Introduction
Overexpression of human epidermal growth factor
receptor 2 (HER2) occurs in approximately 20–25% of invasive ductal
breast carcinomas (BCa). It is associated with increased metastatic
potential and poor prognosis (1).
HER2 belongs to the receptor tyrosine kinases (RTK) HER family that
comprises three other members (HER1/EGFR, HER3, HER4), which
require specific ligand binding for activation. In contrast, no
ligand has been identified for HER2 yet. Overexpressed HER2 was
found to be constitutively phosphorylated in both BCa cell lines
and tumours (2). HER2 forms
homodimers or heterodimers with other ligand-activated members of
the HER family (3). HER2/HER3
heterodimer has been demonstrated as the most potent oncogenic unit
in HER2-positive BCa (4).
Herceptin (Trastuzumab) is a humanized antibody
directed against the extracellular domain of HER2 and routinely
used for the treatment of HER2-overexpressing BCa patients. The
mechanism of Herceptin-mediated cell death is complex and involves
antibody-dependent cell-mediated cytotoxicity, induction of
apoptosis, inactivation of HER2 homodimerization and abrogation of
HER2-triggered cell signalling (5–7). Clinical
data showed that some patients either originally do not respond to
Herceptin or become resistant during the treatment (8,9). There is
a growing evidence demonstrating the interaction between HER2
signalling and estrogen receptor (ER) pathway (10). PR (progesterone receptor), one of the
ER-dependent genes, together with its cognate ligand-progesterone
(PG), play a critical role in breast cancer development and
progression (11–13). A cross-talk between steroid hormones
and RTK (e.g., HER receptor)-initiated signalling has a
bidirectional nature. Steroid hormone receptors may activate either
RTKs or their downstream signalling pathways (14,15).
Balañá et al (16)
demonstrated in MPA (medroxyprogesterone acetate-synthetic
progestin)-induced mice mammary adenocarcinomas, an interaction
between progestins- and heregulin (HRG) (HER1/EGFR and HER3
ligand)-dependent signalling. Conversely, RTK-triggered pathways
are able to modulate steroid receptor's activity (17). HER2 overexpression has been linked
with resistance to endocrine therapies both in vitro and
in vivo (10). Consistently,
there are studies showing that ER activity can function as an
escape pathway for ER+/HER2+ cells exposed to anti-HER2 treatment
(18). The role of PG/PR in the
process of resistance to anti-HER2 therapies remains elusive.
Taking into consideration reciprocal interactions between steroid
hormone receptors and HER2-mediated signalling, we hypothesised
that PG may affect anti-proliferative effect of Herceptin.
Herein we showed for the first time that PG may
attenuate the efficacy of HER2-targeted anticancer compounds. We
demonstrated that PG impaired Herceptin-mediated anti-proliferative
action in HER2-overexpressing cell lines and led to activation of
HER2/HER3-triggered signalling. Moreover, PG reversed
Herceptin-induced cell cycle arrest in G0/G1
cell cycle phase with concomitant upregulation of cyclin-dependent
kinase 2 (CDK2) and downregulation of CDK inhibitor
p27Kip1. These findings indicate complexity of the
mechanism responsible for resistance to Herceptin and suggest that
targeting of multiple signalling pathways may result in better
therapeutic effects.
Materials and methods
Cell lines, antibodies, reagents
BT474 (cat. no. HTB-20™) and MCF7 (cat. no. HTB-22™)
cell lines were obtained from ATCC, cells from passages 86 through
106 and 71 through 91, respectively, were used in these
investigations. BT474 cells were maintained in RPMI-1640
supplemented with 5 µg/ml insulin, whereas MCF7 cells were grown in
DMEM. Media contained 10% of FBS and penicillin/streptomycin (100
U/ml/100 µg/ml). All cell culture reagents were purchased from
Sigma-Aldrich (St. Louis, MO, USA) or HyClone (Logan, UT, USA).
Cells were cultured for a maximum of 3–4 months post resuscitation
and regularly tested for mycoplasma contamination.
Mouse monoclonal antibody against β-actin (A5316,
dilution 1:1,000) was obtained from Sigma-Aldrich. All the
remaining antibodies were from Cell Signaling Technology, Inc.
(Danvers or Beverly, MA, USA): Rabbit monoclonal anti-CDK2 (no.
2546, dilution 1:1,000), rabbit monoclonal anti-HER2/ErbB2 (no.
