Introduction
Loss-of-function analysis is a crucial issue in
reverse genetic studies. In the past decade, RNA interference
(RNAi) has been widely used to knockdown gene expression (1,2). RNAi is
based on the binding of small interfering (si)RNAs to target
transcripts leading to either its degradation or its inhibition at
the translational level. siRNAs are either transiently delivered to
cells or stably produced from small hairpin (sh)RNAs that are
transcribed by polymerase III promoters. Albeit both siRNAs and
shRNAs allow specific silencing of target genes (3), knockdowns by RNAi are mostly incomplete,
vary between experiments and have unpredictable off-target effects
(4,5).
New technologies have recently become available to
generate knockouts rather than knockdowns of target genes in
cultured cells. Among those techniques are transcription
activator-like effector nucleases (TALENs) that use a pair of
artificial DNA-binding domains fused to the catalytic domain of
restriction endonuclease FokI which causes a double-strand break
(DSB) at the targeted genomic locus stimulating DNA repair
(6,7).
Yet, TALEN is labor-intensive and works with low efficiency
(8).
A more recently discovered technique of genomic
engineering is clustered regulatory interspaced short palindromic
repeats (CRISPR)/CRISPR associated 9 (Cas9), which is a unique
mechanism of bacteria and archea to protect themselves against
foreign DNA penetration (9). This
prokaryotic system has been adapted using a Cas9 endonuclease from
Streptococcus pyrogenes that is guided to the target
sequence by a guide RNA (gRNA) chimera that includes a protospacer
adjacent motif. To reduce off-target effects, a mutant Cas9 termed
nickase can be used which requires a pair of gRNAs to introduce
site-specific single strand breaks, called nicks, that are together
equivalent to a DSB (10). Of note,
the use of two gRNAs and the nickase doubles the number of bases
that need to be specifically recognized at the target locus and
thereby significantly increases specificity.
DSBs introduced by TALEN or CRISPR/Cas9 at the
targeted genomic locus are either repaired by the error prone
non-homologous end joining (NHEJ) or by homology-directed repair
(HDR). NHEJ leads to small insertions or deletions (InDels) that
can result in a knockout of gene function due to frameshift
mutations (11). The co-delivery of
locus-specific homology arms with the site-specific nuclease
triggers HDR-mediated genetic alterations and allows efficient
integration of transgenes into an endogenous gene locus. First
proof-of-principle studies showed that Cas9 can be successfully
targeted to endogenous genes in bacteria (12), human pluripotent stem cells (13), as well as in whole organisms such as
zebrafish (14), yeast (15), fruit flies (16), mice (17), rats (18) and rabbits (19). In addition, a haploid human cell line
named engineered-HAPloid cells has been generated by megabase-scale
deletion using CRISPR/Cas9 (20).
An important step in the use of genomic editing
techniques is the confirmation of the knockout events. To analyze
the targeted genomic locus, the target sequence is amplified by
PCR, subcloned into a plasmid vector and subjected to sequencing
(21). Another approach uses direct
sequencing of the PCR products and analysis by ‘Tracking InDels by
Decomposition’ (TIDE) which quantifies the editing efficacy and
identifies predominant types of InDels in the targeted pool of
cells (22). Other methods analyzing
the efficiency of the Cas9-mediated DNA cleavage include
heteroduplex formation that is examined either by high resolution
melting analysis, heteroduplex mobility assay or T7 endonuclease I
cutting. Using these methods, the ratio of homo- to heteroduplexes
can be determined in order to estimate the nuclease efficiency.
However, the latter method fails to accurately detect InDels
(23).
Contrary to applications of CRISPR/Cas9 in haploid
or diploid cells, genomic editing is more challenging when applied
to hyperdiploid genomes as in the case of most cancer cells. In
particular, all functional copies of the target gene must be edited
in cancer cell lines to accomplish a complete knockout situation
(24). As NHEJ works in a random
fashion, there may arise altered structures without gene
inactivation along NHEJ repair events. These insufficient knockout
events, often combined with cellular heterogeneity, enhance the
probability to generate partial knockouts that still harbor alleles
coding for functional gene products or gene products with altered
functionality (24). Hence, the
determination of target gene copy number and cellular heterogeneity
is essential in cancer cell populations to allow generation of
solid CRISPR/Cas9-mediated knockouts and to correctly interpret the
subsequent confirmation of knockout events.
The increase in aberrant ploidy levels and
karyotypic complexity correlates with the progression of tumor
cells from a benign neoplasm to malignant cancer. Chromosomal
abnormalities occur in 75% of blood cancers and in more than 90% of
solid tumors including hepatocellular carcinoma (HCC) (25,26). The
overexpression of the receptor tyrosine kinase Axl along with the
release of soluble Axl (sAxl) was detected in HCC and correlated
with poor survival of HCC patients (27,28). In
this study, we have generated knockouts of Axl in hepatoma cell
lines using NHEJ-mediated CRISPR/Cas9 knockout technology. Our data
show the dynamics of CRISPR/Cas9-mediated editing of the AXL
locus in hyperdiploid hepatoma cell lines and reveal the drawbacks
of the CRISPR/Cas9 technique in cancer cells.
Materials and methods
Cell culture
The human hepatoma cell lines SNU449 and HLF were
cultured in RPMI-1640 supplemented with 10% fetal calf serum (FCS;
Sigma, St. Louis, MO, USA) and DMEM plus 10% FCS, respectively. All
cells were kept at 37°C and 5% CO2 and routinely
screened for the absence of mycoplasma. All hepatoma cell lines
were validated by short tandem repeat analysis.
CRISPR/Cas9-mediated genomic editing
of the AXL locus
The AXL gene was disrupted in hepatoma cell
lines using the human Axl gRNA CRISPR lentivirus set (K0161411,
ABM, Milton, Ontario, Canada). HLF and SNU449 cells were infected
with ready-to-use lentiviral stocks. Two or 6 µl of the gRNA pair
and 2 or 6 µl of Cas9 nickase-encoding virus were added in a ratio
of 1:2 to the cells. The medium was exchanged with standard culture
medium after overnight incubation and cells were selected for
stable expression of Cas9 for at least one week with 1 µg/ml
puromycin. Single cell clones were generated by limiting dilution
in 96-well plates and expanded to 100 mm culture plates. The single
cell clones were further processed for sequence analysis of the
AXL locus.
Sequence analysis of the genomic AXL
locus
The gRNA binding region was amplified by polymerase
chain reaction (PCR), cloned into a pGEM-T easy vector (A1360;
Promega Corp., Madison, WI, USA) and transformed into E.
coli DH5α competent bacteria for blue-white screening. The
plasmid DNA of 20 bacterial colonies was sequenced by Sanger method
and analyzed for sequence variations in the gRNA binding region.
Single cell clones showing InDels in all 20 colonies were
considered to be Axl-deficient (Axl−).
