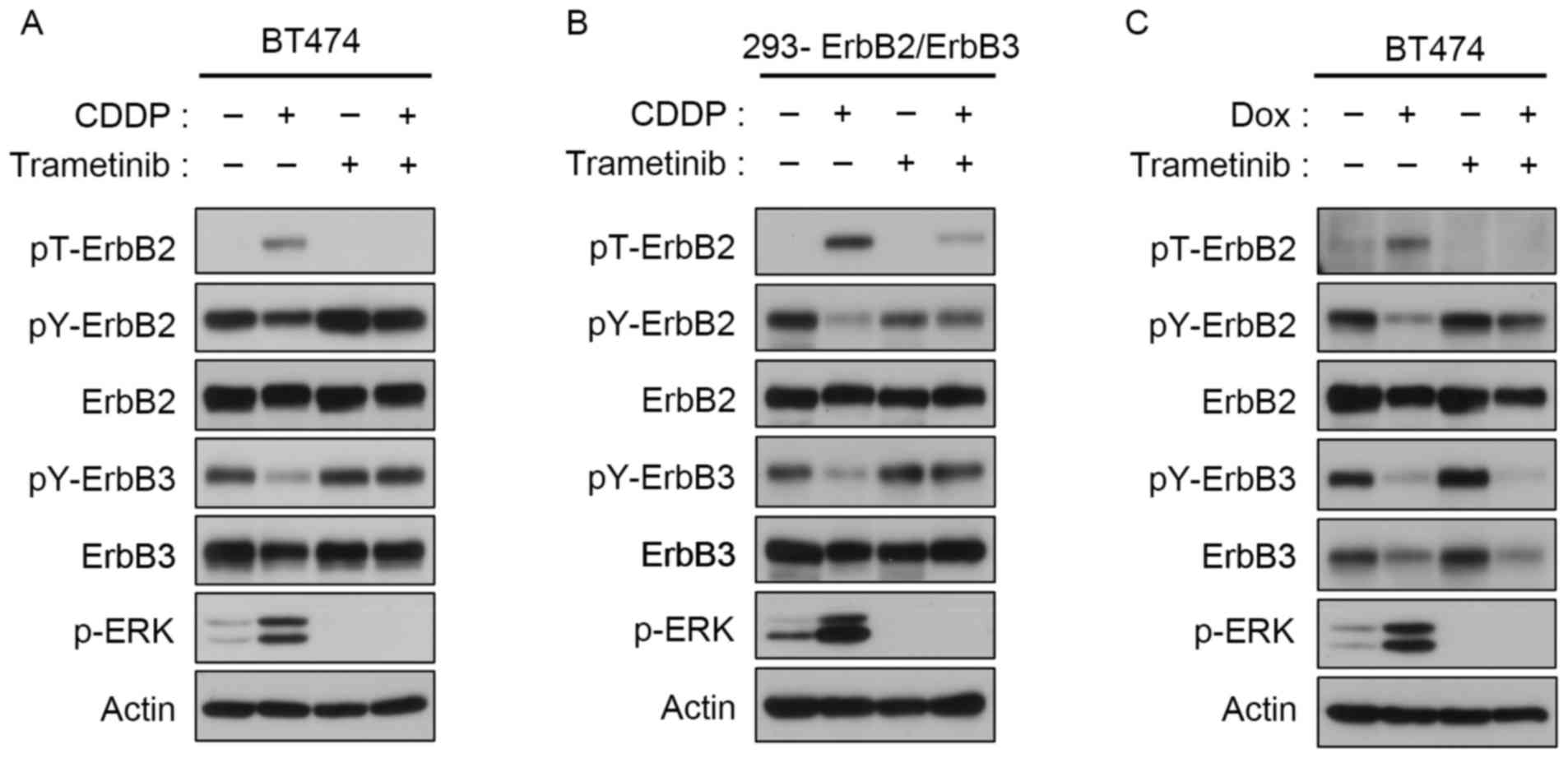
Feedback inhibition of ErbB2 and ErbB3
phosphorylation via ERK activation
ERK-mediated threonine phosphorylation of EGFR and
ErbB2 is a negative feedback mechanism that regulates their
tyrosine autophosphorylation (10,11,24). To
confirm the role of ERK activation in ErbB2 phosphorylation at
Thr-677, trametinib, a mitogen-activated protein kinase (MAPK)/ERK
(MEK) inhibitor approved for use in the treatment of melanoma, was
used. Trametinib suppressed ERK activation in
ErbB2/ErbB3-expressing BT474 cells and in ErbB2/ErbB3-transfected
293 cells initially induced by CDDP or DOX (Fig. 2). The inhibition of ERK activation was
associated with downregulation of Thr-677, and the subsequent
restoration of tyrosine phosphorylation of ErbB2 and ErbB3
(Fig. 2).
