Introduction
Celastrol is a pharmacologically active compound
that was originally isolated from Thunder God Vine (Tripterygium
wilfordii; traditional Chinese medicine). Celastrol was
identified to be a pentacyclic triterpenoid, belonging to a small
category of natural products of triterpene quinine methides
(1,2).
Containing electrophilic sites within the rings of quinone methide
structure, celastrol is able to form covalent Michael adducts with
the nucleophilic thiol groups of cysteine residues (3,4). This
appears to be the mechanism underlying celastrol-mediated effects
on the functions of various proteins, although the structural
determinants in proteins can also regulate their interaction with
celastrol and the covalent adducts formation (5).
Numerous previous studies have indicated that
celastrol may protect against a variety of inflammatory diseases in
animal models (6,7). Furthermore, celastrol is a promising
anticancer drug, which can suppress the proliferation of various
cancer cells and prevent their malignant tissue invasion and
obstruct angiogenesis (8–10). Certain therapeutic studies also
demonstrated that celastrol can sensitize resistant melanoma cell
to temozolomide treatment and potentiate radiotherapy of prostate
cancer cells in combination therapy (11,12).
Signal transducer and activator of transcription 3
(STAT3) has been identified as a key molecular target of celastrol
(13,14); however, the mechanism underlying the
effects of celastrol on STAT3 remains to be elucidated. In
therapeutic investigations of celastrol, certain studies suggest
that celastrol may induce apoptosis and repress invasion of cancer
cells by regulation of microRNA (miRNA) expression levels (15,16).
Therefore, the present study investigated whether the STAT3
signaling pathway is associated with the expression levels of
miRNAs. The present study further investigated whether celastrol
inhibited lung cancer apoptosis via STAT3-associated miRNAs.
Materials and methods
Cell culture
A549 human lung adenocarcinoma cells purchased from
Shanghai Institute of Pharmaceutical Industry (Shanghai, China)
were cultured in RPMI-1640 supplemented with 10% fetal bovine serum
(both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
and 100 U/ml penicillin-streptomycin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) and maintained at 37°C in a humidified
incubator containing 5% CO2. Cells (5×105) in
the logarithmic growth phase were seeded in 6-well plates, and
celastrol (Sigma-Aldrich; Merck KGaA) was dissolved in dimethyl
sulfoxide (DMSO) and added to the plate in complete RPMI-1640
medium when the cell confluence arrived at 70%. Cells were
incubated for 48 h at 37°C. LTEP-a-2, another human lung
adenocarcinoma cell line also came from Shanghai Institute of
Pharmaceutical Industry, were used under the same culture condition
as A549 cells.
MTT assay
Cells (1×104) were cultured into 96-well
plates. Various concentrations of celastrol (0, 1.5, 3 and 4.5 µM)
were added to the medium and 6 replicates were performed for each
concentration. Cells were cultivated for 48 h at 37°C, MTT assays
were performed to determine the cell viability and the growth
inhibition rate of A549 cells. A total of 10 µl MTT (5 mg/ml) was
added to each well and the supernatant was removed by pipette after
another 4 h of incubation at 37°C. Subsequently, 100 µl DMSO
(Sigma-Aldrich; Merck KGaA) was added to dissolve the crystals
produced by MTT at room temperature for 10 min. The optical density
(OD) value was determined using an ELISA reader (ELx800; Bio-Tek
Instruments, Inc., Winooski, VT, USA) at 570 nm. Cell growth
inhibition rate = (OD control - OD sample)/OD control ×100 (%)
(17,18).
Detection of apoptosis
Cells were cultured into 6-well plates and celastrol
was added to the plate following the aforementioned concentration
gradient when the cell confluence was 70% on the following day.
Detection of apoptosis was performed after 48 h incubation at 37°C,
according to the Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) apoptosis detection kit protocol (BD
Biosciences, Franklin Lakes, NJ, USA). Firstly, A549 cells were
gently washed with PBS twice. Subsequently, the cells were
centrifuged for 5 min at 650 × g at room temperature, the
supernatant was discarded and 100 µl 1X binding buffer was added to
each tube for re-suspension at room temperature. Next, 5 µl annexin
V-FITC and 5 µl PI were added and cells were incubated at room
temperature for 15 min. The cells were then analyzed using a flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
A549 cells were treated with the effective
concentration of celastrol (3 µM) for 48 h at 37°C and the control
group was established using an equivalent concentration of DMSO.
miRNA from these cells was isolated using RNAiso (Takara Bio, Inc.,
Otsu, Japan) according to the manufacturer's instruction.