Western blot analysis
Immunoblotting was done as described previously
(29). The antibodies used were
anti-Axl, 1:1,000 (AF154; R&D Systems, Inc., Minneapolis, MN,
USA) and anti-Actin, 1:2,500 (A2066; Sigma-Aldrich, St. Louis, MO,
USA).
Enzyme-linked immunosorbent assay
(ELISA)
Soluble Axl (sAxl) levels were assessed by ELISA
using the DuoSet ELISA Development System Human Axl (DY154; R&D
Systems, Inc.) from supernatants (SNs) of HLF and SNU449 cells.
1.5×106 HLF or 0.8×106 SNU449 cells were each
seeded on 60 mm tissue culture plates. 24 h after plating, the
cells were incubated in serum-free medium for further 24 h.
Whole-cell lysates of the same cells were measured by ELISA using a
protein concentration of 0.1 mg/ml. The samples of parental cells
were measured in 1:10 and 1:20 dilutions. To ensure the detection
of Axl in the knockout situation, the SNs and protein lysates of
Axl− cells were measured undiluted and at a dilution of
1:5. The concentrations of sAxl and Axl were normalized to
1.0×106 cells. All values below the standard curve were
considered as noise and were therefore excluded. A seven-point
standard curve was generated for each plate and quantification was
performed using GraphPad Prism 5.01 software (GraphPad Software,
Inc., La Jolla, CA, USA).
Cell migration
Cells (1×106)were seeded in 6-well tissue
culture plates. Cells were scratched with a sterile pipette tip to
generate artificial wounds. Phase contrast images were taken using
the microscope Nikon-Eclipse Ti-S (Nikon Corporation, Tokyo, Japan)
to monitor wound closure. The second set of phase contrast images
was taken after 24 h of migration. The area of migration into the
artificial wound was analyzed using ImageJ 1.44p.
Array comparative genomic
hybridization (aCGH)
Genomic DNA was isolated from SNU449 and HLF
hepatoma cells including parental cells, Axl− single
cell clones and a control clones expressing nickase without gRNAs
using the QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany)
according to the manufacturer's protocol. Direct and indirect aCGH
analyses were performed using human whole genome
oligonucleotide-based microarrays (SNU449: Cancer Research Array +
single nucleotide polymorphism, 2×400K; HLF: 4×44K; both arrays
from Agilent Technologies, Inc., Santa Clara, CA, USA). Normal
human male DNA (Agilent Technologies, Inc.) or parental
SNU449-derived DNA were used as reference samples in case of direct
and indirect aCGH experiments, respectively. Labeling and
hybridization was carried out following the protocols provided by
the manufacturer. Slides were scanned with a G2600D Microarray
Scanner (Agilent Technologies, Inc.). Feature extraction and data
analysis were performed using the Feature Extraction and Agilent
Genomic Workbench software (Agilent Technologies, Inc.),
respectively.
Statistics
Data were expressed as means ± standard deviations.
The statistical significance of differences was evaluated using a
one-way analysis of variance followed by a Tukey's post hoc test.
*P<0.05, **P<0.01 and ***P<0.005 were considered to
indicate a statistically significant difference.
Results
Sequence analysis of the AXL locus in
CRISPR/Cas9-edited hepatoma cells
To address the role of Axl signaling in HCC cells,
we aimed at generating Axl-deficient HLF and SNU449 hepatoma cells.
Both HLF and SNU449 cells were previously shown to exhibit a
hyperdiploid DNA status (30,31). To accomplish this task, we performed
CRISPR/Cas9-mediated genomic editing using a pair of gRNAs
targeting the genomic AXL locus together with the expression
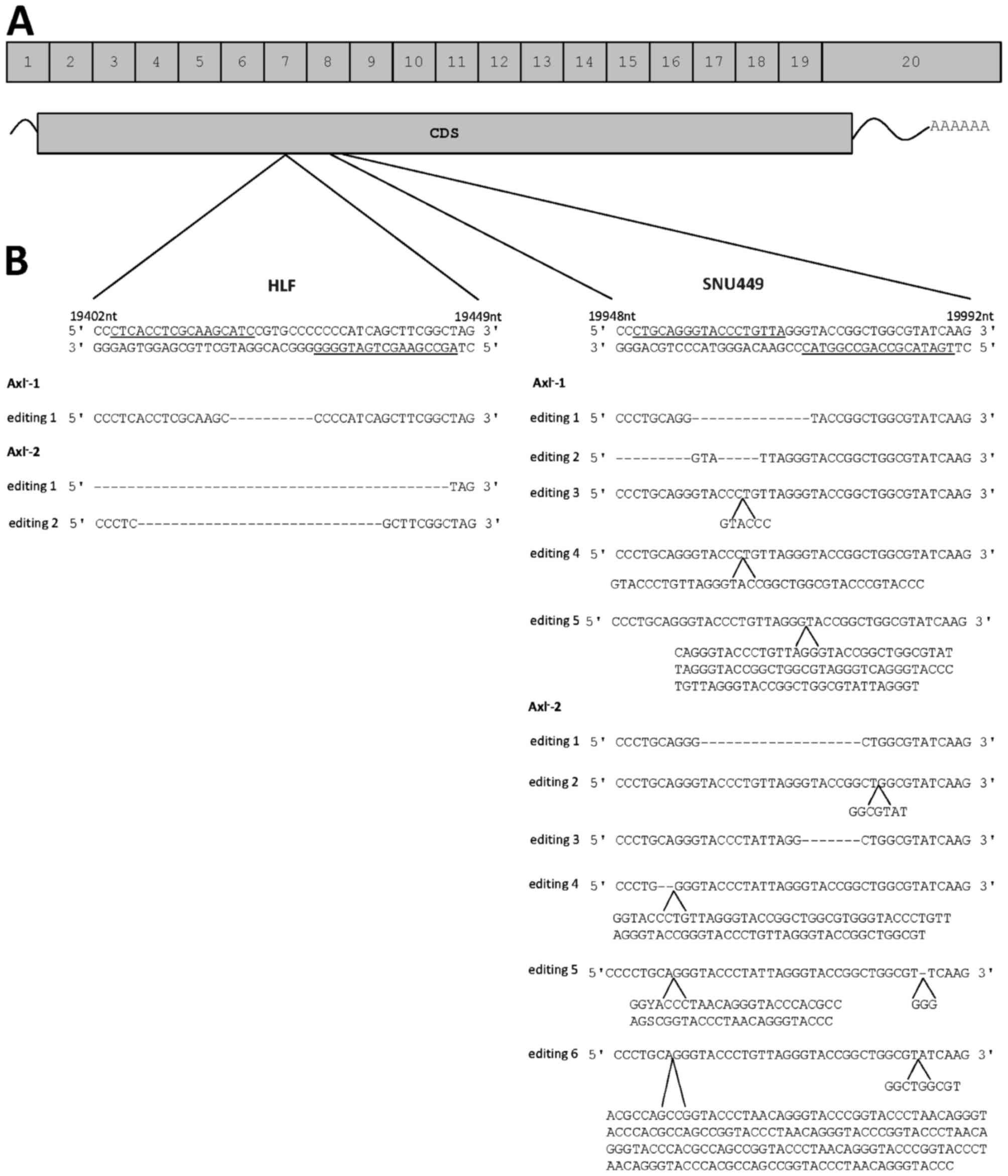
of nickase. Genomic editing was induced in exon 7 or 8 depending on
the pair of gRNAs used (Fig. 1A). The
genomic editing events of two single cell clones each from HLF and
SNU449 cells were analyzed after PCR amplification of the
AXL locus by sequencing of 20 bacterial colonies each. In
HLF cells, a gRNA pair was used that binds to DNA in Exon 7
(Fig. 1A and B). By analyzing the two
HLF single cell clones, the first supposed Axl-negative clone,
designated HLF-Axl−-1, showed one genomic editing event
with one defined deletion (Fig. 1B).
