Introduction
Cervical cancer is the third-leading cause of
cancer-associated mortality among young women worldwide; it is a
severe health threat in developing countries, which is often caused
by a persistent infection with the human papillomavirus (HPV)
(1,2).
However, owing to an increased rate of tumor recurrence and
metastasis, there are no sufficient treatment options at present
other than surgical resection.
The primary cause of cancer-associated mortality is
due to the fact that tumor cells are able to disseminate to distant
sites (3,4). However, the mechanism by which
metastasis occurs remains controversial. Studies have indicated
that the epithelial-mesenchymal transition (EMT) is a necessary
prerequisite for tumor metastasis (5–7). Owing to
the clinical importance of metastasis, a number of studies have
focused on elucidating the mechanisms of EMT (8,9).
Transforming growth factor-β1 (TGF-β1) serves a key role in the
process of EMT and is regarded as a driver of EMT (10,11).
However, the mechanisms underlying the regulation of TGF-β1 in
cervical cancer cells remain elusive.
Probable adenosine triphosphate (ATP)-dependent RNA
helicase DDX5 (also known as p68) was initially identified through
immunological cross-reactivity against the anti-simian virus 40
large T-monoclonal antibody (12). It
has been reported that p68 knockout mice are embryonically lethal
(embryonic day, 11.5), indicating that it serves a key function in
the developmental process (13). p68
is involved in the processing of RNA secondary structures, which
participates in a variety of biological processes, including cell
proliferation and organ differentiation (14–16). At
present, p68 has been identified to activate the transcription of
estrogen receptor, androgen receptor and tumor suppressor p53,
myoblast determination protein and β-catenin (16,17).
Overexpression of p68 has been documented in various types of
cancer including colon, breast and prostate cancer (18–20).
However, to the best of our knowledge, no study has been conducted
on the specific role of p68 in cervical cancer.
In the present study, the expression and potential
mechanism of p68 in the development of cervical cancer was
investigated. To the best of our knowledge, for the first time, it
was demonstrated that p68 was markedly upregulated in cervical
cancer cells. Furthermore, it was demonstrated that p68 may
transcriptionally activate the expression of TGF-β1, thereby
prompting the EMT process of cervical cancer cells.
Materials and methods
Cell culture
The cervical cancer CaSki, HeLa (HPV-18-positive),
SiHa (HPV-16-positive) and C-33A (HPV-negative) cell lines were
compared with the human keratinocyte cell HaCaT line and obtained
from the American Type Culture Collection (Manassas, VA, USA) and
cultured in RPMI-1640 (GE Healthcare Life Sciences, Little
Chalfont, UK). All cultures were supplemented with 10% fetal bovine
serum (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), streptomycin (100 mg/ml; GE Healthcare Life Sciences) and
penicillin (100 IU/ml; GE Healthcare Life Sciences) at 37°C in a
humidified atmosphere containing 5% CO2.
Small interfering RNA
(siRNA)transfection
The p68 siRNA oligonucleotide was purchased from
Shanghai GenePharma Co., Ltd. (Shanghai, China); the sequence was
5′-GCUGAAUAUUGUCGAGCUU-3′. Briefly, CaSki cells were seeded at
1×106 cells/well in the 6-well plates in the presence or
absence of 20 nM TGF-β. The p68-siRNA or a non-specific negative
control (NC) siRNA (5′-TTCTCCGAACGTGTCACGT-3′) were mixed with
HiperFect transfection reagent at a final concentration of 50 nM
(Qiagen, Inc., Valencia, CA, USA) and incubated at room temperature
for 10 min. Next, the complex was added to the culture medium of
cells for 48 h, after which the subsequent experiments were
conducted.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The total RNA from cultured cells was isolated using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's protocol. The total RNA was
reverse transcribed at 72°C for 10 min and 42°C for 60 min into
cDNA with TaqMan RNA Reverse Transcription kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.). qPCR was performed using
SYBR-Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
in a BIO-RAD iCycleriQ real-time PCR detection system, as described
previously (21). Details of PCR
procedures were as follows: 95°C for 10 min followed by 50 cycles
of 95°C for 10 sec, 55°C for 10 sec, 72°C for 5 sec, 99°C for 1
sec, 59°C for 15 sec, 95°C for 1 sec. The primers used were listed
as follows: p68 forward, 5-AGAGGTTCAGGTCGTTCCAGG-3 and reverse,
5-GGAATATCCTGTTGGCATTGG-3; GAPDH forward, 5-CACCCAGAAGACTGTGGATGG-3
and reverse, 5-GTCTACATGGCAACTGTGAGG-3. Relative mRNA expression
was normalized against the endogenous control, GAPDH, using the
2−ΔΔCq method (22).