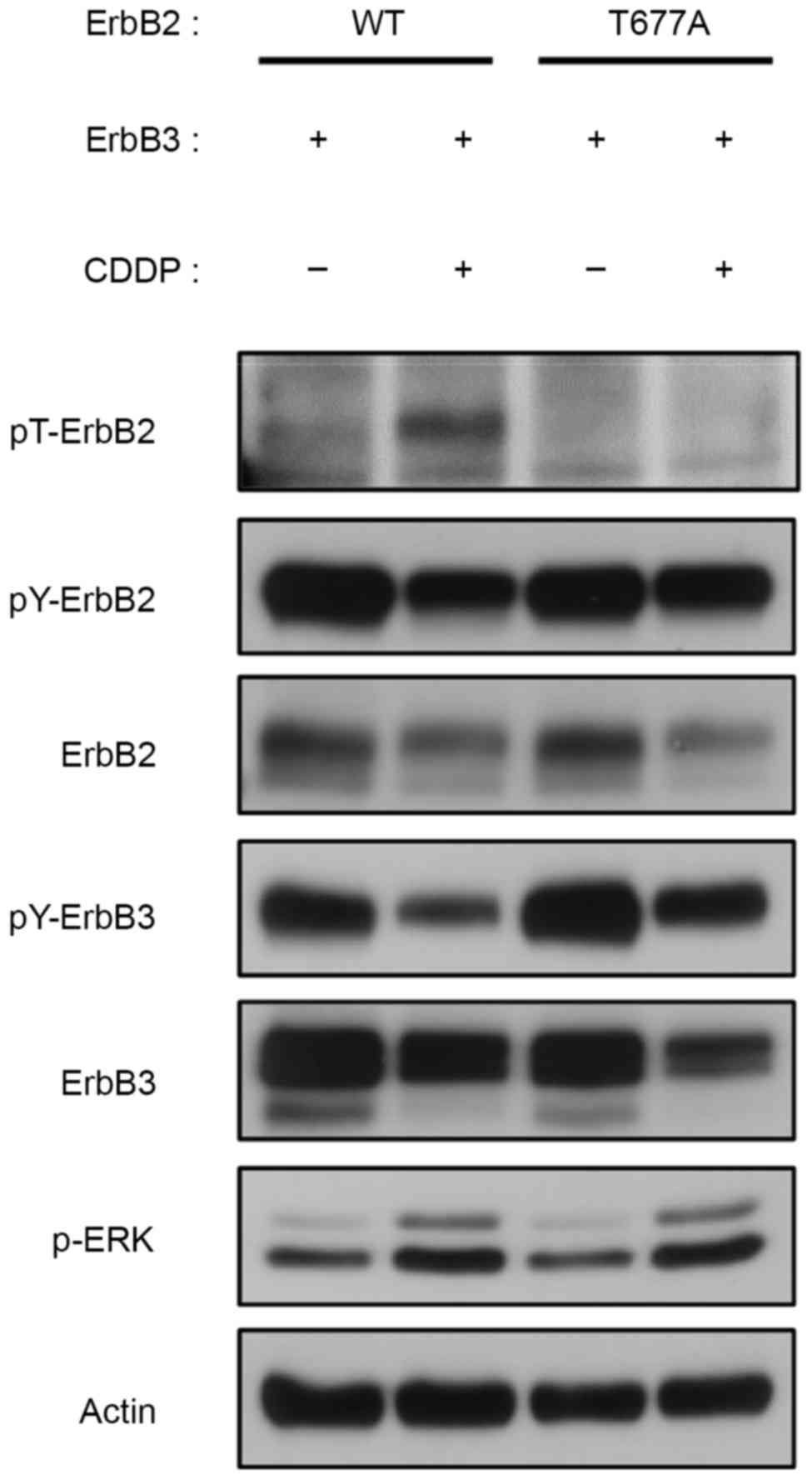
Role of ErbB2 Thr-677 in feedback
inhibition of ErbB2 and ErbB3
A previous study demonstrated the role of
TPA-induced ERK phosphorylation of ErbB2 at Thr-677 on the feedback
regulation of ErbB2/ErbB3 heterodimer (11). In light of these findings, the
feedback inhibition of the ErbB2/ErbB3 heterodimer by Thr-677
phosphorylation in CDDP-treated 293 cells was then investigated.