Sequencing of the second Axl− clone
(HLF-Axl−-2) revealed two genomic editing events
corresponding to deletions of various lengths (Fig. 1B). In SNU449 cells, the used gRNA pair
bound to exon 8 of the AXL gene (Fig. 1A and B). Notably, one Axl−
clone (SNU449-Axl−-1) showed five genomic editing
events, where two events were deletions and the other three ones
resulted in insertions (Fig. 1B). The
second Axl− clone (SNU449-Axl−-2) exhibited
six different genomic editing events consisting of two deletions,
two insertions and two events resulting in a combination of InDels
(Fig. 1B). From these data we
conclude that multiple genomic editing events occurred at the
AXL loci that were differentially affected by independent
gRNA/Cas9 complexes.
Mutations of the AXL locus in
CRISPR/Cas9-edited cells
As a next step we addressed the question of the
consequences of these genomic editing events on Axl protein
expression. All InDels that are not a multiple of three resulted in
frameshift mutations, and as a consequence in premature stops of
translation (Table I). However, the
genomic editing event not resulting in a frameshift and premature
stop was editing event 3 of SNU449-Axl−-1, where six
basepairs were inserted into the genomic AXL locus, which
resulted in an insertion of two amino acids in the extracellular
domain of Axl (Fig. 1B, Table I). In summary, these data suggest that
CRISPR/Cas9-mediated genomic editing may result in the expected
protein deficiency (HLF-Axl−-1, HLF-Axl−-2
and SNU449-Axl−-2) or in the incomplete knockout of the
Axl protein expression based on a single allele editing event
(SNU449-Axl−-1).
 | Table I.Genomic editing by CRISPR/Cas9 in HLF
and SNU449 cells. |
Table I.
Genomic editing by CRISPR/Cas9 in HLF
and SNU449 cells.
| Cell type | Editing event | Insertion (bp) | Deletion (bp) | Effect on
protein |
|---|
|
HLF-Axl−-1 | Editing 1 |
| 10 |
Frameshift-premature stop |
|
HLF-Axl−-2 | Editing 1 |
| 73 |
Frameshift-premature stop |
|
| Editing 2 |
| 29 |
Frameshift-premature stop |
|
SNU449-Axl−-1 | Editing 1 |
| 14 |
Frameshift-premature stop |
|
| Editing 2 |
| 24+5 |
Frameshift-premature stop |
|
| Editing 3 | 6 |
| 2 AA insertion |
|
| Editing 4 | 37 |
|
Frameshift-premature stop |
|
| Editing 5 | 95 |
|
Frameshift-premature stop |
|
SNU449-Axl−-2 | Editing 1 |
| 19 |
Frameshift-premature stop |
|
| Editing 2 | 7 |
|
Frameshift-premature stop |
|
| Editing 3 |
| 7 |
Frameshift-premature stop |
|
| Editing 4 | 77 | 2 |
Frameshift-premature stop |
|
| Editing 5 | 49+3 | 1 |
Frameshift-premature stop |
|
| Editing 6 | 176+9 |
|
Frameshift-premature stop |
Expression of Axl in
CRISPR/Cas9-edited cells
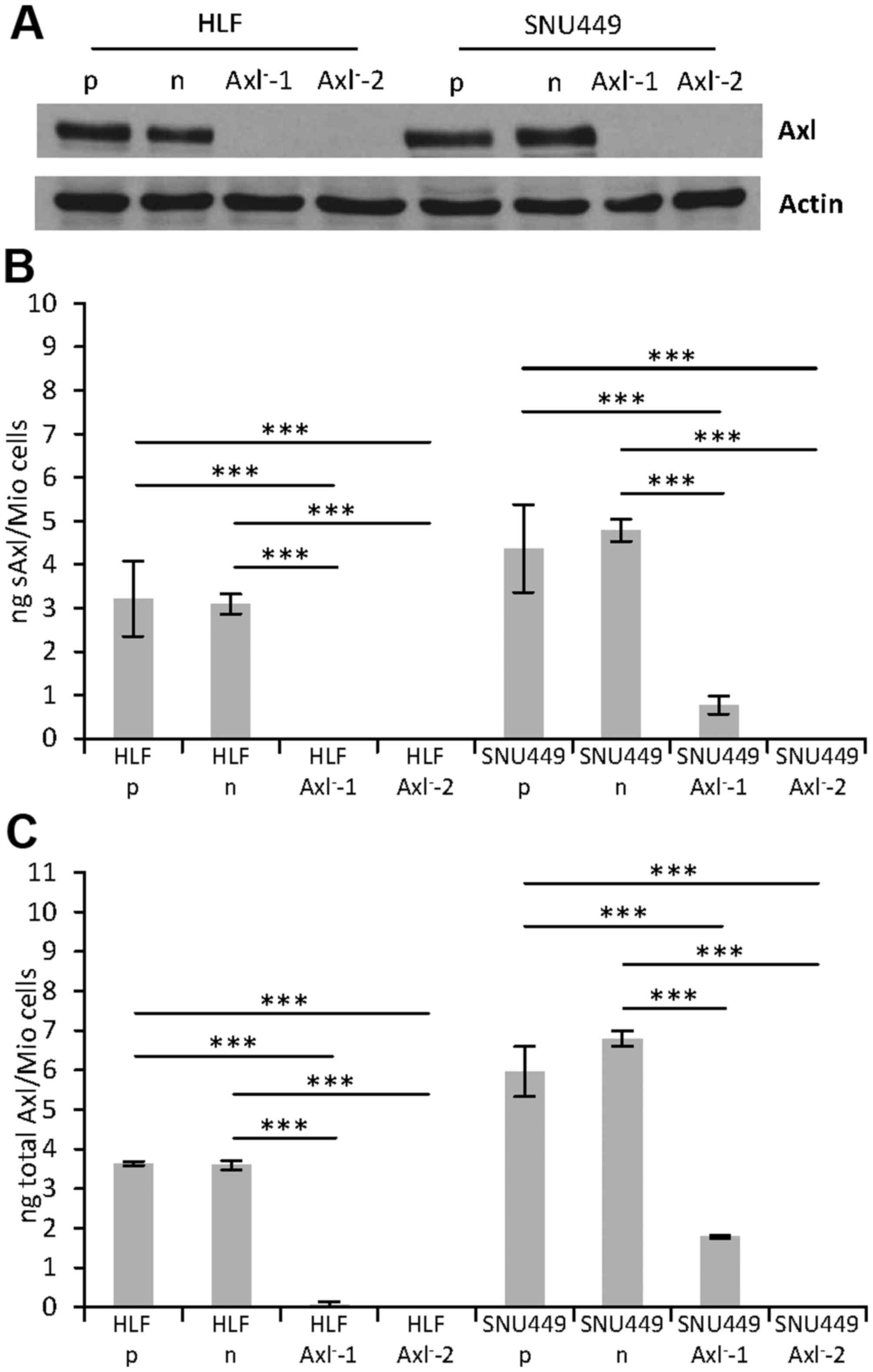