Protein extraction and western blot
analysis
Proteins samples were extracted in RIPA buffer (1%
TritonX-100, 15 mmol/l NaCl, 5 mmol/l EDTA, and 10 mmol/l Tris-HCl;
pH 7.0; Beijing Solarbio Science and Technology Co., Ltd., Beijing,
China) supplemented with a protease and phosphatase inhibitor
cocktail (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and then
20 µg protein loaded per lane was separated by SDS-PAGE (10% gel),
followed by electrophoretic transfer to a polyvinylidene fluoride
membrane. Following incubation with 8% non-fat milk in PBST (pH
7.5) for 2 h at room temperature, membranes were incubated with the
following primary antibodies: Antibodies against decapentaplegic
homolog 2 (Smad2; 1:1,000; cat. no. #8685; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-E-cadherin (1:1,000; cat.
no. 3199; Cell Signaling Technology, Inc.), anti-α-smooth muscle
actin (α-SMA; 1:1,000; cat. no. 19245; Cell Signaling Technology,
Inc.), fibronectin (FN; 1:1,000; cat. no. ab2413; Abcam, Cambridge,
UK), vimentin (Vi; 1:1,000; cat. no. 5714; Cell Signaling
Technology, Inc.) and anti-GAPDH (1:1,000; cat. no. 5174; Cell
Signaling Technology, Inc.). Following several washes with TBST,
the membranes were incubated with HRP-conjugated goat anti-rabbit
IgG (1:5,000; cat. no. ZB-2306; Zhongshan Gold Bridge Biological
Technology Co., Beijing, China) for 2 h at room temperature and
then washed. Immunodetection was performed by enhanced
chemiluminescence detection system (EMD Millipore, Billerica, MA,
USA) according to the manufacturer's protocol. The housekeeping
gene GAPDH was used as the internal control. The protein levels
were quantified using density analysis according to the
manufacturer's protocol (ImageJ version 1.8.0; National Institutes
of Health, Bethesda, MD, USA).
Adenoviral vector construction and
transfection
Recombinant adenoviruses expressing p68 (ad-p68) or
a negative control adenovirus vector containing green fluorescent
protein (ad-NC) were purchased from Shanghai GeneChem Co., Ltd.
(Shanghai, China). In brief, CaSki cells were seeded at
1×106 cells/well in 6-well plates. After 24 h, the
ad-p68 or ad-NC vectors were transfected into the cells at a
multiplicity of infection of 25 and the cells were collected 48 h
later for experimentation.
Migration assay
Cell migration was assessed using in vitro
scratch assays. Firstly, cells were cultured at 1×105
cells/well in 12-well plates for 24 h. Next, a 10 µ1 pipette tip
was used to create an artificial gap in the confluent cell
monolayer. Following transfection with ad-P68 or ad-NC for 48 h,
the cells were washed with pre-warmed PBS three times to remove the
debris. The initial images of the scratch (0 h) and final images of
the scratch (48 h) were captured with an inverted light microscope.
The migratory abilities were quantified by measuring the area of
the scratched regions using the ImagePro Plus 4.5 software (Media
Cybernetics, Inc., Rockville, MD, USA).
Observation of cell morphology
In brief, CaSki cells were cultured at
1×105 cells/well in 12-well plates for 24 h. Then, the
cells were transfected with ad-P68 or ad-NC for 48 h, the cells
were washed with pre-warmed PBS three times to remove the debris.
Subsequently, cell morphology was observed under an inverted light
microscope (magnification, ×400).
Statistical analysis
Data are presented as the mean ± standard deviation
following 3 independent experiments. SPSS (version 13.0; SPSS,
Inc., Chicago, IL, USA) was used to perform statistical analyses.