Cells were transiently transfected with either wild-type or
T677A-mutated ErbB2 and wild-type ErbB3, and then treated with CDDP
for 9 h. Notably, mutated ErbB2-T677A/ErbB3 did not notably respond
to CDDP treatment in terms of threonine/tyrosine modulation, yet
ERK was activated (Fig. 3).
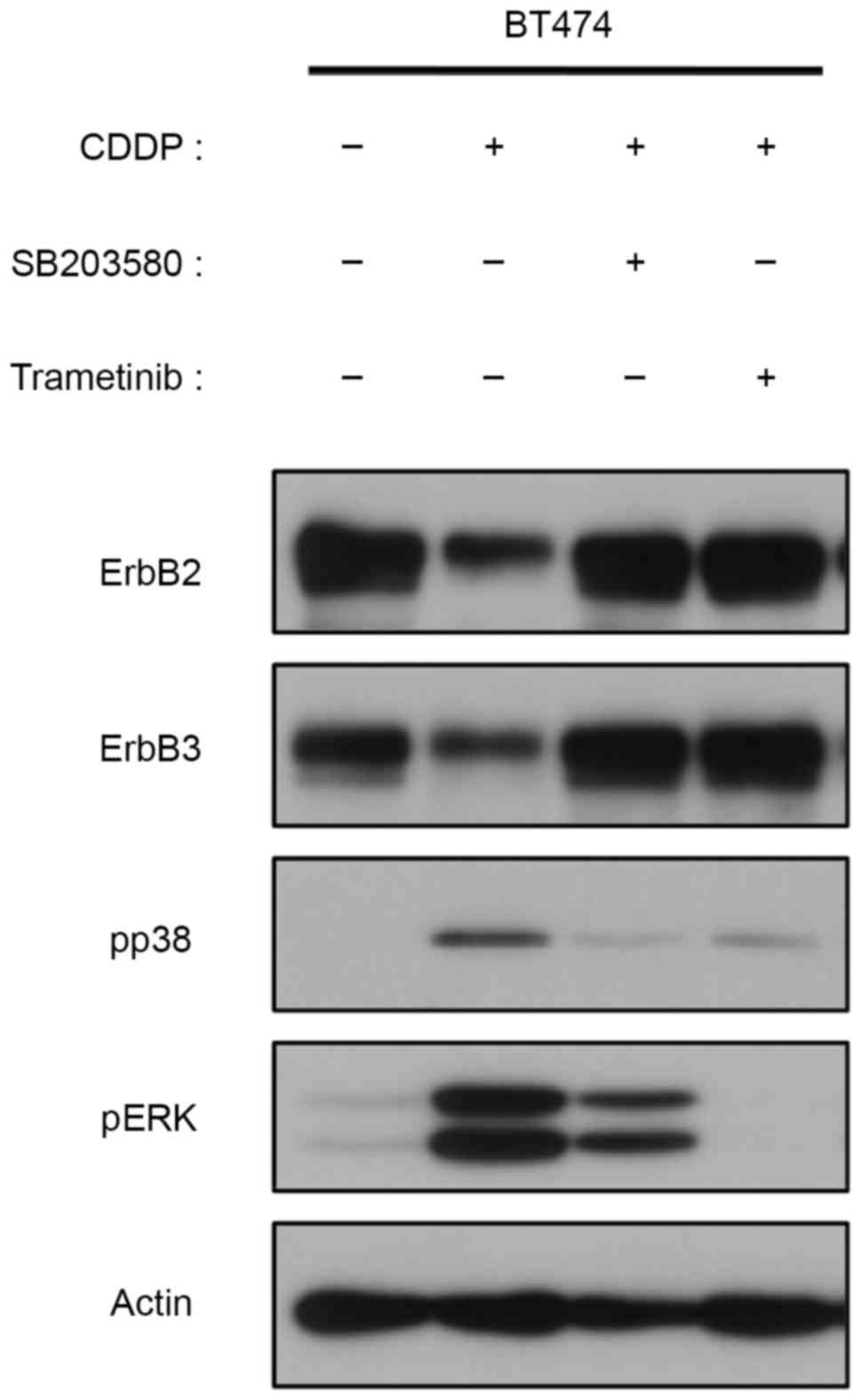
Potential role for MAPKs in the
CDDP-induced degradation of ErbB2 and ErbB3
Western blotting results demonstrated that ErbB2 and
ErbB3 are degraded following CDDP or DOX treatments, as
demonstrated by the decreased phosphorylation observed (Fig. 1A and B). Previous studies have
demonstrated that CDDP can cause EGFR degradation, and suggested
that this occurs through p38 activation (25,26). In
the current study, CDDP induced Thr-677 phosphorylation of ErbB2
through the activation of ERK, and subsequently inhibited tyrosine
phosphorylation of ErbB2 and ErbB3. However, the cause of ErbB2 and
ErbB3 degradation remains unclear, particularly in the later phase
of CDDP treatment. The effects of MAPK inhibitors on CDDP-treated
cells were also examined. Degradation of ErbB2 and ErbB3 following
9 h CDDP treatment was recovered upon the inhibition of p38 with
SB203580 (Fig. 4). Notably,
trametinib, which is primarily used to inhibit ERK phosphorylation,
exerted a similar effect on recovering CDDP-induced degradation of
ErbB2/3 (Fig. 4). These results
suggest that MAPK-mediated protein degradation contributes to the
CDDP-induced inactivation of ErbB2 and ErbB3 in
ErbB2-overexpressing breast cancer cells.
Discussion
Phosphoproteomic analyses have identified numerous
non-canonical phosphorylation sites in the intracellular domains of
RTKs (27); however, their
pathophysiological roles in cancer cells remain unclear. Recent
studies have investigated the biological significance of the
serine/threonine phosphorylation of RTKs in human cancer cells
(10,11,22,24). In
addition, a previous study demonstrated that chemotherapeutic
agents are important regulators of the non-canonical
phosphorylation of EGFR through MAPK signaling pathways (22). Therefore, in the current study the
regulation of ErbB2 and ErbB3 was investigated.
Similar to previous findings on the feedback