4290, dilution 1:1,000), rabbit polyclonal anti-HER2/ErbB2-Tyr877
(no. 2241, dilution 1:1,000), rabbit polyclonal
anti-HER2/ErbB2-Tyr1221/1222 (no. 2249, dilution 1:1,000), rabbit
polyclonal anti-HER2/ErbB2-Tyr1248 (no. 2247, dilution 1:1,000),
rabbit monoclonal anti-HER3/ErbB3 (no. 4754, dilution 1:1,000),
rabbit monoclonal anti-HER3/ErbB3-Tyr1289 (no. 4791, dilution
1:1,000), rabbit polyclonal anti-heregulin (no. 2573, dilution
1:1,000) and rabbit monoclonal anti-p27Kip1 (no. 3686,
dilution 1:1,000). PG was purchased from Sigma-Aldrich. Herceptin
was obtained from Genetech.
Western blot analysis
Cells were grown in monolayer to 60–70% confluency,
scraped in cold PBS and lysed with Laemmli buffer (2X concentrated)
supplemented with: 2 mM PMSF, 10 µg/ml aprotinin, 10 µg/ml
leupeptin, 5 mM EGTA, 1 mM EDTA, 2 mM
Na4P2O7, 5 mM NaF and 5 mM
Na3VO4. Samples containing equal amounts of
protein per lane were loaded, resolved in SDS-PAGE and then
transferred onto nitrocellulose membrane. The membranes were
incubated for 1 h in 5% skimmed milk and probed overnight with
specific primary antibodies at 4°C. Secondary goat anti-rabbit
(A9169, 1:20,000) or rabbit anti-mouse (A9044, 1:10,000) antibodies
conjugated with HRP (Sigma-Aldrich) and Western Lightning Plus-ECL
(PerkinElmer, Inc., Waltham, MA, USA) were used to visualize
specific proteins.
Cell growth in three-dimensional
Matrigel
The three-dimensional cell growth assay was
performed in a Matrigel matrix (BD Biosciences, Heidelberg,
Germany) as previously described (19). Briefly, 1×103 cells were
resuspended in 40 µl of Matrigel (~2 mg of total protein/ml),
placed into 12-well tissue culture plates followed by 30 min
incubation at 37°C for Matrigel to solidify. 3D cultures were then
covered with regular medium supplemented, when appropriate, with PG
(100 nM) and/or Herceptin (10 µg/ml). Media were refreshed every 3
days. To evaluate cell growth, a mean colony diameter was measured
for, at least, 50 randomly chosen colonies after 10 days of culture
with ZEISS PrimoVert microscope and ImageJ software and the mean
colony volume was determined. Each experiment was repeated, at
least, three times.
Development of HER2 overexpression in
MCF7 cells
MCF7 cells were plated in 60 mm plates and grown in
the monolayer to approximately 50% confluency. Medium was refreshed
1 h before transfection. Cells were transfected with 1 µg of
pBABEpuro-ERBB2 plasmid (no. 40978; Addgene, Inc., Cambridge, MA,
USA) (20) containing full-length
HER2 cDNA coding region) in serum free DMEM applying TurboFect
reagent, according to the manufacturer's instructions (Thermo
Fisher Scientific, Waltham, MA, USA). Selection of MCF7 stably
expressing ERBB2 was carried out in 2 µg/µl puromycin (Gibco, Grand
Island, NY, USA).
Flow cytometry
Cells were grown in 12-well plates in the monolayer
up to 50% of confluency and serum starved overnight. Then cells
were treated with PG (100 nM) and/or Herceptin (10 µg/ml), 24 h
after stimulation, cells were trypsinized, washed twice with
ice-cold PBS, fixed in 70% ethanol at −20°C for 15 min, resuspended
in RNaseA 1 mg/ml (EURX Ltd. Gdansk, Poland) and stained with
propidium iodide (2,5 µg/ml). Cell cycle was analysed with BD LSR
II flow cytometer (BD Biosciences).
Stimulation with growth factors,
treatment with Herceptin, signalling analyses
For analysis of growth factors-triggered signalling,
cells were serum-starved overnight before growth factors were
added. Cells were stimulated with PG (100 nM), Herceptin (10 µg/ml)
for indicated periods of time.
Statistical analysis
Data are expressed as means ± SD from at least three
independent experiments. Comparative data were analysed with the
unpaired Student's t-test using the STATISTICA software (v.10;
StatSoft, Inc., Tulsa, OK, USA). Two-sided P<0.05 was considered
to indicate a statistically significant difference.