We next performed western blot analysis to detect
the effects of the genomic editing events on Axl protein
expression. Interestingly, no protein expression was observed in
each of the HLF-Axl− and SNU449-Axl− single
cell clones as compared to the HLF and SNU449 parental cells (p) or
control HLF and SNU449 cells (n) expressing nickase without gRNAs
(Fig. 2A). Furthermore, enzyme-linked
immunosorbent assays (ELISAs) were performed to detect very low
levels of Axl protein expression that could not be detected by the
less sensitive western blot analysis. Thereby, we measured both the
cleaved extracellular domain of Axl, termed soluble Axl (sAxl) in
supernatants (Fig. 2B) as well as the
full length Axl receptor in protein lysates (total Axl; Fig. 2C). Most notably, while no protein
expression was observed in HLF-Axl−-1, Axl−-2
and SNU449-Axl−-2 cells, significant expression of sAxl
and total Axl was detected in SNU449-Axl−-1 cells. Axl
expression in this single cell clone confirmed the incomplete
knockout (Fig. 1B, Table I), resulting in a 4-fold reduced
expression to 24.7% of combined Axl values (sAxl + total Axl) as
compared to parental SNU449-p cells (Table II). Noteworthy, all other
CRISPR/Cas9-edited HLF and SNU449 single cell clones displayed no
Axl expression at all (HLF-Axl−-2, and
SNU449-Axl−-2) or showed very low Axl levels that were
below the detection limit and thus considered as signal noise
(HLF-Axl−-1; Table II).
Together, these data demonstrate that the genomic editing is
incomplete in multi-allelic hepatoma cell lines, as three of four
clones show deficiency of Axl and one clone maintains Axl
expression despite genomic editing.
| Table II.Axl protein levels in CRISPR/Cas9
edited HLF and SNU449 cells. |
Table II.
Axl protein levels in CRISPR/Cas9
edited HLF and SNU449 cells.
| Cell type | sAxl (%) | Total Axl (%) | Combined Axl
(%) |
|---|
| HLF-p | 100 | 100 | 100 |
| HLF-n | 96.18 | 98.83 | 97.58 |
|
HLF-Axl−-1 | 0 | 1.93 | 1.02 |
|
HLF-Axl−-2 | 0 | 0 | 0 |
| SNU449-p | 100 | 100 | 100 |
| SNU449-n | 109.56 | 113.93 | 112.08 |
|
SNU449-Axl−-1 | 17.66 | 29.88 | 24.7 |
|
SNU449-Axl−-2 | 0 | 0 | 0 |
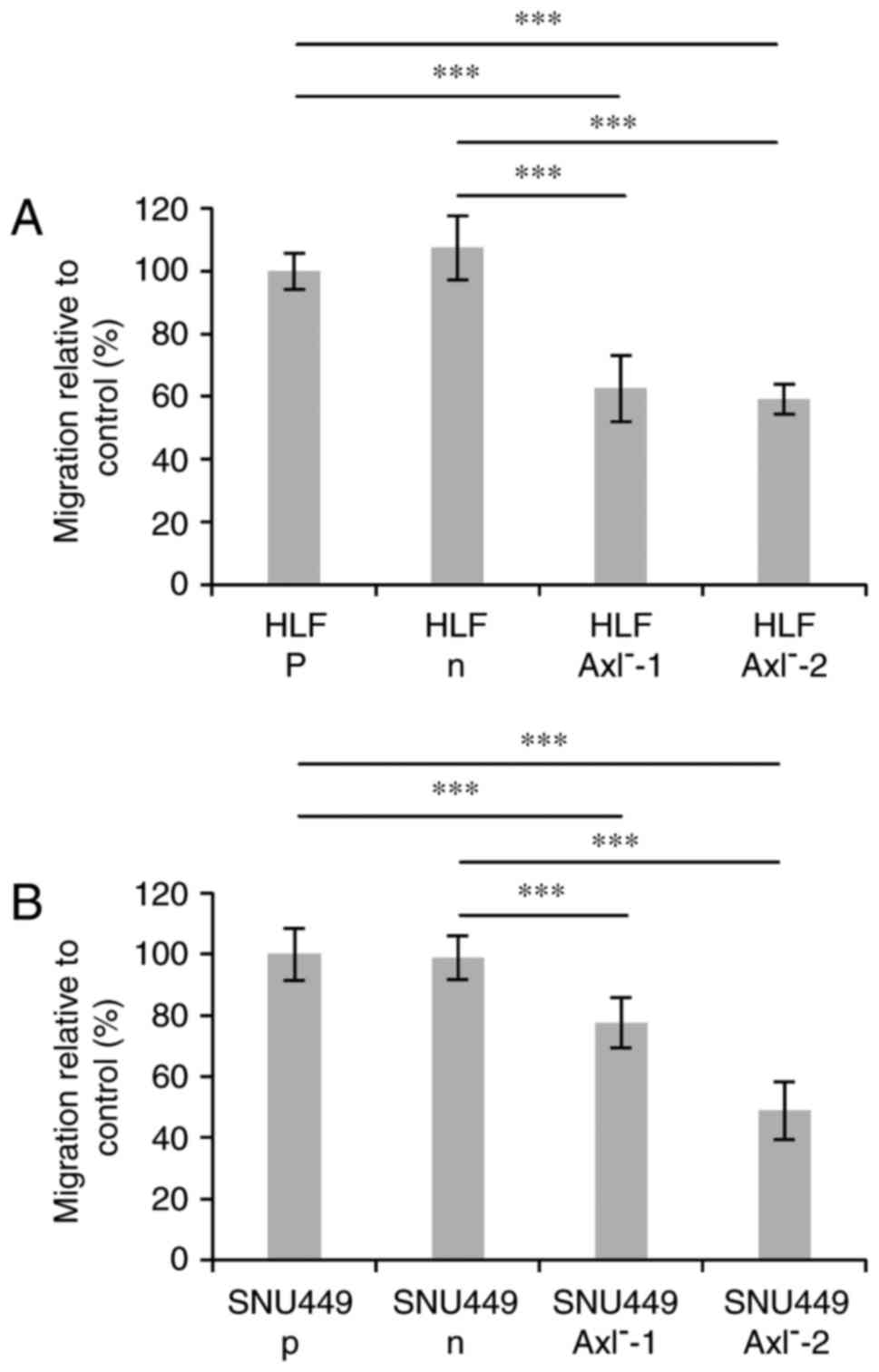
Migration of CRISPR/Cas9-edited
hepatoma cells
Our recent data demonstrated that the siRNA-mediated
loss of Axl expression caused a decrease of cell motility of human
hepatoma cells (27). Therefore, we
next analyzed the effects of the genomic editing events on the
migratory cell behavior by performing a wound healing assay. A
significant difference in migration was observed between the
parental cells (p) or cells expressing nickase without gRNAs (n)
and the Axl− clones in both, HLF and SNU449 cells
(Fig. 3A and B).