inhibition of EGFR in lung cancer cells (22), the current study revealed that the
ERK-mediated phosphorylation of a conserved threonine residue of
ErbB2 in the juxtamembrane domain is a common feedback inactivation
mechanism of ErbB2/ErbB3 heterodimers in breast cancer cells
targeted with DNA-damaging agents. These agents induce the
suppression of protein levels of ErbB2 and ErbB3 through the ERK
and p38 MAPK signaling pathways, suggesting a novel non-canonical
mechanism of ErbB downregulation. Ahsan et al (28) previously reported that EGFR
degradation serves a role in cisplatin-induced cytotoxicity in head
and neck cancer. Preliminary results using a Phos-tag have
demonstrated that CDDP induces phosphorylation of ErbB members at
multiple non-canonical residues (Park, unpublished); therefore, the
identification of novel serine/threonine sites that regulate the
degradation of ErbB receptors is essential for understanding the
underlying molecular mechanisms of chemotherapy-induced
inactivation of these receptors.
Notably, relatively high concentrations of CDDP and
DOX were used in the present study; however, this is similar to
previous studies, of which several also used longer treatment
durations (22,25,29–34). Data
from a previous study (11), in
addition to titration data from the present study, provided the
optimum conditions for the phosphorylation of ERK in addition to
ErbB2/3. However, further research is required in clinical models
to verify these results.
In conclusion, data from the present study
demonstrated that DNA-damaging agents cause the inactivation of
ErbB2 and ErbB3 via MAPK-mediated ErbB2 phosphorylation and
degradation. Trametinib suppressed the CDDP-induced negative
feedback regulation of ErbB2/3, suggesting that MEK inhibitors
affect the effectiveness of chemotherapy in combination with
DNA-damaging agents in clinical settings. These observations of the
association between tyrosine kinase-dependent canonical activation
and serine/threonine phosphorylation-mediated non-canonical
regulation of RTKs may aid in the establishment of more effective
strategies for cancer treatment, including ways to overcome
resistance to anticancer drugs.
