Introduction
Statins are a class of drug used for the treatment
of hypercholesterolaemia. They reduce plasma cholesterol by
competitively inhibiting 3-hydroxy-3-methyl-glutaryl coenzyme A
reductase (HMGCR), an enzyme involved in the mevalonate pathway
that is responsible for cholesterol synthesis. Inhibition of HMGCR
also leads to a reduction in other products of the mevalonate
pathway including farnesyl diphosphate (FPP) and geranylgeranyl
diphosphate (GGPP). These isoprenoids are used in the
post-translational modification and membrane localization of
several important GTPases, such as Ras, Rho, Rac, Rab and Cdc42,
many of which have been identified as oncogenes. Consequently,
statins reduce the recruitment of small GTPases to the cell
membrane. We have previously reported the anti-cancer activity of
statins in ovarian cancer cells, showing that both high
concentrations and continuous exposure to statins were required to
induce cell death (1). More recently,
we showed that pitavastatin, a hydrophobic statin with a relatively
long half-life, demonstrated potent anti-cancer activity,
inhibiting the growth and promoting apoptosis in ovarian cancer
cell lines and causing regression of tumour xenografts (2). Pitavastatin has been evaluated in phase
II clinical trials for the treatment of hypercholesterolaemia at
doses up to 64 mg daily, with dose-limiting toxicities observed
after 2–4 weeks reversible within 2 weeks of discontinuing therapy
(3). The concentrations of
pitavastatin required to cause cell death [0.2–7.6 µM depending on
cell line (2)] are similar to the
expected plasma concentration in vivo following
administration of 64 mg pitavastatin [Cmax, ~3 µM,
assuming linear pharmacokinetics and using data from (4–6)]. Despite
this, the anti-cancer activity of pitavastatin can be suppressed by
exposure to geranylgeraniol, an isoprenoid found in many common
foodstuffs, thereby raising the possibility that dietary
isoprenoids may impede the effectiveness of statins in clinical
trials (2). One possible solution is
to control patients' diet in oncology clinical trials. However, the
potential for pitavastatin to cause myopathy, particularly at high
doses, makes it desirable to identify drugs which could be used in
combination with pitavastatin to reduce the dose required and
potentially reduce the incidence of adverse drug effects.
The BH3 mimetics ABT-737 and obatoclax have been
employed to overcome the pro-survival effects of anti-apoptotic
proteins by competitively binding to and inhibiting the Bcl-2
family of proteins (7). We have
previously shown that ABT-737 and the orally bioavailable analogue,
ABT-263, can enhance the cell death induced by carboplatin or
paclitaxel in ovarian cancer cells (8,9). A closely
related selective Bcl-2 inhibitor, venetoclax, has been approved
for the treatment of chronic lymphocytic leukemia. However, we have
shown that inhibitors of Bcl-xL, a member of the Bcl-2
family, are likely to be needed for the treatment of ovarian cancer
(10). These observations suggest
that BH3 mimetics which inhibit Bcl-xL may be useful in
combination with statins, which have also be shown to induce
apoptotic cell death (1,2,11,12).
The phosphatidylinositol 3-kinase (PI3K) pathway
plays an important role in cell survival, proliferation, migration
and metabolism, and has been recently reported to be frequently
activated in advanced epithelial ovarian cancers (13,14).
Pictilisib is an orally active PI3K inhibitor which is more than
100 times more potent against class I PI3K compared to class II,
III and IV family members (15).
Statins have also been shown to interfere with PI3K signalling by
inhibiting NFκB, and consequently increasing transcription of PTEN
and reducing Akt phosphorylation (11). This suggests that pitavastatin in
combination with PI3K inhibitors could synergistically inhibit PI3K
signalling, leading to an increase in cell death.
To evaluate whether ABT-737, obatoclax or pictilisib
could potentiate the activity of pitavastatin, we evaluated the
anti-cancer activity of pitavastatin alone and in combination with
these drugs. We found that ABT-737 and pictilisib combined
additively with pitavastatin in cell growth assays, and potentiated
the cell death induced by pitavastatin, in several ovarian cancer
cell lines.
Materials and methods
Cell culture
Human ovarian cancer cells (A2780, Ovcar-3, Ovcar-8
and Igrov-1; American Type Culture Collection, Manassas, VA, USA)
were cultured in Roswell Park Memorial Institute (RPMI 1640; Lonza
Group, Ltd., Basel, Switzerland) supplemented with 10% fetal bovine
serum (FBS), 50 U/ml penicillin/streptomycin and 2 mM glutamine. In
addition, Ovcar-3 cells were supplemented with 0.11 g/l sodium
pyruvate and 0.01 mg/ml insulin. Cells were incubated at 37°C and
in a humidified 5% CO2 atmosphere.