HLF-Axl−-1 and HLF-Axl−-2 cells showed
comparable levels of migration which is around 40% reduced when
compared to HLF-n or HLF-p cells. Interestingly, migration of
SNU449-Axl−-1 cells was reduced by approximately 20%,
whereas SNU449-Axl−-2 cells displayed half of the
migration level of SNU449-n or SNU-449-p cells. In conclusion, the
CRISPR/Cas9-edited hepatoma cells show a clear phenotype of
diminished migration, thus confirming recent data obtained with
siRNA. In addition, the incompletely edited
SNU449-Axl−-1 cells display an inhibition of migration
with a lesser extent than those cells with a complete knockout
(HLF-Axl−, SNU449-Axl−-2), indicating a close
correlation between the genotype and the phenotype.
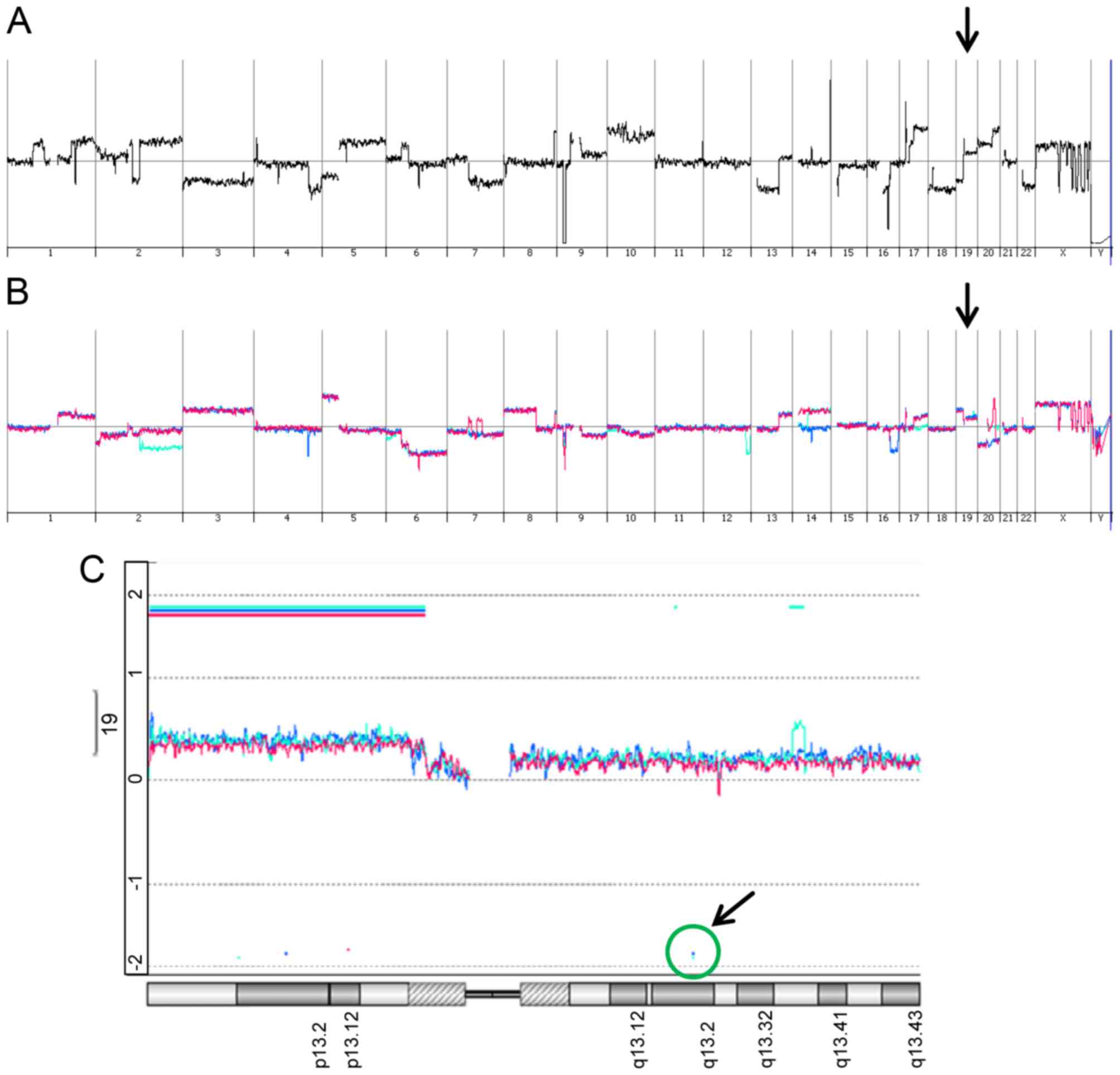
Genomic changes in CRISPR/Cas9-edited
hepatoma cells
We further addressed the question on the gain and
loss of genomic DNA in parental SNU449-p, control SNU449-n cells
and SNU449-Axl− single cell clones by employing direct
(SNU449-p) and indirect (control SNU449-n,
SNU449-Axl−-1, SNU449-Axl−-2) array
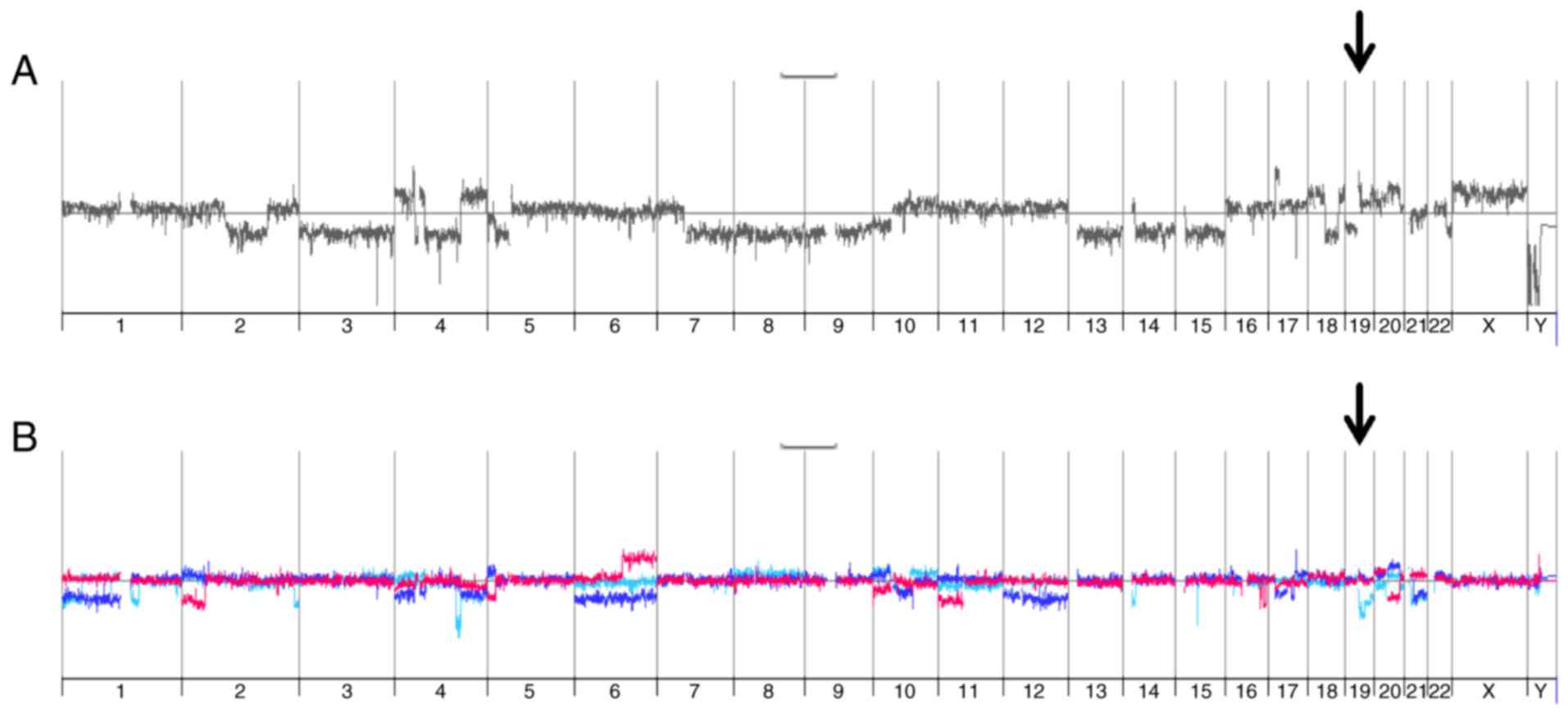
comparative genomic hybridization (aCGH) analyses. As expected,
SNU449-p cells showed vast changes in genomic DNA as compared to
the diploid reference DNA by multiple gains and losses (Fig. 4A). In more detail, these alterations
included on the one hand focal deletions/amplifications such as
e.g. deletions at chromosome 9p21 (CDKN2A) and 16q21 (CDH8, CDH11)
and amplifications at 17p11.2 (MAPK7, MFAP4). On the other hand,
these genomic changes involved gains and losses of whole
chromosomes/chromosome arms such as loss of chromosome 3, loss of
chromosome 7q, and gain of chromosome 10. Importantly, SNU449-n
cells showed multiple differences in genomic DNA alterations as
compared to SNU449-p cells (Fig. 4B,
red line), suggesting that the constitutive expression of nickase
induced changes in the genomic DNA despite the absence of gRNAs. A
very similar pattern of genomic alterations was observed in both
SNU449-Axl−-1 and SNU449-Axl−-2 cells
expressing gRNAs (Fig. 4B, turquoise
and blue lines, respectively), suggesting that a multitude of
genomic changes in control SNU449-n, SNU449-Axl−-1 and
SNU449-Axl−-2 cells is primarily caused by nickase
activity.
SNU449-p cells showed a gain at the AXL locus
with a mean log2 ratio of 0.3 (data not shown).
SNU449-n, SNU449-Axl−-1 and SNU449-Axl−-2
cells each displayed an additional low level gain of chromosome 19q
at the AXL locus (Fig. 4C).
However, both SNU449-Axl−-1 and SNU449-Axl−-2
cells displayed the expected deletion of the AXL locus as a
decrease in AXL-specific genomic DNA could be detected