Two-tailed unpaired Student's t-tests were used for comparisons of
two groups. Analysis of variance followed by Turkey's post hoc test
was used for comparisons of two more groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
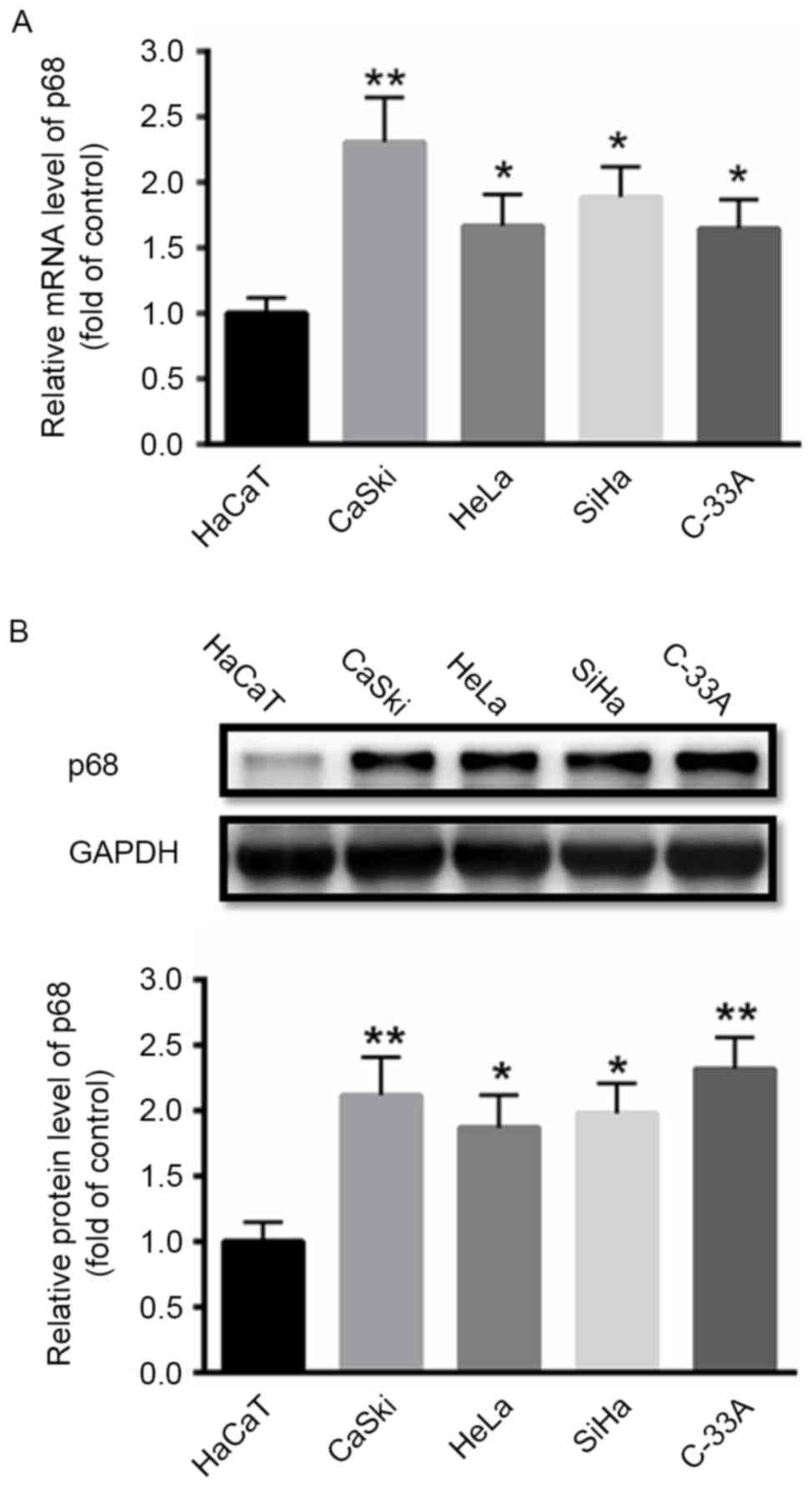
Upregulation of p68 in cervical cancer
cells
First, the expression of p68 was investigated in
cervical cancer cells. RT-qPCR and western blot analyses
demonstrated that the mRNA and protein levels of p68 were
significantly enhanced in cervical cancer CaSki, HeLa, SiHa, and
C-33A cell lines compared with a human keratinocyte HaCaT cell line
(Fig. 1).
p68 enhances EMT in CaSki cells
The effect of p68 on the process of EMT in CaSki
cells, which serves a key function in cancer cell migration, was
investigated. First, ad-p68 or ad-NC was transfected into CaSki
cells. At 48 h later, western blot analysis demonstrated that
transfection with ad-p68 significantly enhanced the protein
expression of p68 compared with NC-treated cells. Furthermore,
overexpression of p68 induced the expression of the mesenchymal
markers α-SMA, vimentin and fibronectin, whereas the epithelial
marker E-cadherin was significantly decreased (Fig. 2A). CaSki cells exhibited an elongated
and spindle-shaped morphology following transfection with ad-p68
(Fig. 2B). In addition, the role of
p68 on CaSki cell migration was also investigated. As presented in
Fig. 2C, the in vitro scratch
assay demonstrated that overexpression of p68 markedly enhanced
CaSki cell migratory capacity at 24 and 48 h (Fig. 2C).