Subsequently, poly(A) was added using poly(A) polymerase (Ambion;
Thermo Fisher Scientific, Inc.). miRNA extraction and adding
poly(A) should follow the respective protocols. Complementary DNA
was synthesized using RT primer 5′-AACATGTACAGTCCATGGATGd(T)30(A,
G, C or T)-3′. miR primers were as follows: miR-24 forward,
5′-CTCCGGTGCCTACTGAGCTGA-3′ and reverse,
5′-AACATGTACAGTCCATGGATG-3′; miR-181b forward,
5′-GGTCACAATCAACATTCATTG-3′ and reverse,
5′-AACATGTACAGTCCATGGATG-3′. Human 5S rRNA was used as reference
gene, and primers were as follows: Forward,
5′-GCCATACCACCCTGAACG-3′ and reverse, 5′-AACATGTACAGTCCATGGATG-3′.
SYBR® Premix Ex Taq™ kit (Takara Bio, Inc.) was used
according to the manufacturer's instructions. The expression levels
of miRNA was assessed using the RG3000 system (Qiagen, Inc.,
Valencia, CA, USA) as follows: Initiation with 3 min of
denaturation at 95°C, followed by 40 cycles of amplification with
20 sec of denaturation at 95°C, 20 sec at 56°C for annealing and 20
sec of extension at 72°C. Fluorescence was detected at 585 nm using
the RG3000 system (Qiagen, Inc.). The above process was repeated 3
times in triplicate. The results ware quantified using the
2−ΔΔCq method as before (19).
Western blotting
Cells were lysed using cold lysis buffer (RIPA;
Beyotime Institute of Biotechnology, Haimen, China) for 30 min on
ice and protein concentration of cell lysis was determined by a BCA
assay (cat no. PC0020; Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China). The experiment followed the
manufacturer's instructions. Subsequently, each 35 µg protein
sample was subjected to SDS-PAGE (12% separating gel, 5%
concentrating gel) for electrophoresis and transferred to
polyvinylidene difluoride membranes. Following transfer, membranes
were incubated in a blocking buffer (5% non-fat milk TBST solution,
TBST: NaCl 0.8%, 0.02% KCl, Tris-base 0.3%, Tween-20 0.1%, pH 7.4)
for 2 h at room temperature. Subsequently, the membranes were
washed 3 times with TBST (10 min each time). Next, the membranes
were immunoblotted with antibodies against p-STAT-3 (1:800;
sc-81523) and STAT-3 (1:800; sc-8019) (both from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), Bcl-2 (1:500; BS1031), BAX