(Fig. 4C), thus confirming
CRISPR/Cas9-specific genomic editing events. In particular, five
and four AXL-specific oligonucleotides indicated a loss of
genomic DNA in SNU449-Axl−-1 and
SNU449-Axl−-2 cells between nucleotide 20198 and 21255
as well as 21179 and 22539 of the AXL gene, respectively.
Both regions encompassing the AXL-specific oligonucleotides
were located downstream of the gRNA binding sites.
We performed additional aCGH analyses of parental
HLF cells, one HLF clone derived from nickase-treated cells and two
Axl knockout clones. Comparable to the results obtained in SNU449
cells, parental HLF cells showed a multitude of gains and losses
which is characteristic for cancer cells, thus mirroring a highly
aberrant and re-arranged genome (Fig.
5A). In these cells as well, the chromosomal region 19q13.2
containing the AXL locus showed a low level gain (about
1.2-fold) as compared to normal diploid cells (data not shown).
Interestingly, both Axl knockout clones as well as the clone
derived from the nickase treated cells displayed various new,
additional changes as compared to parental HLF cells (Fig. 5B). Together, these data provide
evidence that high resolution aCGH detects CRISPR/Cas9-mediated
genomic editing of AXL. Furthermore, the expression of
nickase alone rather than the CRISPR/Cas9-dependent editing of the
AXL locus causes pleiotropic gene-dose changes in the genome
of established HCC cell lines.
Discussion
Despite the extraordinary achievements of the
CRISPR/Cas9 technology in genome engineering, its application still
poses a challenging task in hyperdiploid cancer cell models. The
data obtained in this study strongly suggest that ploidy and gene
copy number play an important role in the efficacy of the
CRISPR-dependent genomic editing. Two recent studies demonstrated
that CRISPR/Cas9 is not suitable in cellular cancer models showing
gene amplifications, as overrepresented genomic regions cause false
positive results. Aguirre et al (32) provided evidence that DNA breaks
generated by the CRISPR/Cas9 system lead to gene-independent cell
toxicity and the number of DNA cuts rather than the gene knockout
predicts the cellular response. Munoz et al (33) found a higher number of lethal genes
when using CRISPR/Cas9 as compared to RNAi. In aneuploid cancer
cell models, the genes located in amplified regions scored as
lethal, indicating false positive results as already described by
Aguirre et al (32). Thus, the
number of loci representing the gene of interest in a cancer cell
line under investigation is of crucial importance for the
physiological response to CRISPR/Cas9 and for the experimental
interpretation.