Results
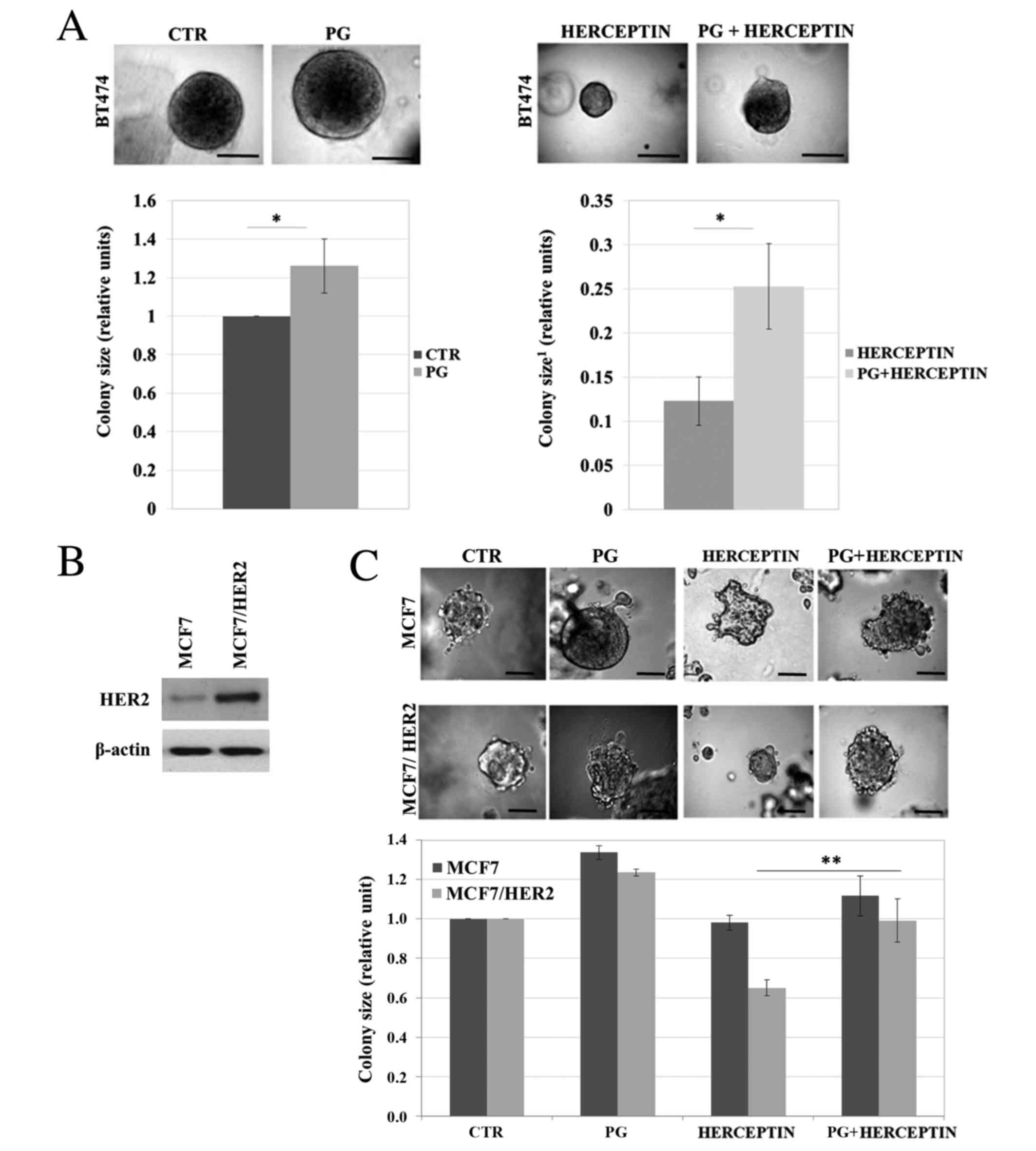
PG impairs Herceptin effect on
HER2-overexpressing cells growth
To investigate the potential impact of PG on
HER2-overexpressing cells response to Herceptin treatment we
evaluated BT474 BCa cells (PR+, HER2++) growth in three-dimensional
Matrigel. We found a modest (~28%) PG-triggered stimulation of cell
proliferation (reflected in colony size) (Fig. 1A, left panel). As expected Herceptin
significantly inhibited colonies growth (~88%, P<0,05).
Importantly, Herceptin-mediated inhibition of growth was impaired
by PG (Fig. 1A, right panel). To
confirm these results we developed a HER2-overexpressing variant
(MCF/HER2) of MCF7 BCa cells (representing luminal A subtype,
ER+/PR+/HER2low) by transient
transfection with plasmid coding erbB2 gene (Fig. 1B). Growth analysis in
three-dimensional Matrigel revealed that overexpression of HER2
resulted in sensitization of MCF7 cells to Herceptin (Fig. 1C). Although PG had modest growth
stimulatory effect on both MCF7 and MCF7/HER2 cells, it clearly
exerted a significant protective effect against Herceptin treatment
(*P<0,05).