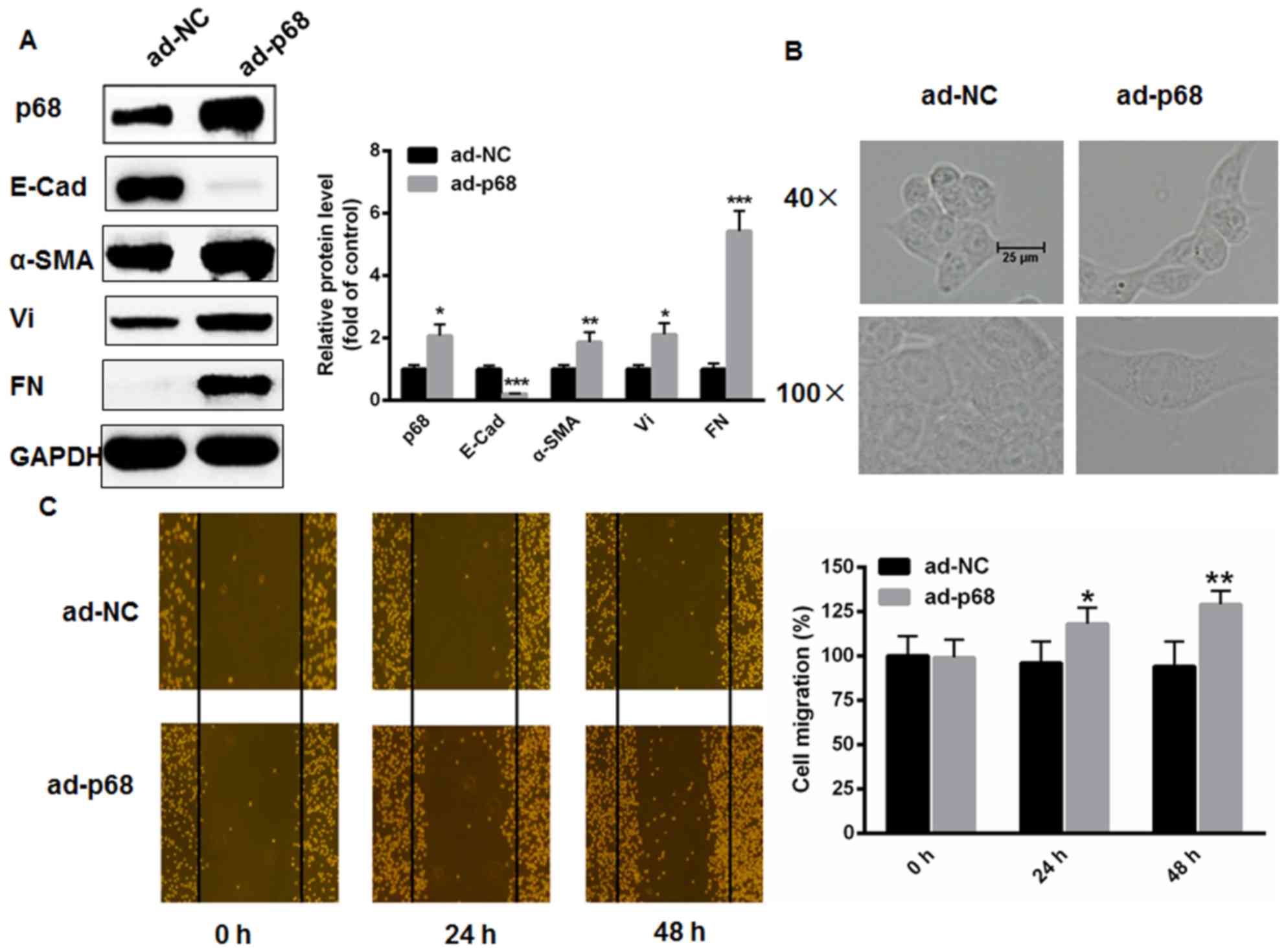 | Figure 2.p68 enhances EMT in CaSki cells. (A)
The changes of EMT markers were analyzed using western blot
analysis following adenoviral transfection with p68. (B) CaSki
cells exhibited an elongated, spindle-shaped morphology following
transfection with p68 for 48 h. (C) In vitro scratch test
demonstrated that overexpression of p68 markedly enhanced CaSki
cell migration capacity at 24 and 48 h (n=3). *P<0.05,
**P<0.01, vs. ad-NC. EMT, epithelial-mesenchymal transition;
E-Cad, E-cadherin; α-SMA, α-smooth muscle actin; Vi, vimentin; FN,
fibronectin; ad-NC, adenoviral-transfected negative control;
ad-p68, adenoviral-transfected p68. |
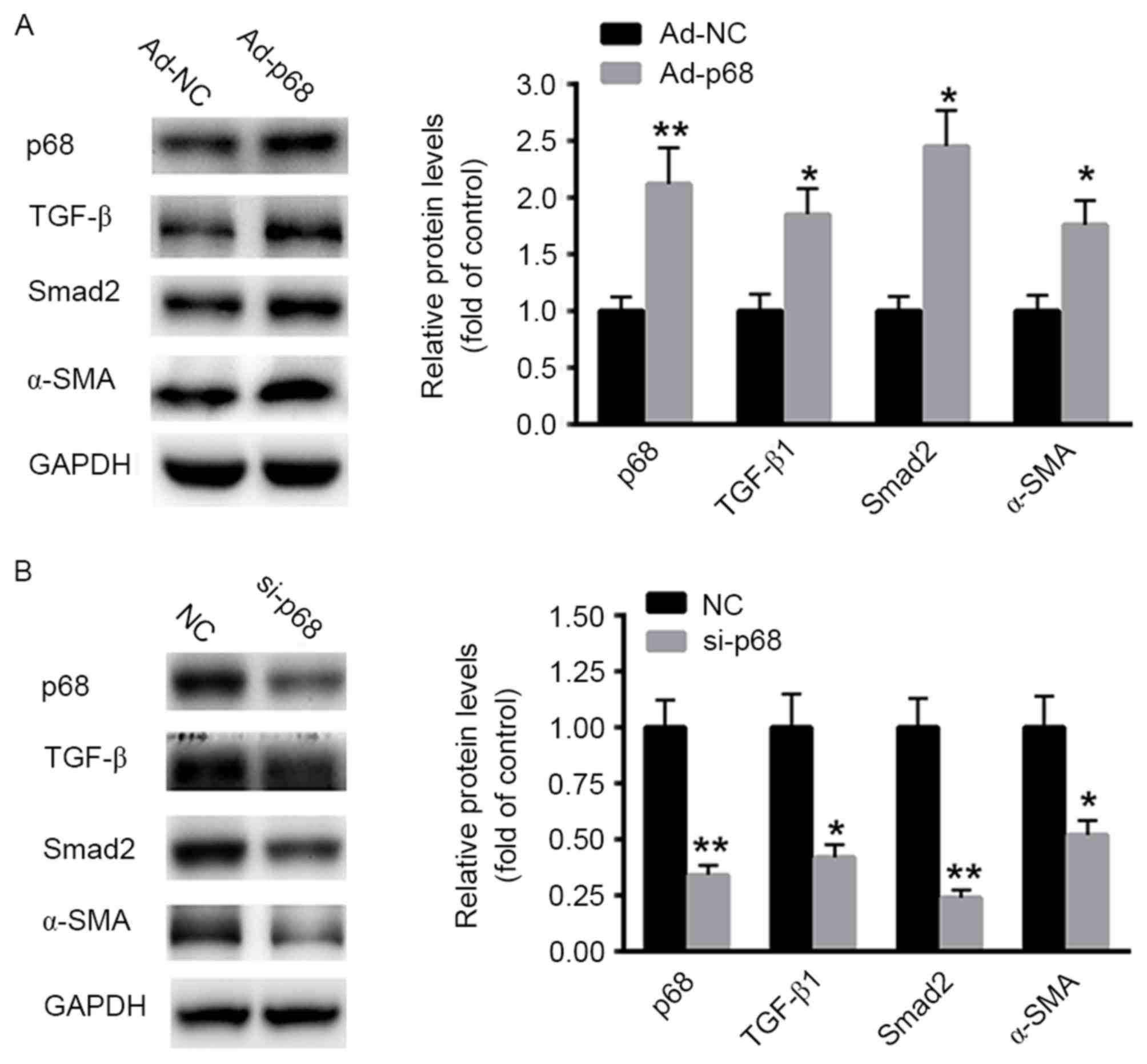
p68 stimulates the expression of
TGF-β1 in CaSki cells
Next, ad-p68 was transfected into CaSki cells for 48
h. Western blot analysis revealed that overexpression of p68
significantly enhanced the expression TGF-β1 (Fig. 3A). Expression of downstream effectors,
including Smad2 and α-SMA, was also significantly upregulated
(Fig. 3A). By contrast, silencing of
p68 with a specific siRNA significantly suppressed the protein
expression of TGF-β1 as well as the downstream effectors Smad2 and
α-SMA compared with the NC siRNA (Fig.
3B). These data indicated that p68 stimulates the expression of
TGF-β1, inducing downstream signaling in CaSki cells.
Silence of p68 partially abolishes
TGF-β1-induced EMT process in CaSki cells
To determine whether p68 prompts the EMT process in
CaSki cells by stimulating TGF-β1 expression, CaSki cells with
siRNA-p68, TGF-β1, either alone or together. Silencing of p68
significantly suppressed the TGF-β1 signaling pathway. By
comparison, treatment with TGF-β1 markedly activated the TGF-β1
signaling pathway, including upregulation of Smad2, α-SMA, FN, Vi
and downregulation of E-cad (Fig.