Cell growth/survival assays
ABT-737 (Abbott Laboratories, Chicago, IL, USA) and
obatoclax (Active Biochem, Maplewood, NJ, USA) were prepared as 10
and 5 mM solutions respectively in dimethyl sulfoxide (DMSO).
Pitavastatin (Sequoia Research Products, Pangbourne, UK) and
pictilisib (LC Laboratories, Woburn, MA, USA) were prepared as 20
mM solutions in DMSO. Single-agent and combination studies were
completed as previously reported (8).
Fixed concentrations of ABT-737, which had been determined to
inhibit cell growth by 5% (A2780, 3 µM; Ovcar-3, 1 µM; Ovcar-8, 1
µM; Igrov-1, 0.6 µM), were added to 18 different concentrations of
pitavastatin in cell growth assays (8). Obatoclax or pictilisib and pitavastatin
were combined at a fixed ratio of their IC50 values as
determined from single-agent studies. Combination indicies
(16) were calculated to measure the
combined effect of pitavastatin with ABT-737, pictilisib or
obatoclax, and quoted at a fraction affected of 0.5 or 0.75, which
is the concentration of the drug combination that inhibited 50 or
75% of cell growth respectively.
Cell death assays
Ovcar-3 and Igrov-1 cells were incubated with DMSO,
pitavastatin (12 and 6 µM respectively), ABT-737 (1 and 0.6 µM),
obatoclax (2 and 3 µM), pictilisib (2 and 0.7 µM) alone or in
combination with pitavastatin for 48 h (caspase-3/7 assay) or 72 h
(trypan blue assay). Cells were collected by centrifugation (150 g,
3 min), and resuspended in phosphate-buffered saline (PBS)
containing 0.2% trypan blue (Sigma-Aldrich, St. Louis, MO, USA).
Cells which did not exclude trypan blue were considered dead.
Alternatively, an equal volume of caspase-Glo-3/7 substrate
(Promega Corporation, Madison, WI, USA) was added directly to the
cells, and after 30 min incubation, a microplate reader was used to
measure luminescence, as per the manufacturers protocol. For
caspase-3/7 assays, a paired t-test was used to compare the
effect of the drug combination to the effect of single agent
pitavastatin. For trypan blue assays, a paired t-test was used to
compare the observed effect of the drug combination to the expected
additive effect calculated using the Bliss independence criterion
(17).
Western blotting
For western blotting, Igrov-1 cells were exposed to
pitavastatin or ABT-737 or solvent (DMSO) alone or in combination.
After 48 h, cell lysates were prepared as described (18) and total protein was quantified using
the Bicinchoninic acid (BCA) protein assay kit (Sigma-Aldrich).
Membranes were incubated with rabbit anti-human polyclonal PARP
antibody (1:1,000) (#9542), rabbit anti-human polyclonal Bim
antibody (1:1,000) (#2819), rabbit anti-human polyclonal
Bcl-xL antibody (1:1,000) (#2762), or rabbit anti-human
monoclonal Mcl-1 (D35A5) antibody (1:1,000) (#5453; all from Cell
Signaling Technology, Inc., Danvers, MA, USA), and mouse anti-human
monoclonal GAPDH antibody (1:5,000) (MAB374; EMD Millipore,
Billerica, MA, USA) as a loading control. Bands were quantified
using AlphaView SA (ProteinSimple, San Jose, CA, USA) and
normalized to GAPDH. The amount of cleaved PARP was expressed as a
fraction of uncleaved-PARP, and all other proteins were expressed
as a fraction of the solvent control.
Statistical analysis
Data are presented as the mean ± standard deviation.
Differences were analysed using a t-test (Microsoft Excel, 2010;
Microsoft Corporation, Redmond, WA, USA). At least three
experimental replicates were performed. P<0.05 was considered to
indicate a statistically significant difference.
Results
ABT-737, obatoclax or pictilisib
additively combine with pitavastatin in ovarian cancer cells
To investigate the activity of drug combinations
with pitavastatin, the potencies of pitavastatin, ABT-737,
obatoclax and pictilisib as single agents in cell growth assays
were first determined in a panel of four ovarian cancer cell lines.
Pitavastatin (IC50=0.26–5.8 µM), ABT-737
(IC50=2.1–4.0 µM), obatoclax (IC50=0.15–0.36
µM) and pictilisib (IC50=0.072–0.88 µM) inhibited the
growth of ovarian cancer cell lines (Fig.
1A).