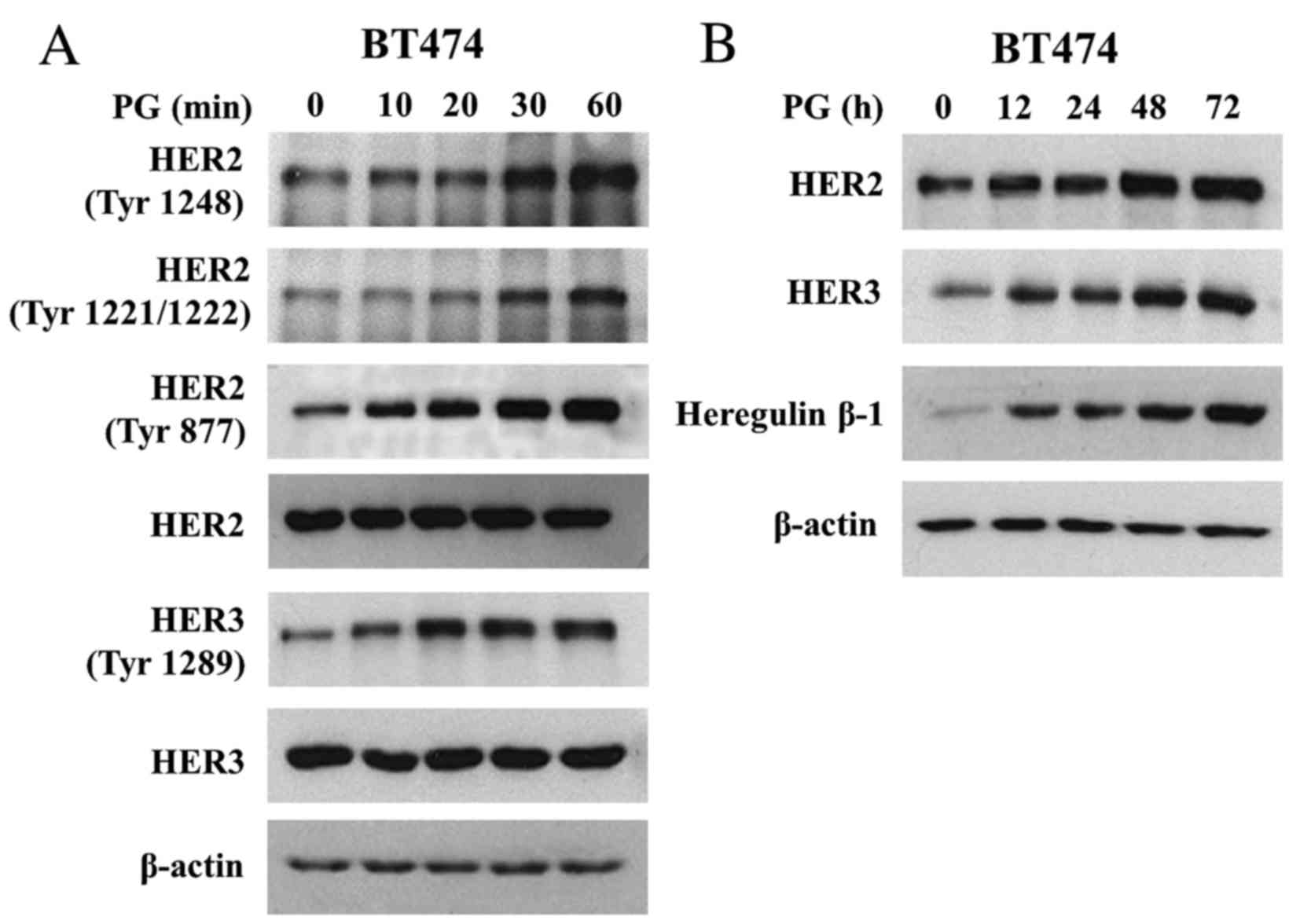
PG induces both activation and
expression of HER2/HER3 signalling
To analyse mechanisms of PG action on HER2 and HER3
function we treated BT474 cells with PG (up to 60 min) and analysed
HER2/HER3 activation. It was observed that PG triggered rapid
phosphorylation of Tyr1221/1222, Tyr1248 and Tyr877 of HER2
(Fig. 2A). In addition, PG induced
Tyr1289 HER3 phosphorylation. Prolonged exposure of BT474 cells to
PG (up to 72 h) showed that PG not only regulated the activation of
HER2 and HER3 but also caused a gradual increase in their
expression (Fig. 2B). In addition, PG
enhanced the expression of heregulin β-1, a HER3 ligand, the
binding of which is known to promote HER2/HER3 heterodimerization
(21). Heregulin β-1 reached the peak
of expression after 48 h of exposure to PG (Fig. 2B).