4A). Notably, knockdown of p68 partially reversed
TGF-β1-treatment-induced changes to the expression of EMT markers
(Fig. 4A). The morphological changes
of CaSki cells were also determined. As presented in Fig. 4B, TGF-β1-induced cell morphological
changes, including elongated and spindle-shaped morphology, were
partially reversed by knockdown of p68. These data indicated that
p68 prompted CaSki cell malignancies, primarily by stimulating the
expression of TGF-β1.
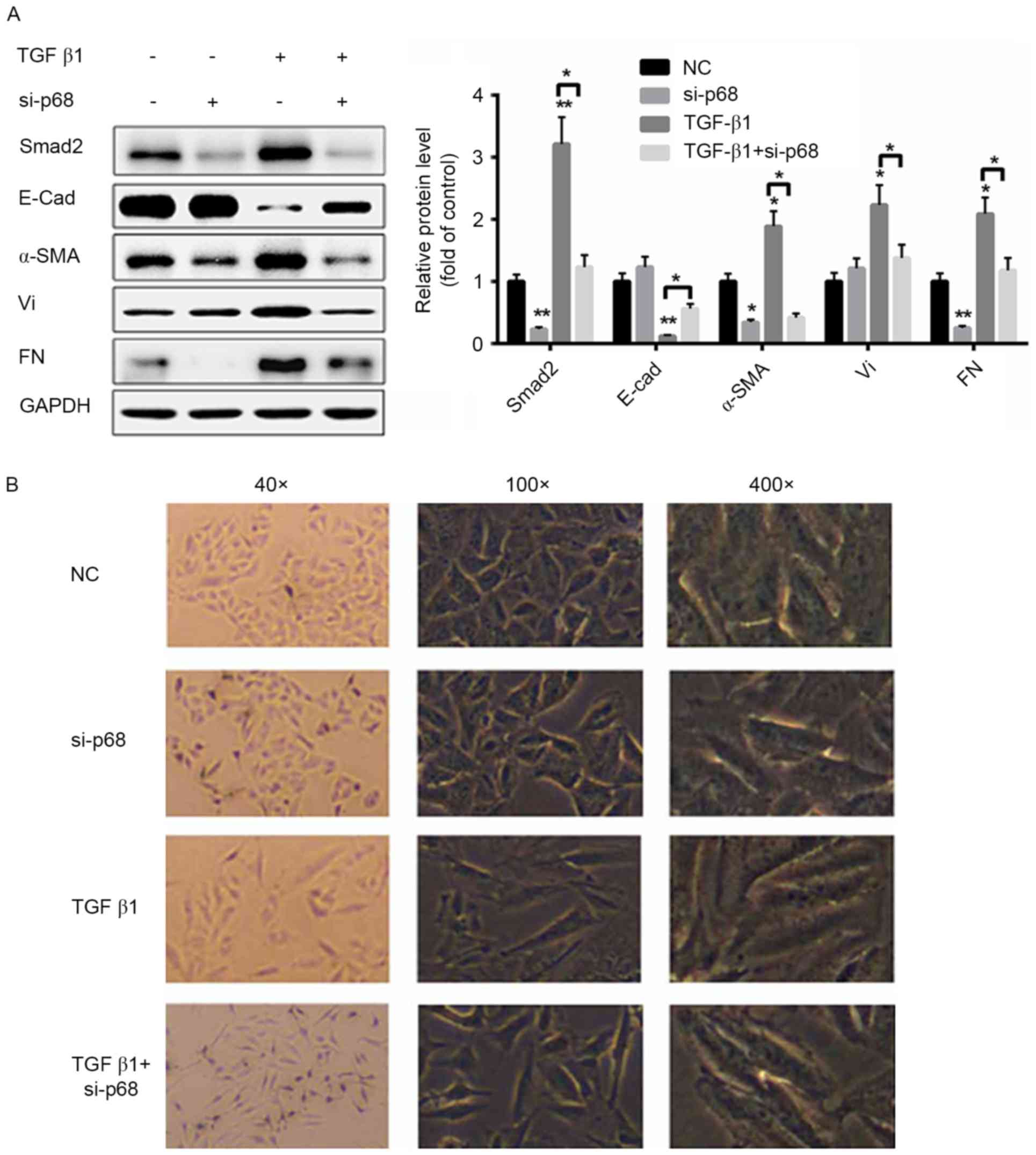 | Figure 4.Silencing of p68 partially abolishes
TGF-β1-induced the epithelial-mesenchymal transition process in
CaSki cells. (A) Western blot analysis of the TGF-β1 signaling
pathway when CaSki cells were treated with si-p68 and/or TGF-β1.