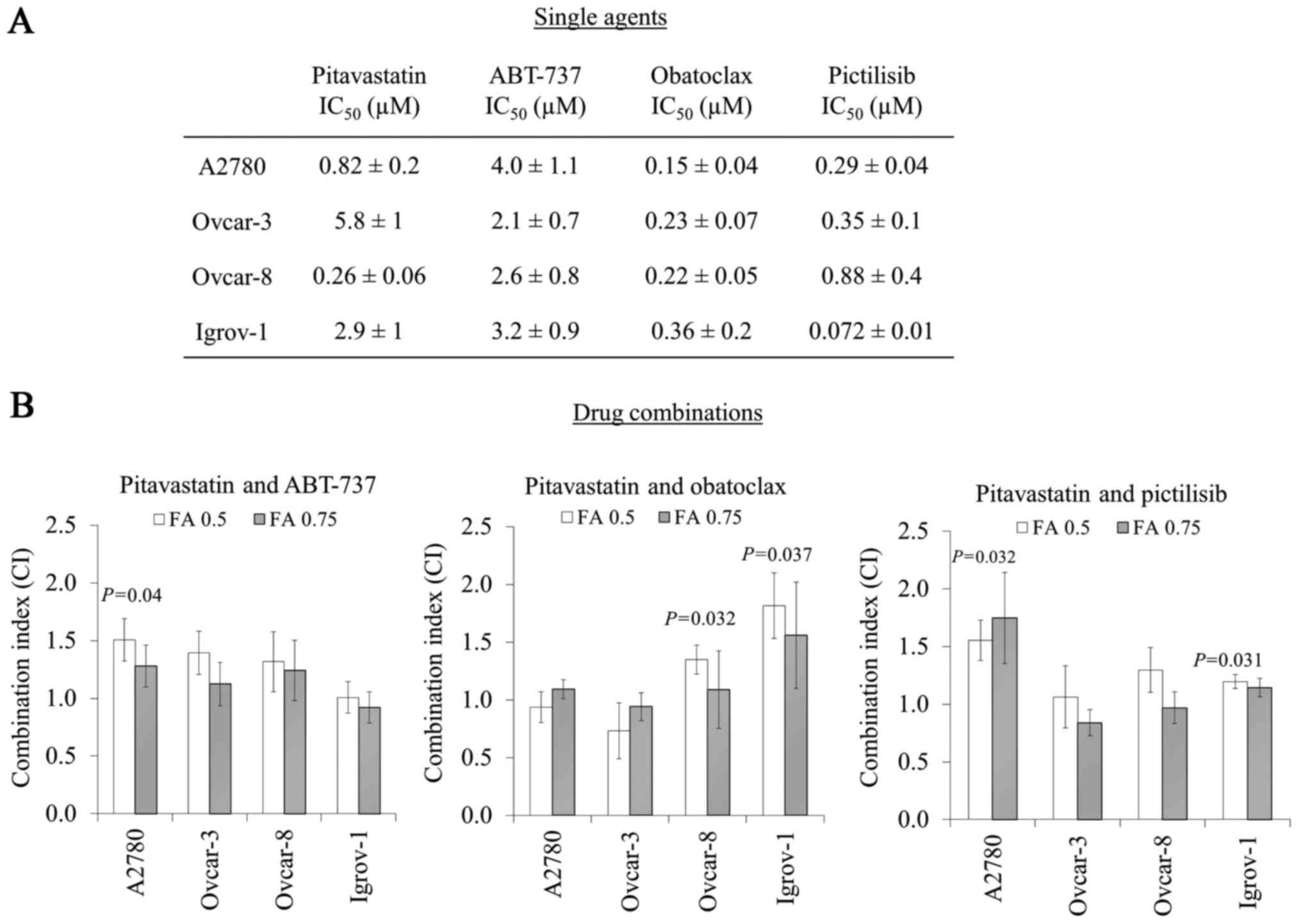 | Figure 1.Potency of pitavastatin, ABT-737,
obatoclax and pictilisib as single agents or in combination in cell
growth assays. (A) The potency of pitavastatin, ABT-737, obatoclax
and pictilisib as single agents were measured in cell
proliferation/growth assays (columns 1–4, mean ± standard
deviation; n=3–9). (B) To measure the activity of pitavastatin in
combination with ABT-737, obatoclax or pictilisib, cells were
exposed to a range of concentrations of pitavastatin and either, a
fixed concentration of ABT-737, or a range of concentrations of
either obatoclax or pictilisib, combined at a ratio of their single
agent IC50s. CIs (mean ± standard deviation; n=3–9) are
quoted at the indicated FA and differed significantly from unity
where indicated (paired t-test). IC50, half maximal
inhibitory concentration; CIs, combination indices; FA, fraction
affected. |
Concentrations of ABT-737 above 20 µM have
previously been shown to inhibit cell growth independently of
Bcl-xL (8). Thus, drug
combination experiments using ABT-737 were performed using a fixed
concentration of ABT-737 which inhibits cell growth by <10%.
When combined with pitavastatin, ABT-737 either caused additive
effects or in A2780 cells, an antagonistic interaction was
observed. Although synergy was not observed between these drugs in
any of the cell lines evaluated, an additive interaction was
obtained in Igrov-1 cells (Fig.
1B).