(1:500; BS2538) (both from Bioworld Technology, Inc., St. Louis
Park, MN, USA) and GAPDH (1:3,000; TA309157; Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd., Beijing, China), which was
used as the control. Immunoblotting was for 16–18 h at 4°C and then
washing was performed with TBST 3 times (10 min each time).
Membranes were then incubated with secondary antibodies conjugated
to horseradish peroxidase (1:3000; BS13271 and BS12471; Bioworld
Technology, Inc.). Blots were quantified densitometrically using
Quantity One software (V4.62; Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
miRNAs prediction
The webpage (http://www.microrna.org/microrna/home.do) was opened,
input the name of protein of interest was inputted interested in
the item ‘Target mRNA’ and miRNAs with potential interaction with
the protein and could be confirmed by RT-qPCR were searched
for.
Statistical analysis
Quantitative results are presented as the mean ±
standard deviation. Comparisons of parameters between the two
groups were made using an unpaired Student's t-test or a
Mann-Whitney U test. Statistical significance was evaluated with
GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Celastrol inhibits proliferation and
promotes apoptosis of A549 cells
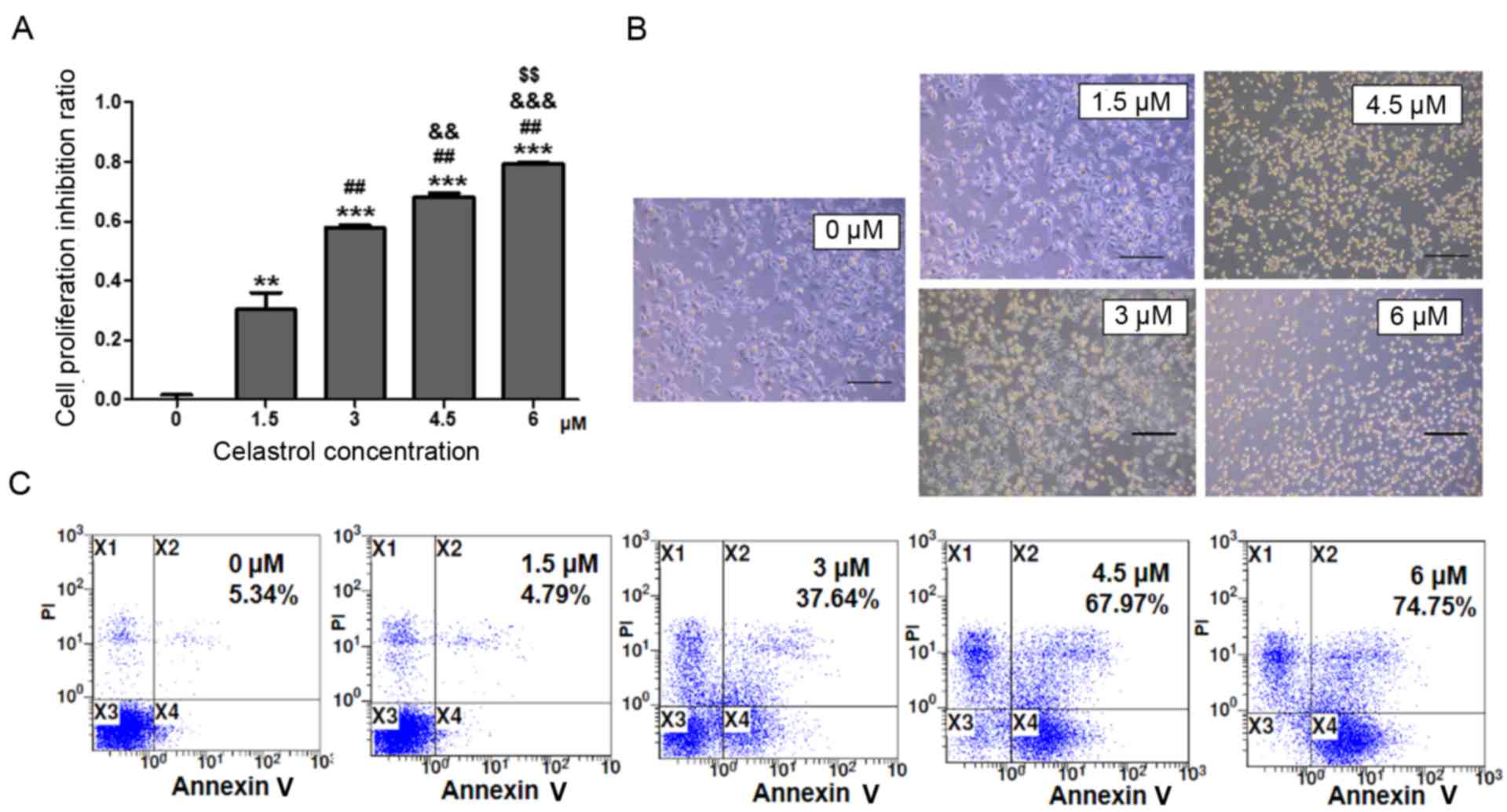
To investigate the effects of celastrol on A549 cell
growth, A549 cells were treated with various concentrations of
celastrol for 24 h. The MTT assay indicated that A549 cell
proliferation was suppressed by celastrol in a dose-dependent
manner. There was a higher level of proliferation suppression of
A549 cells with a higher concentration of celastrol (Fig. 1A). The microcopy examination results
confirmed these findings. There were more dead and floating cells
in the higher celastrol concentration-treated cultures compared
with other cultures (Fig. 1B). In
order to determine whether the decrease of cell number was due to
apoptosis induced by celastrol, Annexin V-FITC and PI staining was
performed. The results revealed that celastrol induced A549 cell
apoptosis in a dose-dependent manner (Fig. 1C). In the concentration cascade, 3 µM
celastrol was the closest to ID50, thus 3 µM celastrol was selected
as the most reasonable concentration in this study.