Acknowledgements
The present study was supported by Grants-in-Aid for
Scientific Research (grant no. 16H04694), the Ministry of
Education, Culture, Sports, Science and Technology (grant no.
16K08366), and a grant from the Tamura Science and Technology
Foundation and the Smoking Research Foundation (grant no.
1904).
References
|
1
|
Roskoski R Jr: The ErbB/HER family of
protein-tyrosine kinases and cancer. Pharmacol Res. 79:34–74. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sebastiana S, Settlemanb J, Reshkinc SJ,
Azzaritia A, Bellizzia A and Paradisoa A: The complexity of
targeting EGFR signalling in cancer: From expression to turnover.
Biochim Biophys Acta. 1766:120–139. 2006.PubMed/NCBI
|
|
3
|
Schneider MR and Wolf E: The epidermal
growth factor receptor ligands at a glance. J Cell Physiol.
218:460–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh AB and Harris RC: Autocrine,
paracrine and juxtacrine signaling by EGFR ligands. Cell Signal.
17:1183–1193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sorkin A and Goh LK: Endocytosis and
intracellular trafficking of ErbBs. Exp Cell Res. 314:3093–3106.
2008.PubMed/NCBI
|
|
6
|
Avraham R and Yarden Y: Feedback
regulation of EGFR signalling: Decision making by early and delayed
loops. Nat Rev Mol Cell Biol. 12:104–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pines G, Köstler WJ and Yarden Y:
Oncogenic mutant forms of EGFR: Lessons in signal transduction and
targets for cancer therapy. FEBS Lett. 584:2699–2706. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park JW, Neve RM, Szollosi J and Benz CC:
Unraveling the biologic and clinical complexities of HER2. Clin
Breast Cancer. 8:392–401. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ménard S, Tagliabue E, Campiglio M and
Pupa SM: Role of HER2 gene overexpression in breast carcinoma. J
Cell Physiol. 182:150–162. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sato K, Shin MS, Sakimura A, Zhou Y,
Tanaka T, Kawanishi M, Kawasaki Y, Yokoyama S, Koizumi K, Saiki I
and Sakurai H: Inverse correlation between Thr-669 and constitutive
tyrosine phosphorylation in the asymmetric epidermal growth factor
receptor dimer conformation. Cancer Sci. 104:1315–1322. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kawasaki Y, Sakimura A, Park CM, Tomaru R,
Tanaka T, Ozawa T, Zhou Y, Narita K, Kishi H, Muraguchi A and
Sakurai H: Feedback control of ErbB2 via ERK-mediated
phosphorylation of a conserved threonine in the juxtamembrane
domain. Sci Rep. 6:315022016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dasari S and Tchounwou PB: Cisplatin in
cancer therapy: Molecular mechanisms of action. Eur J Pharmacol.
740:364–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: Correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Holbro T, Beerli RR, Maurer F, Koziczak M,
Barbas CF III and Hynes NE: The ErbB2/ErbB3 heterodimer functions
as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor
cell proliferation. Proc Natl Acad Sci USA. 100:pp. 8933–8938.
2003; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valabrega G, Montemurro F and Aglietta M:
Trastuzumab: Mechanism of action, resistance and future
perspectives in HER2-overexpressing breast cancer. Ann Oncol.
18:977–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bailey TA, Luan H, Clubb RJ, Naramura M,
Band V, Raja SM and Band H: Mechanisms of Trastuzumab resistance in
ErbB2-driven breast cancer and newer opportunities to overcome
therapy resistance. J Carcinog. 10:282011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bezlera M, Hengstlerb JG and Ullricha A:
Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances
apoptosis of ovarian cancer cells. Mol Oncol. 6:516–529. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin A, Ozawa T, Tajiri K, Obata T, Kondo
S, Kinoshita K, Kadowaki S, Takahashi K, Sugiyama T, Kishi H and
Muraguchi A: A rapid and efficient single-cell manipulation method
for screening antigen-specific antibody-secreting cells from human
peripheral blood. Nat Med. 15:1088–1092. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ozawa T, Piao X, Kobayashi E, Zhou Y,
Sakurai H, Andoh T, Jin A, Kishi H and Muraguchi A: A novel rabbit
immunospot array assay on a chip allows for the rapid generation of
rabbit monoclonal antibodies with high affinity. PLoS One.