Obatoclax in combination with pitavastatin did not
show a significant synergistic interaction in any of the cell lines
tested (measured at fraction affected 0.5) (Fig. 1B). However, in Ovcar-8 and Igrov-1
cells, there was significant antagonism (P=0.032 and P=0.037
respectively, fraction affected, 0.5), which was reduced at higher
drug concentrations (fraction affected, 0.75; Fig. 1B). Pictilisib and pitavastatin were
additive in Ovcar-3 and Ovcar-8 cells, even at higher drug
concentrations (fraction affected, 0.75). However, significant
antagonism was observed in A2780 and Igrov-1 cells (P=0.032 and
P=0.031, fraction affected, 0.5, Fig.
1B).
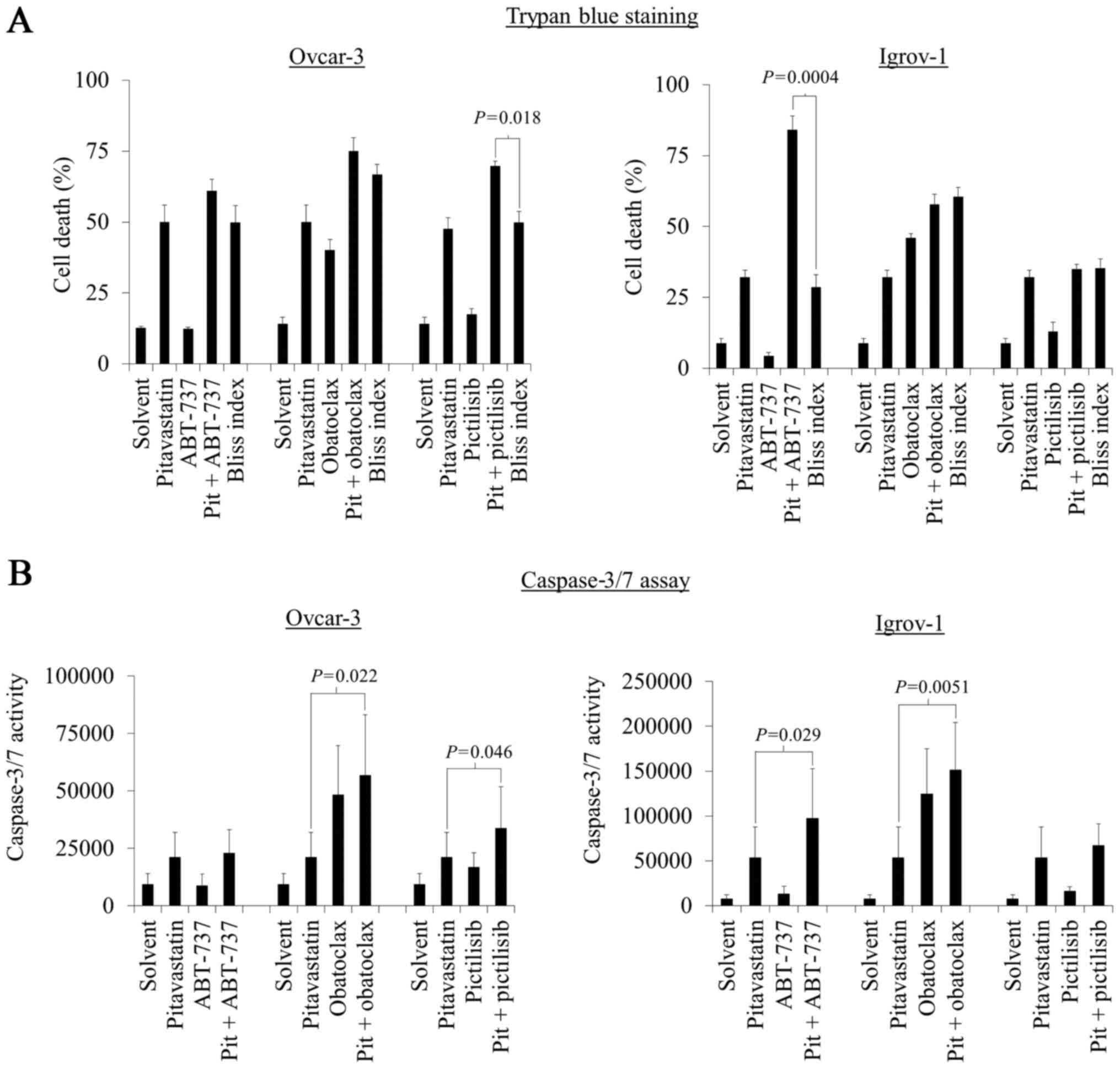
Pitavastatin in combination with
ABT-737 or pictilisib increases cell death in Igrov-1 or
Ovcar-3
The activity of the most promising of these drug
combinations was evaluated in cell death assays. Ovcar-3 or Igrov-1
cells were exposed to pitavastatin in combination with ABT-737, and
the Bliss independence criterion was used to estimate the effect of
the drugs. In Igrov-1 cells, but not Ovcar-3 cells, significantly
more cell death was observed by trypan blue staining (P=0.0004)
than would have been expected if the drugs had demonstrated an
additive effect. (Fig. 2A). However,
in Ovcar-3 cells, the combination of pitavastatin and pictilisib
resulted in significantly more cell death than would be expected
from an additive combination (P=0.018, Fig. 2A).