Established cancer cell lines frequently represent a
pool of genetically heterogeneous subclones with varying numbers of
chromosomes and different physiological behavior. These progressive
genomic alterations and aneuploidy might drive a high degree of
genomic instability leading to new mutations and gene-dose
alterations over time and thus give rise to new subclones (34). The predominance of such subclones can
change depending on time and conditions of cultivation. Upon
selection of CRISPR/Cas9-positive cells, expansion of single cells
harbor the risk of developing a phenotype not specific for the
knockout as cells go through multiple cell divisions in which
compensatory mechanisms could arise due to the loss of the target
protein. Even without any genetic manipulation, single cell clones
can behave differently from the original cancer cell population and
might not be representative for a certain cancer cell model. Thus,
the use of CRISPR/Cas9 might have drawbacks in cancer cell lines as
the reliable evaluation of experiments is less predictable in the
absence of data about target gene copies and the dynamic modulation
of subclone heterogeneity also under the stress of clonal
selection.
Notably, the expression of nickase in the absence of
gRNAs in SNU449 and HLF cells causes multiple genomic changes
comparable to those in CRISPR/Cas9-processed cells in the presence
of gRNAs (Figs. 4B and 5B). These data show that gRNA expression
results in specific editing of the targeted AXL locus and
further suggest that cells expressing nickase without gRNAs
represent the most suitable control for CRISPR/Cas9-edited cells.
In this context, however, it is an open issue whether the genomic
changes observed in nickase-only expressing cells are due to the
pleiotropic activity of nickase or due to the selection of a
favorable subclone from heterogenous SNU449-p or HLF-p cells. Thus,
further aCGH profiling of nickase-only expressing cells vs.
representative subclones of parental hepatoma cells could clarify
the impact of clonal selection vs. nickase activity and the
evolution of genomic alterations as compared to the parental cell
pool.
In order to reduce unspecific genomic changes by the
nickase, CRISPR-Cas9 inhibitory proteins could be expressed to
inhibit the activity of Cas9 after genomic editing (35,36). The
prophage-encoded inhibitor proteins AcrIIA2 and AcrIIA4, which
allow phages to evade the bacterial host's CRISPR/Cas immune
system, were found to inhibit Cas9-based targeting in their native
host Listeria monocytogenes, as well as Cas9 of
Streptococcus pyogenes in bacteria and human cells. Another
approach for minimizing off-target effects employs the delivery of
Cas9 protein/gRNA ribonucleoprotein complexes (37). When delivering Cas9 protein directly,
cleavage occurs only temporarily, as the Cas9 protein is rapidly
degraded in cells (38).
The confirmation of the desired gene knockout
requires the determination of target protein levels in a large
number of single cell clones using immunoblotting and the more
sensitive ELISA. Interestingly, this study showed different levels
of Axl protein in SNU449-Axl−-1 cells when detected
either by immunoblotting or ELISA (Fig.
2), albeit the same polyclonal Axl-specific antibody was
employed in both methods. We speculate that the insertion of two
additional amino acids into the AXL locus of
CRISPR/Cas9-edited SNU449-Axl−-1 cells (Fig. 1B, Table
I) accounts for changes in the protein structure or the
stability of the Axl protein allowing detection in its native form
by ELISA but no detection of the reduced form by immunoblotting.
Obviously one incompletely edited copy of AXL was enough to
produce considerable amount of protein that is detectable by ELISA.
In accordance with these data, both SNU449-Axl− clones
exhibited a significant reduction of the migratory phenotype, which
was less pronounced in incompletely edited SNU449-Axl−-1
cells (Fig. 3).
Furthermore, 20 bacterial colonies of the
AXL-specific PCR-amplified region were each analyzed by
sequencing in order to verify the complete knockout. As 20 analyses
might not cover all genomic changes, we suggest applying next
generation sequencing (NGS) methods to identify all InDels in the
targeted gene locus. NGS analysis could be even employed at various
passage numbers during selection in order to examine the dynamics
of subclonal evolution. In case of many alleles and an incomplete
knockout, one intact and functional target gene copy, even if not
detected in the beginning, could provide a particular growth
advantage in culture over time.
Loss-of-function studies employing CRISPR/Cas9,
TALEN or stable RNAi involve selection of edited cells through
multiple cell divisions in which compensatory mechanisms could
arise due to the loss/knockdown of the target protein, resulting in
altered cellular phenotypes. Notably, transient RNAi by siRNA
transfection could overcome such drawbacks as cells are not
subjected to selection and might not adapt to environmental
conditions. Thus, siRNA-induced changes are more likely due to the
downregulation of the target protein rather than to an artifact
derived from clonal selection. Since transient RNAi is only used
for short-term analysis, we suggest to compare e.g. the CRISPR/Cas9
phenotype with the one obtained by transient RNAi, both of which
must coincide in phenotypical characteristics in vitro.
In conclusion, the CRISPR/Cas9 system has the
advantage of generating a complete and stable knockout which can be
successfully used in cancer cell lines by considering the above
mentioned aspects. Once the knockout is confirmed and selection
artefacts can be excluded, a robust and trustworthy system has been
established.
Acknowledgements
The present study was supported by the Austrian
Science Fund, FWF, P25356 and the Herzfelder Family Foundation.
Glossary
Abbreviations
Abbreviations:
|
aCGH
|
array comparative genomic
hybridization
|
|
Cas9
|
CRISPR associated 9
|
|
CDS
|
coding sequence
|
|
CRISPR
|
clustered regularly interspaced short
palindromic repeats
|
|
DSB
|
double-strand break
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
gRNA
|
guide ribonucleic acid
|
|
HDR
|
homology-directed repair
|
|
HCC
|
hepatocellular carcinoma
|
|
InDels
|
insertions and deletions
|
|
NGS
|
next generation sequencing
|
|
NHEJ
|
non-homologous end joining
|
|
ORF
|
open reading frame
|
|
PCR
|
polymerase chain reaction
|
|
RNAi
|
ribonucleic acid interference
|
|
sAxl
|
soluble Axl
|
|
shRNA
|
small hairpin ribonucleic acid
|
|
siRNA
|
small interfering ribonucleic acid
|
|
SN
|
supernatant
|
|
TALEN
|
transcription activator-like effector
nucleases
|
References
|
1
|
Davidson BL and Paulson HL: Molecular
medicine for the brain: Silencing of disease genes with RNA
interference. Lancet Neurol. 3:145–149. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xia H, Mao Q, Paulson HL and Davidson BL:
siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol.
20:1006–1010. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miller VM, Xia H, Marrs GL, Gouvion CM,
Lee G, Davidson BL and Paulson HL: Allele-specific silencing of
dominant disease genes. Proc Natl Acad Sci USA. 100:7195–7200.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gaj T, Gersbach CA and Barbas CF III: ZFN,
TALEN, and CRISPR/Cas-based methods for genome engineering. Trends
Biotechnol. 31:397–405. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liberali P, Snijder B and Pelkmans L:
Single-cell and multivariate approaches in genetic perturbation
screens. Nat Rev Genet. 16:18–32. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Joung JK and Sander JD: TALENs: A widely
applicable technology for targeted genome editing. Nat Rev Mol Cell
Biol. 14:49–55. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller JC, Tan S, Qiao G, Barlow KA, Wang
J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al: A TALE
nuclease architecture for efficient genome editing. Nat Biotechnol.