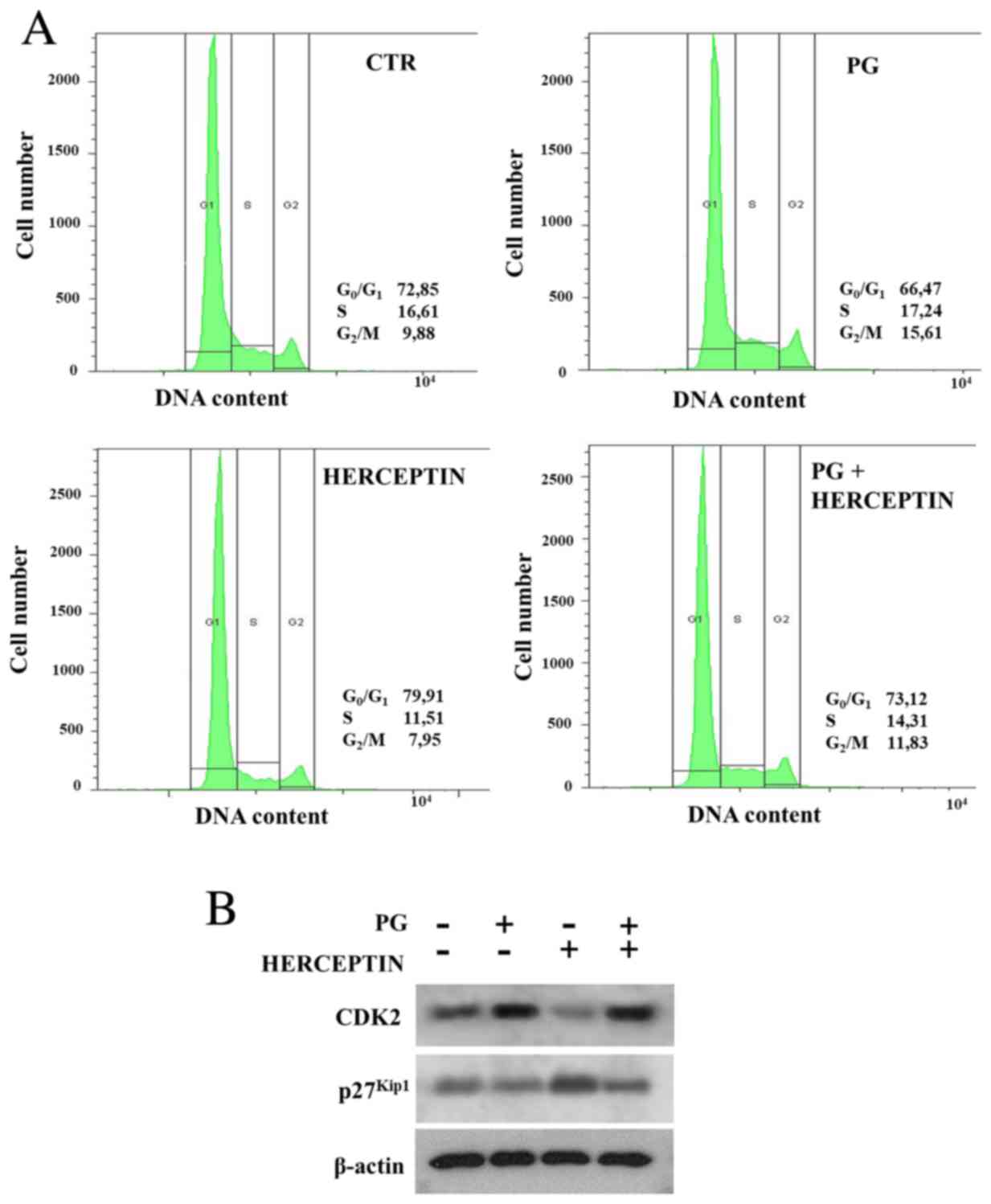
PG reverses Herceptin-induced cell
cycle arrest
To further assess the impact of PG on cell response
to Herceptin, cell cycle was analysed. Herceptin promoted the
accumulation of cells in G0/G1 phase
(Fig. 3A; Table I), which was reverted by the PG
treatment (observed as a shift towards S and G2/M
phase). Analysis of PG-mediated mechanism of cell transition
through G1 to S demonstrated that PG attenuated both
Herceptin-induced upregulation of p27Kip1 and
downregulation of CDK2 (Fig. 3B).