To confirm that the drug combinations resulted in
apoptosis, activation of caspase-3/7 was measured. Directly
reflecting the results obtained in the trypan blue assays, there
was also a significant increase in caspase-3/7 activity in Ovcar-3
and Igrov-1 cells exposed to pitavastatin and pictilisib or ABT-737
respectively compared to pitavastatin alone (P=0.046 and P=0.029),
suggesting that apoptosis contributes to the cell death caused by
these drug combinations (Fig. 2B).
Unsurprisingly, pitavastatin in combination with obatoclax resulted
in a significant increase in caspase-3/7 activity in ovarian cancer
cells compared to single agent pitavastatin (P=0.022 and P=0.0051,
Fig. 2B); obatoclax as a single agent
also significantly activated caspase-3/7 activity, suggesting that
this was not a synergistic interaction.
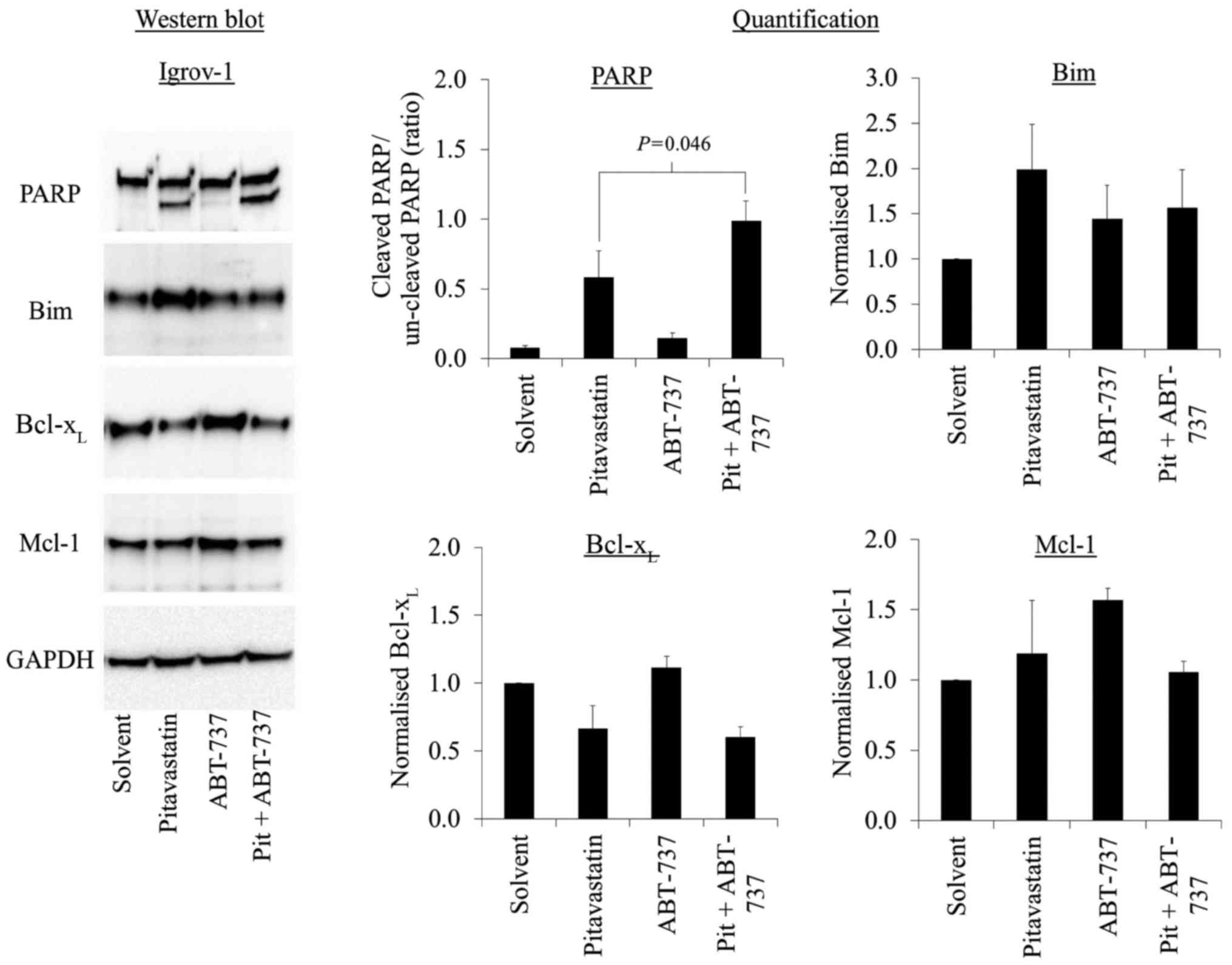
The synergy observed in the trypan blue studies was
most striking in the Igrov-1 cells exposed to ABT-737 and
pitavastatin, so further studies focused on this drug combination.