29:143–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Harrison MM, Jenkins BV, O'Connor-Giles KM
and Wildonger J: A CRISPR view of development. Genes Dev.
28:1859–1872. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jinek M, Chylinski K, Fonfara I, Hauer M,
Doudna JA and Charpentier E: A programmable dual-RNA-guided DNA
endonuclease in adaptive bacterial immunity. Science. 337:816–821.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hryhorowicz M, Lipinski D, Zeyland J and
Slomski R: CRISPR/Cas9 immune system as a tool for genome
engineering. Arch Immunol Ther Exp (Warsz). 65:233–240. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lopes R, Korkmaz G and Agami R: Applying
CRISPR-Cas9 tools to identify and characterize transcriptional
enhancers. Nat Rev Mol Cell Biol. 17:597–604. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang W, Bikard D, Cox D, Zhang F and
Marraffini LA: RNA-guided editing of bacterial genomes using
CRISPR-Cas systems. Nat Biotechnol. 31:233–239. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mali P, Yang L, Esvelt KM, Aach J, Guell
M, DiCarlo JE, Norville JE and Church GM: RNA-guided human genome
engineering via Cas9. Science. 339:823–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai
SQ, Sander JD, Peterson RT, Yeh JR and Joung JK: Efficient genome
editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol.
31:227–229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
DiCarlo JE, Norville JE, Mali P, Rios X,
Aach J and Church GM: Genome engineering in Saccharomyces
cerevisiae using CRISPR-Cas systems. Nucleic Acids Res.
41:4336–4343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bassett AR, Tibbit C, Ponting CP and Liu
JL: Highly efficient targeted mutagenesis of Drosophila with the
CRISPR/Cas9 system. Cell Rep. 4:220–228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang H, Yang H, Shivalila CS, Dawlaty MM,
Cheng AW, Zhang F and Jaenisch R: One-step generation of mice
carrying mutations in multiple genes by CRISPR/Cas-mediated genome
engineering. Cell. 153:910–918. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu
M, Li Y, Gao N, Wang L, Lu X, et al: Heritable gene targeting in
the mouse and rat using a CRISPR-Cas system. Nat Biotechnol.
31:681–683. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang D, Xu J, Zhu T, Fan J, Lai L, Zhang J
and Chen YE: Effective gene targeting in rabbits using RNA-guided
Cas9 nucleases. J Mol Cell Biol. 6:97–99. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Essletzbichler P, Konopka T, Santoro F,
Chen D, Gapp BV, Kralovics R, Brummelkamp TR, Nijman SM and
Bürckstümmer T: Megabase-scale deletion using CRISPR/Cas9 to
generate a fully haploid human cell line. Genome Res. 24:2059–2065.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brinkman EK, Chen T, Amendola M and van
Steensel B: Easy quantitative assessment of genome editing by
sequence trace decomposition. Nucleic Acids Res. 42:e1682014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nemudryi AA, Valetdinova KR, Medvedev SP
and Zakian SM: TALEN and CRISPR/Cas genome editing systems: Tools
of discovery. Acta Naturae. 6:19–40. 2014.PubMed/NCBI
|
|
24
|
Boettcher M and McManus MT: Choosing the
right tool for the job: RNAi, TALEN, or CRISPR. Mol Cell.
58:575–585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roschke AV and Rozenblum E: Multi-layered
cancer chromosomal instability phenotype. Front Oncol. 3:3022013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Teoh NC, Dan YY, Swisshelm K, Lehman S,
Wright JH, Haque J, Gu Y and Fausto N: Defective DNA strand break
repair causes chromosomal instability and accelerates liver
carcinogenesis in mice. Hepatology. 47:2078–2088. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reichl P, Dengler M, van Zijl F, Huber H,
Führlinger G, Reichel C, Sieghart W, Peck-Radosavljevic M,
Grubinger M, Mikulits W, et al: Axl activates autocrine
transforming growth factor-β signaling in hepatocellular carcinoma.
Hepatology. 61:930–941. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reichl P, Fang M, Starlinger P, Staufer K,
Nenutil R, Muller P, Greplova K, Valik D, Dooley S, Brostjan C, et
al: Multicenter analysis of soluble Axl reveals diagnostic value
for very early stage hepatocellular carcinoma. Int J Cancer.
137:385–394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Petz M, Them N, Huber H, Beug H and
Mikulits W: La enhances IRES-mediated translation of laminin B1
during malignant epithelial to mesenchymal transition. Nucleic
Acids Res. 40:290–302. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hwang HJ, Kim GJ, Lee GB, Oh JT, Chun YH
and Park SH: A comprehensive karyotypic analysis on Korean
hepatocellular carcinoma cell lines by cross-species color banding
and comparative genomic hybridization. Cancer Genet Cytogenet.
141:128–137. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dor I, Namba M and Sato J: Establishment
and some biological characteristics of human hepatoma cell lines.
Gan. 66:385–392. 1975.PubMed/NCBI
|
|
32
|
Aguirre AJ, Meyers RM, Weir BA, Vazquez F,
Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et
al: Genomic copy number dictates a gene-independent cell response
to CRISPR/Cas9 targeting. Cancer Discov. 6:914–929. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Munoz DM, Cassiani PJ, Li L, Billy E, Korn
JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G, et al: CRISPR
screens provide a comprehensive assessment of cancer
vulnerabilities but generate false-positive hits for highly
amplified genomic regions. Cancer Discov. 6:900–913. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hastings RJ and Franks LM: Cellular
heterogeneity in a tissue culture cell line derived from a human
bladder carcinoma. Br J Cancer. 47:233–244. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rauch BJ, Silvis MR, Hultquist JF, Waters
CS, McGregor MJ, Krogan NJ and Bondy-Denomy J: Inhibition of
CRISPR-Cas9 with bacteriophage proteins. Cell. 168(150–158):
e1102017.
|
|
36
|
Pawluk A, Amrani N, Zhang Y, Garcia B,
Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL
and Davidson AR: Naturally occurring off-switches for CRISPR-Cas9.
Cell. 167:1829–1838.e9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang X, Potter J, Kumar S, Zou Y,
Quintanilla R, Sridharan M, Carte J, Chen W, Roark N and
Ranganathan S: Rapid and highly efficient mammalian cell
engineering via Cas9 protein transfection. J Biotechnol. 208:44–53.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim S, Kim D, Cho SW, Kim J and Kim JS:
Highly efficient RNA-guided genome editing in human cells via
delivery of purified Cas9 ribonucleoproteins. Genome Res.
24:1012–1019. 2014. View Article : Google Scholar : PubMed/NCBI
|