These findings indicate that Herceptin-triggered cell cycle arrest
at G1 phase, which is most likely mediated by
p27Kip1, is abrogated by PG.
 | Table I.Effect of PG and/or Herceptin
treatment on cell cycle. |
Table I.
Effect of PG and/or Herceptin
treatment on cell cycle.
| BT474 | % of cells in
G0-G1 |
|---|
| CTR | 72,85±2,31 |
| PG | 66,47±3,80 |
| HERCEPTIN | 79,91±1,63 |
| PG+HERCEPTIN | 73,12±2,51 |
Discussion
HER2 gene amplification and protein overexpression
have been found to be an adverse prognostic factor in invasive
ductal breast cancer. Herceptin, a monoclonal antibody directed
against domain IV of HER2, was approved in 1998 for the treatment
of HER2-amplified cancers (8,9). Despite the undisputed benefits of
Herceptin-based therapy, clinical data show that the development of
resistance to the drug remains an unsolved problem. Herceptin
anticancer mechanism is complex and not fully elucidated. It has
been well documented that resistance to Herceptin arises from the
activation of alternative pathways, including ER-dependent
signalling, which becomes the dominant driver of cell proliferation
and survival (22–25). Wang et al (18) demonstrated that in ER+/HER2+ tumor
cells, increased expression of ER as well as its downstream target
Bcl2, was associated with resistance to anti-HER2 therapy. ER was
proved to enhance PR expression (26)
and physically interact with PR, which promoted breast cancer cells
proliferation (27). As ER and PR
share similar signalling pathways (28), we hypothesized that PR activity may
affect Herceptin anti-proliferative action. Herein, we demonstrated
for the first time that Herceptin-mediated growth inhibition was
significantly impaired by PG. We observed that a short stimulation
with PG (up to 60 min) led to HER2/HER3 activation in BT474 cells.
On the other hand, prolonged stimulation (up to 72 h) induced not
only the expression of both receptors (i.e., HER2 and HER3) but
also that of heregulin, a ligand for HER3. Our results are in
accordance with the data presenting upregulation of heregulin in
MPA (synthetic PG)-induced mammary tumours (16) and its impact on cell proliferation.
Taken together, it can be speculated that PG action towards the
anti-proliferative effect of Herceptin may involve PG-promoted
increase of expression of heregulin, which, by binding to HER3,
induces HER2/HER3 heterodimerization and subsequent activation of
downstream signals. Our data demonstrated that PG also abolished
Herceptin-mediated cell-cycle arrest in G0-G1
phase. PG-promoted cell shift towards S and G2/M phase
was observed with a concomitant upregulation of CDK2 and
downregulation of p27Kip1. There seems to be a
reciprocal regulation of PR-CDK2 activities, as CDK2 was shown to
phosphorylate PR (8 out of 14 PR phosphorylation sites are known to
be targeted by CDK2) (29,30) and increase PR transcriptional function
(31).
Steroid hormones play a critical role in breast
carcinogenesis (32–34). Although the relationship between the
level of circulating estrogen/PG and breast cancer risk has been
extensively studied, the effect of steroids on the efficacy of
anti-HER2 BCa therapy has not been greatly explored. Our findings
provide support for the hypothesis that, in steroid hormone
receptors-positive BCa, acquisition of resistance to Herceptin
might be triggered by PG. This finding seems to be important
especially for premenopausal women, who are exposed to periodically
increased level of PG. Our results reveal a new level of complexity
of resistance to Herceptin mechanisms and suggest that combined
therapy involving Herceptin and PR antagonists may have potential
benefits for HER2-overexpressing BCa in premenopausal patients.
Acknowledgements
pBABEpuro-ERBB2 (Addgene plasmid no. 40978) was a
gift from Matthew Meyerson (Dana-Farber Cancer Institute). The
research leading to these results has received funding from
National Science Centre-UMO-2012/06/M/NZ3/00023 (to RS), the
European Union Seventh Framework Programme (grant agreement no
316094-MOBI4Health project) and Polish Ministry of Science and
Higher Education from science funds 2013–2016 within co-funded
projects. Kamila Kitowska has been supported by the MOBI4health
project.
References
|
1
|
Gutierrez C and Schiff R: HER2: Biology,
detection, and clinical implications. Arch Pathol Lab Med.
135:55–62. 2011.PubMed/NCBI
|
|
2
|
DiGiovanna MP, Chu P, Davison TL, Howe CL,
Carter D, Claus EB and Stern DF: Active signaling by HER-2/neu in a
subpopulation of HER-2/neu-overexpressing ductal carcinoma in situ:
Clinicopathological correlates. Cancer Res. 62:6667–6673.