The synergistic activation of caspase-3/7 was confirmed by
measuring the cleavage of its substrate, PARP. ABT-737 on its own
caused no PARP cleavage, but when combined with pitavastatin,
ABT-737 potentiated the PARP cleavage caused by pitavastatin
(Fig. 3).
The mechanism by which ABT-737 potentiated the
apoptosis induced by pitavastatin was explored by measuring the
levels of several Bcl-2 family members. Pitavastatin, when used as
a single agent, increased the level of the pro-apoptotic protein,
Bim, and decreased the level of Bcl-xL (Fig. 3) In contrast, ABT-737, used as a
single agent, resulted in the increase in Bcl-xL and
Mcl-1 (Fig. 3). Overall,
Bcl-xL was reduced in cells exposed to the combination
of pitavastatin and ABT-737 (Fig.
3).
Discussion
We have investigated the potential for ABT-737,
obatoclax and pictisilib to potentiate the activity of
pitavastatin. This is of particular concern because we have
previously shown relatively high doses of statins are likely to be
necessary to treat patients with ovarian cancer (1). Statins have been previously evaluated in
combination with various chemotherapeutic agents including
cisplatin and doxorubicin, resulting in an additive or synergistic
reduction in ovarian cancer cell proliferation (19,20).
ABT-737 or pictilisib combined additively with pitavastatin in cell
growth assays. In shorter term cell death and apoptosis assays,
both drugs potentiated the activity of pitavastatin. This suggests
that these drugs increase the rate of apoptosis caused by
pitavastatin. We have previously observed this phenomenon with
other drug targets. Inhibition of autotaxin speeds up apoptosis
induced by carboplatin, whilst having less pronounced effects in
longer duration cell growth assays (21).
The PI3K pathway contributes to proliferative and
anti-apoptotic effects on tumour cells, and is deregulated in 45%
of high-grade serous ovarian cancers (14). Statins have also been shown to inhibit
PI3K signalling by inhibiting NF-κB, which results in an increase
in the expression of PTEN and a reduction in Akt phosphorylation
(11,22). Dual inhibition of PI3K/Akt/mTOR
signalling with a combination of pictilisib and pitavastatin
increased apoptosis in Ovcar-3 cells, whilst demonstrating
antagonism in A2780 and Igrov-1 cells. Ovcar-3, Ovcar-8, Igrov-1
and A2780 cells have PI3K/Akt pathway alterations consistent with
activation of PI3K/Akt signalling, suggesting that these cell lines
may be particularly sensitive to PI3K pathway inhibition (23,24). This
was supported by the submicromolar IC50 values obtained
for pictilisib in all cell lines tested. The lack of synergy
between pitavastatin and pictisilib in A2780 and Igrov-1 cells may
be due to PTEN deletions in these cell lines, resulting in
low or undetectable levels of PTEN protein (23). This may hyperactivate the Akt pathway
and prevent further inhibition of PI3K signalling through PTEN
modulation by statins.
The activity of ABT-737 has previously been
attributed to inhibition of the pro-apoptotic mediators, Bcl-2,
Bcl-xL or Bcl-w, of which Bcl-xL is
overexpressed in ovarian cancer (8,25). Statins
have been shown to induce apoptosis through a number of pathways,
including suppression of Akt/Erk activation (11,12),
increased phosphorylation of the p38 MAPK pathway (12), and attenuation of Mcl-1, probably
through the inhibition of NF-κB (26). The most synergy between ABT-737 and
pitavastatin was observed in Igrov-1 cells. It is noticeable that,
of a panel of ovarian cancer cell lines, these cells were the ones
in which the most pronounced synergy between ABT-737 and
carboplatin was observed (8). It is
possible that these cells are ‘primed’ for cell death (27). In ‘primed’ cells, ABT-737 prevents
Bcl-xL from sequestering pre-existing pro-apoptotic
mediators, thereby enabling apoptosis to occur more readily. In
contrast to previous reports, pitavastatin did not decrease Mcl-1
levels, but instead reduced the levels of Bcl-xL and
increased Bim. This suggests a mechanism by which the drug
combination is synergistic. Pitavastatin increases the ratio of Bim
to Bcl-xL, facilitating the release of Bim from
Bcl-xL by ABT-737, and consequently activating the
intrinsic apoptosis pathway. Additivity or mild antagonism was
observed in the other cell lines exposed to this drug combination,
and these differences could be related to the expression of
apoptosis inhibitiors. Previous research demonstrated that
expression of Bcl-xL was markedly lower in A2780 cells,
and this, together with increased Mcl-1 levels (8), previously linked to cellular resistance
to ABT-737 (28), could account for
the antagonism observed in this cell line. Mcl-1 can cause
resistance to ABT-737 because it is able to suppress apoptosis by
sequestering pro-apoptotic BH3-only proteins such as Bim, but it is
not inhibited by ABT-737. We have also previously observed
antagonism between ABT-737 and carboplatin in A2780 cells, despite
observing synergy between these drugs in other cell lines (8).