(B) Morphological changes of CaSki cells were determined in CaSki
cells were treated with si-p68 and/or TGF-β1 (n=3). *P<0.05,
**P<0.01, vs. NC. E-Cad, E-cadherin; α-SMA, α-smooth muscle
actin; Vi, vimentin; FN, fibronectin; TGF-β1, transforming growth
factor-β1; Smad2, mothers against decapentaplegic homolog 2;
si-p68, small interfering RNA targeted at p68; NC, negative
control. |
Discussion
Cervical cancer is the third most common type of
cancer among females worldwide (1,2). At
present, the treatment outcome for cervical cancer is
unsatisfactory, particularly when advanced-stage tumors are
considered. It is widely accepted that tumor metastases accounts
for ~90% of all cancer-associated mortalities (23). Therefore, identification of the causes
of metastasis may assist in the development of novel treatment
methods for patients with cervical cancer, and therefore research
in this field is of great importance.
p68 belongs to the Asp-Glu-Ala-Asp (DEAD)-box family
of RNA helicases, with a conserved DEAD peptide sequence. This
family is reported to modulate RNA structure through its
ATP-dependent RNA helicase activity (24). Studies have demonstrated that
DEAD-box-containing proteins serve a key role in ribosome
biogenesis, embryogenesis and cell division (25–27).
Previous studies revealed that p68 activates the expression of
several oncogenes, thereby modulating cancer growth and metastasis
(24,28). It is reported that the upregulation of
p68 serves a key function in cancer progression, particularly in
breast cancer (29). The present
study investigated the expression of p68 in cervical cancer cells,
determining that the expression of p68 was significantly increased
in cervical cancer CaSki, HeLa, SiHa and C-33A cell lines, compared
with a human keratinocyte HaCaT cell line at the transcriptional
and post-transcriptional levels. These results demonstrated the
oncogenic role of p68 in cervical cancer cells.
TGF-β1 signaling serves a key function in tissue
homeostasis and cancer progression (30). It is reported that activation of
TGF-β1 signaling contributes to an abnormal EMT process (10). However, whether p68 regulates the EMT
process has, to the best of our knowledge, never been investigated.
The present study identified that overexpression of p68
significantly enhanced the expression of TGF-β1, thereby
contributing to the EMT process in cervical cancer cells. In line
with these observations, transfection with ad-p68 induced
morphological changes inhuman cervical cancer cells. TGF-β1 is
considered to be a primary driver of EMT processes and tumor
progression. Results reported in the present study determined that
p68 is able to activate TGF-β1 production and downstream
signaling.
To the best of our knowledge, this is the first
study demonstrating that inhibition of p68 reverses TGF-β-induced
EMT in cervical cancer cells by inactivating TGF-β1 signaling. The
results of the present study revealed that silencing of p68
inhibits cell proliferation and reverses EMT. These results provide
novel mechanistic insight into the pro-tumor effects of p68 in
cervical cancer cells.
Acknowledgements
The present study was supported by Science and
Technology Planning Project of Guangdong Province, China (grant no.
2014A020212345) and Medical Science Research Foundation of
Guangdong Province, China (grant no. C2015038).
References
|
1
|
Kim M, Kim YS, Kim H, Kang MY, Park J, Lee
DH, Roh GS, Kim HJ, Kang SS, Cho GJ, et al: O-linked
N-acetylglucosamine transferase promotes cervical cancer
tumorigenesis through human papillomaviruses E6 and E7 oncogenes.
Oncotarget. 7:44596–44607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN, 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caramel J, Papadogeorgakis E, Hill L,
Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J,
Hutchinson P, Tse G, et al: A switch in the expression of embryonic
EMT-inducers drives the development of malignant melanoma. Cancer
Cell. 24:466–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fenouille N, Tichet M, Dufies M, Pottier
A, Mogha A, Soo JK, Rocchi S, Mallavialle A, Galibert MD, Khammari
A, et al: The epithelial-mesenchymal transition (EMT) regulatory
factor SLUG (SNAI2) is a downstream target of SPARC and AKT in
promoting melanoma cell invasion. PLoS One. 7:e403782012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang GM, Xie WY, Wang HS, Du J, Wu BP, Xu
W, Liu HF, Xiao P, Liu ZG, Li HY, et al: Curcumin combined with
FAPαc vaccine elicits effective antitumor response by targeting
indolamine-2,3-dioxygenase and inhibiting EMT induced by TNF-α in
melanoma. Oncotarget. 6:25932–25942. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jung HY, Fattet L and Yang J: Molecular
pathways: Linking tumor microenvironment to epithelial-mesenchymal
transition in metastasis. Clin Cancer Res. 21:962–968. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang P, Sun Y and Ma L: ZEB1: At the
crossroads of epithelial-mesenchymal transition, metastasis and
therapy resistance. Cell Cycle. 14:481–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Laurenzana A, Biagioni A, Bianchini F,
Peppicelli S, Chillà A, Margheri F, Luciani C, Pimpinelli N, Del
Rosso M, Calorini L and Fibbi G: Inhibition of uPAR-TGFβ crosstalk
blocks MSC-dependent EMT in melanoma cells. J Mol Med (Berl).