2002.PubMed/NCBI
|
|
3
|
Graus-Porta D, Beerli RR, Daly JM and
Hynes NE: ErbB-2, the preferred heterodimerization partner of all
ErbB receptors, is a mediator of lateral signaling. EMBO J.
16:1647–1655. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holbro T, Beerli RR, Maurer F, Koziczak M,
Barbas CF III and Hynes NE: The ErbB2/ErbB3 heterodimer functions
as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor
cell proliferation. Proc Natl Acad Sci USA. 100:pp. 8933–8938.
2003; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sliwkowski MX, Lofgren JA, Lewis GD,
Hotaling TE, Fendly BM and Fox JA: Nonclinical studies addressing
the mechanism of action of trastuzumab (Herceptin). Semin Oncol.
26(4 Suppl 12): S60–S70. 1999.
|
|
6
|
Baselga J, Albanell J, Molina MA and
Arribas J: Mechanism of action of trastuzumab and scientific
update. Semin Oncol. 28(5 Suppl 16): S4–S11. 2001. View Article : Google Scholar
|
|
7
|
Luque-Cabal M, García-Teijido P,
Fernández-Pérez Y, Sánchez-Lorenzo L and Palacio-Vázquez I:
Mechanisms behind the resistance to trastuzumab in HER2-amplified
breast cancer and strategies to overcome it. Clin Med Insights
Oncol. 10 Suppl 1:S21–S30. 2016.
|
|
8
|
Vogel CL, Cobleigh MA, Tripathy D, Gutheil
JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF,
Burchmore M, et al: Efficacy and safety of trastuzumab as a single
agent in first-line treatment of HER2-overexpressing metastatic
breast cancer. J Clin Oncol. 20:719–726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cobleigh MA, Vogel CL, Tripathy D, Robert
NJ, Scholl S, Fehrenbacher L, Wolter JM, Paton V, Shak S, Lieberman
G and Slamon DJ: Multinational study of the efficacy and safety of
humanized anti-HER2 monoclonal antibody in women who have
HER2-overexpressing metastatic breast cancer that has progressed
after chemotherapy for metastatic disease. J Clin Oncol.
17:2639–2648. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shou J, Massarweh S, Osborne CK, Wakeling
AE, Ali S, Weiss H and Schiff R: Mechanisms of tamoxifen
resistance: Increased estrogen receptor-HER2/neu cross-talk in
ER/HER2-positive breast cancer. J Natl Cancer Inst. 96:926–935.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schairer C, Lubin J, Troisi R, Sturgeon S,
Brinton L and Hoover R: Menopausal estrogen and estrogen-progestin
replacement therapy and breast cancer risk. JAMA. 283:485–491.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soyal S, Ismail PM, Li J, Mulac-Jericevic
B, Conneely OM and Lydon JP: Progesterone's role in mammary gland
development and tumorigenesis as disclosed by experimental mouse
genetics. Breast Cancer Res. 4:191–196. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Graham JD and Clarke CL: Physiological
action of progesterone in target tissues. Endocr Rev. 18:502–519.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Béguelin W, Díaz Flaqué MC, Proietti CJ,
Cayrol F, Rivas MA, Tkach M, Rosemblit C, Tocci JM, Charreau EH,
Schillaci R and Elizalde PV: Progesterone receptor induces ErbB-2
nuclear translocation to promote breast cancer growth via a novel
transcriptional effect: ErbB-2 function as a coactivator of Stat3.
Mol Cell Biol. 30:5456–5472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stoica GE, Franke TF, Wellstein A,
Czubayko F, List HJ, Reiter R, Morgan E, Martin MB and Stoica A:
Estradiol rapidly activates Akt via the ErbB2 signaling pathway.
Mol Endocrinol. 17:818–830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Balañá ME, Lupu R, Labriola L, Charreau EH
and Elizalde PV: Interactions between progestins and heregulin
(HRG) signaling pathways: HRG acts as mediator of progestins
proliferative effects in mouse mammary adenocarcinomas. Oncogene.
18:6370–6379. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Labriola L, Salatino M, Proietti CJ, Pecci
A, Coso OA, Kornblihtt AR, Charreau EH and Elizalde PV: Heregulin
induces transcriptional activation of the progesterone receptor by
a mechanism that requires functional ErbB-2 and mitogen-activated
protein kinase activation in breast cancer cells. Mol Cell Biol.