Obatoclax is a pan-Bcl-2 inhibitor which also
inhibits Mcl-1, and therefore, may overcome the resistance to
ABT-737. However, obatoclax in combination with pitavastatin was
additive at best in A2780 and Ovcar-3 cells, with antagonism
observed in Ovcar-8 and Igrov-1 cells. We have previously shown
that obatoclax has off-target effects. It accumulates in lysosomes
causing their alkalinisation (29).
This may have contributed to the antagonism observed in these and
previous combination studies (30).
Taken together, these results suggest that the combination of
obatoclax and pitavastatin may be of limited value in a clinical
setting.
Taken together, pictilisib or ABT-737 in combination
with pitavastatin could be used in a subset of ovarian tumours in a
clinical setting. This has several implications. Although the
synergy between these drugs may reduce the dose of statin that is
necessary to treat patients, consideration should be given to
identifying which patient groups may benefit most from these
combination treatments. ‘BH3 profiling’ is one method that can be
used to determine the sensitivity of cancer cells to Bcl-2
inhibitors by determining the effects of the drug or related
peptide on mitochondria isolated from the cancer cells (31). For the PI3K inhibitor and pitavastatin
combination, measurement of PTEN expression and other pathway
markers may help to select which patients may respond to this
combination.
In conclusion, these promising results demonstrate
that combinations of pitavastatin and BH3 mimetics or inhibitors of
the PI3K pathway warrant further studies as potential therapeutics
for ovarian cancer. There is a legitimate concern, however, that
the inclusion of pictisilib or a BH3 mimetic in combination with
pitavastatin will result in additional toxicity. Whether the
therapeutic window for the use of the proposed combinations is an
improvement over that with pitavastatin alone is something that can
best be addressed in clinical trials.
Acknowledgements
The present study was funded internally by Keele
University (Stoke-on-Trent, UK).
References
|
1
|
Robinson E, Nandi M, Wilkinson LL,
Arrowsmith DM, Curtis AD and Richardson A: Preclinical evaluation
of statins as a treatment for ovarian cancer. Gynecol Oncol.
129:417–424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Wolf E, Abdullah MI, Jones S, Menezes
K, Moss DM, Drijfhout FP, Hart SR, Hoskins C, Stronach EA and
Richardson A: Dietary geranylgeraniol can limit the activity of
pitavastatin as a potential treatment for drug-resistant ovarian
cancer. Sci Rep. 7:54102017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mukhtar RY, Reid J and Reckless JP:
Pitavastatin. Int J Clin Pract. 59:239–252. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ando H, Tsuruoka S, Yanagihara H, Sugimoto
K, Miyata M, Yamazoe Y, Takamura T, Kaneko S and Fujimura A:
Effects of grapefruit juice on the pharmacokinetics of pitavastatin
and atorvastatin. Br J Clin Pharmacol. 60:494–497. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chung JY, Cho JY, Yu KS, Kim JR, Oh DS,
Jung HR, Lim KS, Moon KH, Shin SG and Jang IJ: Effect of OATP1B1
(SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin
in healthy volunteers. Clin Pharmacol Ther. 78:342–350. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hui CK, Cheung BM and Lau GK:
Pharmacokinetics of pitavastatin in subjects with child-pugh A and
B cirrhosis. Br J Clin Pharmacol. 59:291–297. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Labi V, Grespi F, Baumgartner F and
Villunger A: Targeting the Bcl-2-regulated apoptosis pathway by BH3
mimetics: A breakthrough in anticancer therapy? Cell Death Differ.
15:977–987. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Witham J, Valenti MR, De-Haven-Brandon AK,
Vidot S, Eccles SA, Kaye SB and Richardson A: The Bcl-2/Bcl-XL
family inhibitor ABT-737 sensitizes ovarian cancer cells to
carboplatin. Clin Cancer Res. 13:7191–7198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stamelos VA, Robinson E, Redman CW and
Richardson A: Navitoclax augments the activity of carboplatin and
paclitaxel combinations in ovarian cancer cells. Gynecol Oncol.