93:783–794. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin K, Baritaki S, Militello L, Malaponte
G, Bevelacqua Y and Bonavida B: The role of B-RAF mutations in
melanoma and the induction of EMT via DYsregulation of the
NF-κB/Snail/RKIP/PTEN circuit. Genes Cancer. 1:409–420. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schlegel NC, von Planta A, Widmer DS,
Dummer R and Christofori G: PI3K signalling is required for a
TGFβ-induced epithelial-mesenchymal-like transition (EMT-like) in
human melanoma cells. Exp Dermatol. 24:22–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tulchinsky E, Pringle JH, Caramel J and
Ansieau S: Plasticity of melanoma and EMT-TF reprogramming.
Oncotarget. 5:1–2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lane DP and Hoeffler WK: SV40 large T
shares an antigenic determinant with a cellular protein of
molecular weight 68,000. Nature. 288:167–170. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fukuda T, Yamagata K, Fujiyama S,
Matsumoto T, Koshida I, Yoshimura K, Mihara M, Naitou M, Endoh H,
Nakamura T, et al: DEAD-box RNA helicase subunits of the drosha
complex are required for processing of rRNA and a subset of
microRNAs. Nat Cell Biol. 9:604–611. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abdelhaleem M: RNA helicases: Regulators
of differentiation. Clin Biochem. 38:499–503. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fuller-Pace FV: DExD/H box RNA helicases:
Multifunctional proteins with important roles in transcriptional
regulation. Nucleic Acids Res. 34:4206–4215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Metivier R, Penot G, Hubner MR, Reid G,
Brand H, Kos M and Gannon F: Estrogen receptor-alpha directs
ordered, cyclical, and combinatorial recruitment of cofactors on a
natural target promoter. Cell. 115:751–763. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Clark EL, Coulson A, Dalgliesh C, Rajan P,
Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ, et
al: The RNA helicase p68 is a novel androgen receptor coactivator
involved in splicing and is overexpressed in prostate cancer.
Cancer Res. 68:7938–7946. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shin S, Rossow KL, Grande JP and Janknecht
R: Involvement of RNA helicases p68 and p72 in colon cancer. Cancer
Res. 67:7572–7578. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang L, Lin C and Liu ZR: P68 RNA helicase
mediates PDGF-induced epithelial mesenchymal transition by
displacing Axin from beta-catenin. Cell. 127:139–155. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang L, Lin C, Zhao S, Wang H and Liu ZR:
Phosphorylation of p68 RNA helicase plays a role in
platelet-derived growth factor-induced cell proliferation by
up-regulating cyclin D1 and c-Myc expression. J Biol Chem.
282:16811–16819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo J, Li M, Meng X, Sui J, Dou L, Tang W,
Huang X, Man Y, Wang S, Li J, et al: MiR-291b-3p induces apoptosis
in liver cell line NCTC1469 by reducing the level of RNA-binding
protein HuR. Cell Physiol Biochem. 33:810–822. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gupta GP and Massagué J: Cancer
metastasis: Building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fuller-Pace FV: RNA helicases: Modulators
of RNA structure. Trends Cell Biol. 4:271–274. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schmid SR and Linder P: D-E-A-D protein
family of putative RNA helicases. Mol Microbiol. 6:283–291. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song C, Hotz-Wagenblatt A, Voit R and
Grummt I: SIRT7 and the DEAD-box helicase DDX21 cooperate to
resolve genomic R loops and safeguard genome stability. Genes Dev.
2017. View Article : Google Scholar
|
|
27
|
Lumb JH, Li Q, Popov LM, Ding S, Keith MT,
Merrill BD, Greenberg HB, Li JB and Carette JE: DDX6 represses
aberrant activation of interferon-stimulated genes. Cell Rep.
20:819–831. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mazurek A, Luo W, Krasnitz A, Hicks J,
Powers RS and Stillman B: DDX5 regulates DNA replication and is
required for cell proliferation in a subset of breast cancer cells.
Cancer Discov. 2:812–825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Westphal P, Mauch C, Florin A, Czerwitzki
J, Olligschläger N, Wodtke C, Schüle R, Büttner R and Friedrichs N:
Enhanced FHL2 and TGF-β1 expression is associated with invasive
growth and poor survival in malignant melanomas. Am J Clin Pathol.
143:248–256. 2015. View Article : Google Scholar : PubMed/NCBI
|