23:1095–1111. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang YC, Morrison G, Gillihan R, Guo J,
Ward RM, Fu X, Botero MF, Healy NA, Hilsenbeck SG, Phillips GL, et
al: Different mechanisms for resistance to trastuzumab versus
lapatinib in HER2-positive breast cancers-role of estrogen receptor
and HER2 reactivation. Breast Cancer Res. 13:R1212011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sadej R, Romanska H, Baldwin G,
Gkirtzimanaki K, Novitskaya V, Filer AD, Krcova Z, Kusinska R,
Ehrmann J, Buckley CD, et al: CD151 regulates tumorigenesis by
modulating the communication between tumor cells and endothelium.
Mol Cancer Res. 7:787–798. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Greulich H, Kaplan B, Mertins P, Chen TH,
Tanaka KE, Yun CH, Zhang X, Lee SH, Cho J, Ambrogio L, et al:
Functional analysis of receptor tyrosine kinase mutations in lung
cancer identifies oncogenic extracellular domain mutations of
ERBB2. Proc Natl Acad Sci USA. 109:pp. 14476–14481. 2012;
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weiss FU, Wallasch C, Campiglio M, Issing
W and Ullrich A: Distinct characteristics of heregulin signals
mediated by HER3 or HER4. J Cell Physiol. 173:187–195. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagata Y, Lan KH, Zhou X, Tan M, Esteva
FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al: PTEN
activation contributes to tumor inhibition by trastuzumab, and loss
of PTEN predicts trastuzumab resistance in patients. Cancer Cell.
6:117–127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berns K, Horlings HM, Hennessy BT,
Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM,
Stemke-Hale K, Hauptmann M, et al: A functional genetic approach
identifies the PI3K pathway as a major determinant of trastuzumab
resistance in breast cancer. Cancer Cell. 12:395–402. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nahta R, Yuan LX, Zhang B, Kobayashi R and
Esteva FJ: Insulin-like growth factor-I receptor/human epidermal
growth factor receptor 2 heterodimerization contributes to
trastuzumab resistance of breast cancer cells. Cancer Res.
65:11118–11128. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Scaltriti M, Eichhorn PJ, Cortés J,
Prudkin L, Aura C, Jiménez J, Chandarlapaty S, Serra V, Prat A,
Ibrahim YH, et al: Cyclin E amplification/overexpression is a
mechanism of trastuzumab resistance in HER2+ breast cancer
patients. Proc Natl Acad Sci USA. 108:pp. 3761–3766. 2011;
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Horwitz KB and McGuire WL: Estrogen
control of progesterone receptor in human breast cancer.
Correlation with nuclear processing of estrogen receptor. J Biol
Chem. 253:2223–2228. 1978.PubMed/NCBI
|
|
27
|
Daniel AR, Gaviglio AL, Knutson TP,
Ostrander JH, D'Assoro AB, Ravindranathan P, Peng Y, Raj GV, Yee D
and Lange CA: Progesterone receptor-B enhances estrogen
responsiveness of breast cancer cells via scaffolding PELP1- and
estrogen receptor-containing transcription complexes. Oncogene.
34:506–515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ballaré C, Uhrig M, Bechtold T, Sancho E,
Di Domenico M, Migliaccio A, Auricchio F and Beato M: Two domains
of the progesterone receptor interact with the estrogen receptor
and are required for progesterone activation of the c-Src/Erk
pathway in mammalian cells. Mol Cell Biol. 23:1994–2008. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Knotts TA, Orkiszewski RS, Cook RG,
Edwards DP and Weigel NL: Identification of a phosphorylation site
in the hinge region of the human progesterone receptor and
additional amino-terminal phosphorylation sites. J Biol Chem.
276:8475–8483. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Beck CA, Clement JP IV,
Prendergast P, Yip TT, Hutchens TW, Edwards DP and Weigel NL:
Phosphorylation of human progesterone receptor by cyclin-dependent
kinase 2 on three sites that are authentic basal phosphorylation
sites in vivo. Mol Endocrinol. 11:823–832. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pierson-Mullany LK and Lange CA:
Phosphorylation of progesterone receptor serine 400 mediates
ligand-independent transcriptional activity in response to
activation of cyclin-dependent protein kinase 2. Mol Cell Biol.
24:10542–10557. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bernstein L: Epidemiology of
endocrine-related risk factors for breast cancer. J Mammary Gland
Biol Neoplasia. 7:3–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Campagnoli C, Clavel-Chapelon F, Kaaks R,
Peris C and Berrino F: Progestins and progesterone in hormone
replacement therapy and the risk of breast cancer. J Steroid
Biochem Mol Biol. 96:95–108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou W and Slingerland JM: Links between
oestrogen receptor activation and proteolysis: Relevance to
hormone-regulated cancer therapy. Nat Rev Cancer. 14:26–38. 2014.
View Article : Google Scholar : PubMed/NCBI
|