128:377–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abed MN, Abdullah MI and Richardson A:
Antagonism of Bcl-XL is necessary for synergy between carboplatin
and BH3 mimetics in ovarian cancer cells. J Ovarian Res. 9:252016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ghosh-Choudhury N, Mandal CC,
Ghosh-Choudhury N and Choudhury G Ghosh: Simvastatin induces
derepression of PTEN expression via NFkappaB to inhibit breast
cancer cell growth. Cell Signal. 22:749–758. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qi XF, Zheng L, Lee KJ, Kim DH, Kim CS,
Cai DQ, Wu Z, Qin JW, Yu YH and Kim SK: HMG-CoA reductase
inhibitors induce apoptosis of lymphoma cells by promoting ROS
generation and regulating Akt, Erk and p38 signals via suppression
of mevalonate pathway. Cell Death Dis. 4:e5182013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelman JA, Luo J and Cantley LC: The
evolution of phosphatidylinositol 3-kinases as regulators of growth
and metabolism. Nat Rev Genet. 7:606–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cancer Genome Atlas Research Network, .
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Workman P, Clarke PA, Raynaud FI and van
Montfort RL: Drugging the PI3 kinome: From chemical tools to drugs
in the clinic. Cancer Res. 70:2146–2157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goldoni M and Johansson C: A mathematical
approach to study combined effects of toxicants in vitro:
evaluation of the bliss independence criterion and the loewe
additivity model. Toxicol In Vitro. 21:759–769. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Richardson A, Malik RK, Hildebrand JD and
Parsons JT: Inhibition of cell spreading by expression of the
C-terminal domain of focal adhesion kinase (FAK) is rescued by
coexpression of Src or catalytically inactive FAK: A role for
paxillin tyrosine phosphorylation. Mol Cell Biol. 17:6906–6914.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kato S, Smalley S, Sadarangani A, Chen-Lin
K, Oliva B, Brañes J, Carvajal J, Gejman R, Owen GI and Cuello M:
Lipophilic but not hydrophilic statins selectively induce cell
death in gynaecological cancers expressing high levels of HMGCoA
reductase. J Cell Mol Med. 14:1180–1193. 2010.PubMed/NCBI
|
|
20
|
Taylor-Harding B, Orsulic S, Karlan BY and
Li AJ: Fluvastatin and cisplatin demonstrate synergistic
cytotoxicity in epithelial ovarian cancer cells. Gynecol Oncol.
119:549–556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vidot S, Witham J, Agarwal R, Greenhough
S, Bamrah HS, Tigyi GJ, Kaye SB and Richardson A: Autotaxin delays
apoptosis induced by carboplatin in ovarian cancer cells. Cell
Signal. 22:926–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miraglia E, Högberg J and Stenius U:
Statins exhibit anticancer effects through modifications of the
pAkt signaling pathway. Int J Oncol. 40:867–875. 2012.PubMed/NCBI
|
|
23
|
Hanrahan AJ, Schultz N, Westfal ML, Sakr
RA, Giri DD, Scarperi S, Janakiraman M, Olvera N, Stevens EV, She
QB, et al: Genomic complexity and AKT dependence in serous ovarian
cancer. Cancer Discov. 2:56–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang J, Zhang L, Greshock J, Colligon TA,
Wang Y, Ward R, Katsaros D, Lassus H, Butzow R, Godwin AK, et al:
Frequent genetic abnormalities of the PI3K/AKT pathway in primary
ovarian cancer predict patient outcome. Genes Chromosomes Cancer.
50:606–618. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Robinson E, Fisher N, Stamelos V, Redman C
and Richardson A: New strategies for the treatment of ovarian
cancer. Biochem Soc Trans. 42:125–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu H, Yang J, Yuan Y, Xia Z, Chen M, Xie
L, Ma X, Wang J, Ouyang S, Wu Q, et al: Regulation of Mcl-1 by
constitutive activation of NF-κB contributes to cell viability in
human esophageal squamous cell carcinoma cells. BMC Cancer.
14:982014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Certo M, Del Gaizo Moore V, Nishino M, Wei
G, Korsmeyer S, Armstrong SA and Letai A: Mitochondria primed by
death signals determine cellular addiction to antiapoptotic BCL-2
family members. Cancer Cell. 9:351–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stamelos VA, Redman CW and Richardson A:
Understanding sensitivity to BH3 mimetics: ABT-737 as a case study
to foresee the complexities of personalized medicine. J Mol Signal.
7:122012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stamelos VA, Fisher N, Bamrah H, Voisey C,
Price JC, Farrell WE, Redman CW and Richardson A: The BH3 mimetic
obatoclax accumulates in lysosomes and causes their alkalinization.
PLoS One. 11:e01506962016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stamelos V: Investigation of the
BH3-mimetics navitoclax and obatoclax as potential therapeutics for
ovarian cancer. PhD thesisSchool of Pharmacy Keele University
Staffordshire: pp. 246ProQuest No. 1874909132. 2014
|
|
31
|
Del Gaizo Moore V and Letai A: BH3
profiling-measuring integrated function of the mitochondrial
apoptotic pathway to predict cell fate decisions. Cancer Lett.
332:202–205. 2013. View Article : Google Scholar : PubMed/NCBI
|