7:e523832012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sakurai H, Miyoshi H, Toriumi W and Sugita
T: Functional interactions of transforming growth factor
beta-activated kinase 1 with IkappaB kinases to stimulate NF-kappaB
activation. J Biol Chem. 274:10641–10648. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sakurai H, Suzuki S, Kawasaki N, Nakano H,
Okazaki T, Chino A, Doi T and Saiki I: Tumor necrosis
factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine
536 is mediated through the TRAF2, TRAF5, and TAK1 signaling
pathway. J Biol Chem. 278:36916–36923. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Refaat A, Aminullah, Zhou Y, Kawanishi M,
Tomaru R, Abdelhamed S, Shin MS, Koizumi K, Yokoyama S, Saiki I and
Sakurai H: Role of tyrosine kinase-independent phosphorylation of
EGFR with activating mutation in cisplatin-treated lung cancer
cells. Biochem Biophys Res Commun. 458:856–861. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cunningham D, Allum WH, Stenning SP,
Thompson JN, Van de Velde CJ, Nicolson M, Scarffe JH, Lofts FJ,
Falk SJ, Iveson TJ, et al: Perioperative chemotherapy versus
surgery alone for resectable gastroesophageal cancer. N Engl J Med.
355:11–20. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishimura M, Shin MS, Singhirunnusorn P,
Suzuki S, Kawanishi M, Koizumi K, Saiki I and Sakurai H:
TAK1-mediated serine/threonine phosphorylation of epidermal growth
factor receptor via p38/extracellular signal-regulated kinase:
NF-{kappa}B-independent survival pathways in tumor necrosis factor
alpha signaling. Mol Cell Biol. 29:5529–5539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim KK, Han A, Yano N, Ribeiro JR, Lokich
E, Singh RK and Moore RG: Tetrathiomolybdate mediates
cisplatin-induced p38 signaling and EGFR degradation and enhances
response to cisplatin therapy in gynecologic cancers. Sci Rep.
5:159112015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Frey MR, Dise RS, Edelblum KL and Polk DB:
p38 kinase regulates epidermal growth factor receptor
downregulation and cellular migration. EMBO J. 25:5683–5692. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hornbeck PV, Kornhauser JM, Tkachev S,
Zhang B, Skrzypek E, Murray B, Latham V and Sullivan M:
PhosphoSitePlus: A comprehensive resource for investigating the
structure and function of experimentally determined
post-translational modifications in man and mouse. Nucleic Acids
Res. 40(Database Issue): D261–D270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahsan A, Hiniker SM, Ramanand SG, Nyati S,
Hegde A, Helman A, Menawat R, Bhojani MS, Lawrence TS and Nyati MK:
Role of epidermal growth factor receptor degradation in
cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res.
70:2862–2869. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Benhar M, Engelberg D and Levitzki A:
Cisplatin-induced activation of the EGF receptor. Oncogene.
21:8723–8731. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Winograd-Katz SE and Levitzki A: Cisplatin
induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF
receptor. Oncogene. 25:7381–7390. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boone JJ, Bhosle J, Tilby MJ, Hartley JA
and Hochhauser D: Involvement of the HER2 pathway in repair of DNA
damage produced by chemotherapeutic agents. Mol Cancer Ther.
8:3015–3023. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Campiglio M, Somenzi G, Olgiati C, Beretta
G, Balsari A, Zaffaroni N, Valagussa P and Ménard S: Role of
proliferation in HER2 status predicted response to doxorubicin. Int
J Cancer. 105:568–573. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li X, Lu Y, Liang K, Liu B and Fan Z:
Differential responses to doxorubicin-induced phosphorylation and
activation of Akt in human breast cancer cells. Breast Cancer Res.
7:R589–R597. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
De U, Chun P, Choi WS, Lee BM, Kim ND,
Moon HR, Jung JH and Kim HS: A novel anthracene derivative, MHY412,
induces apoptosis in doxorubicin-resistant MCF-7/Adr human breast
cancer cells through cell cycle arrest and downregulation of
P-glycoprotein expression. Int J Oncol. 44:167–176. 2014.
View Article : Google Scholar : PubMed/NCBI
|