Celastrol decreases STAT3
phosphorylation and Bcl-2/Bax ratio
Previous studies have revealed the role of celastrol
in the prevention of cancer growth, in which numerous targets of
celastrol were identified, including STAT3 (2,9,10,17). To
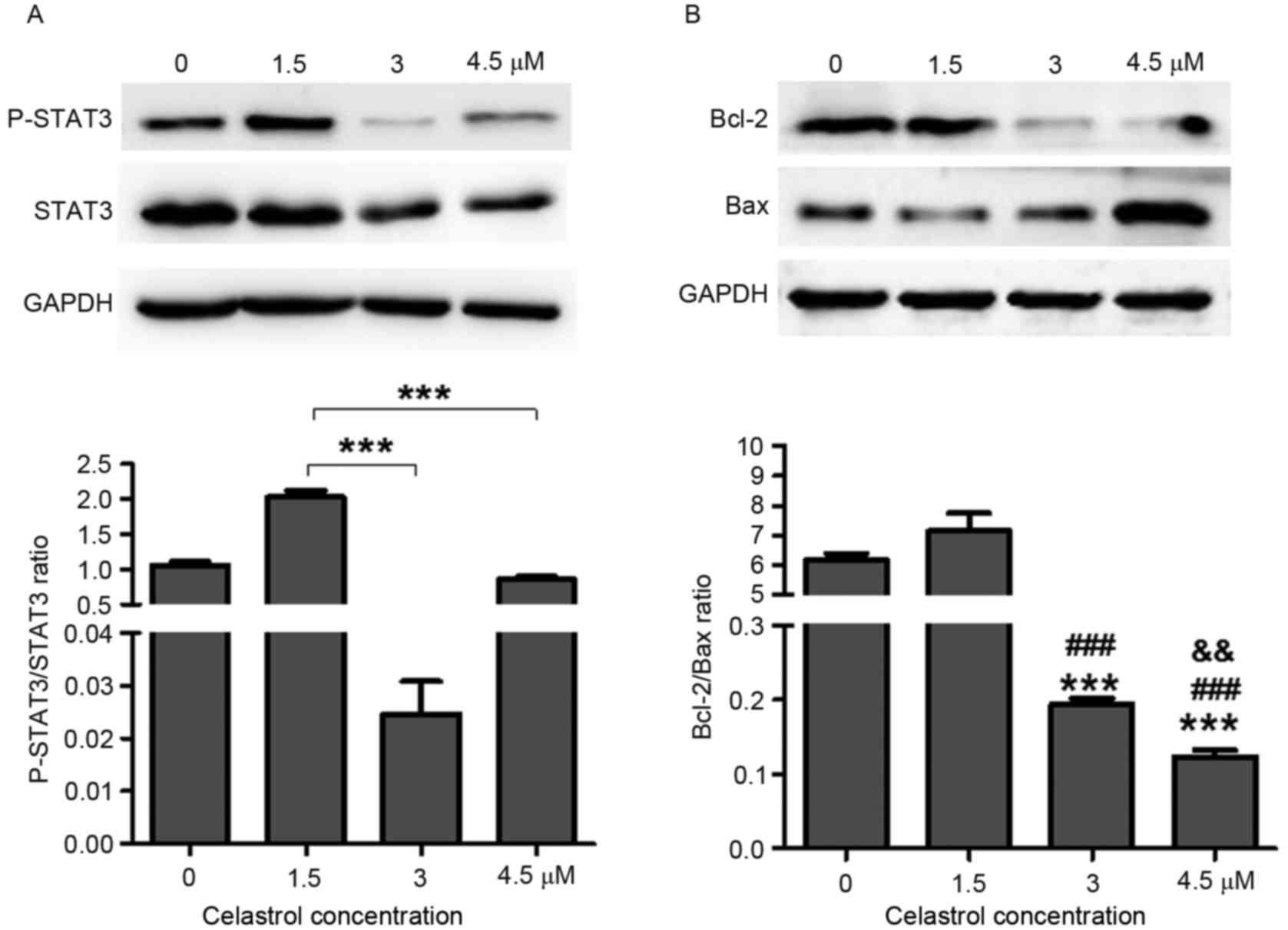
further investigate the role of celastrol in lung cancer, STAT3
expression was detected in A549 cells following treatment with
various concentrations of celastrol. The results demonstrated that
3 µM (P<0.0001) and 4.5 µM (P=0.0003) celastrol treatment
significantly reduced STAT3 phosphorylation in A549 cells compared
with 1.5 µM treatment (Fig. 2A).
STAT3 serves a critical role in cell proliferation
and survival. It has been revealed that deactivation of STAT3
abrogates the anti-apoptotic advantage (such as via radioresistance
and chemoresistance) in various types of tumor cell lines, mainly
due to the attenuation of Bcl-2 (18,20).
Deactivation of the STAT3 signaling pathways by celastrol induced
tumor cell apoptosis (Fig. 1C). The
Bcl-2/Bax ratio was significantly decreased in 3 µM (P<0.0001)
and 4.5 µM (P<0.0001) celastrol-treated cells compared with 0
and 1.5 µM celastrol-treated groups (Fig.
2B). Indeed, 4.5 µM P=0.006) celastrol treatment also could
lead Bcl-2/Bax ratio decreased compared with 3 µM treatment
(Fig. 2B). These results further
suggested higher concentration celastrol treatment attenuated
Bcl-2/Bax ratio more within certain scope. The apoptosis induced by
celastrol may account for deactivation of the STAT3 signaling
pathway.
Celastrol induces the overexpression
of miR-24 and miR-181b
The molecular mechanisms underlying celastrol
induction of apoptosis require further elucidation. Therefore the
mechanisms underlying the effect of celastrol on STAT3
phosphorylation was investigated. Previous studies have revealed
that miRNAs are involved in the antitumor mechanism underlying
celastrol (15,16). Therefore, the present study
investigated whether celastrol induced A549 cell apoptosis through
the regulation of STAT3-associated miRNAs.
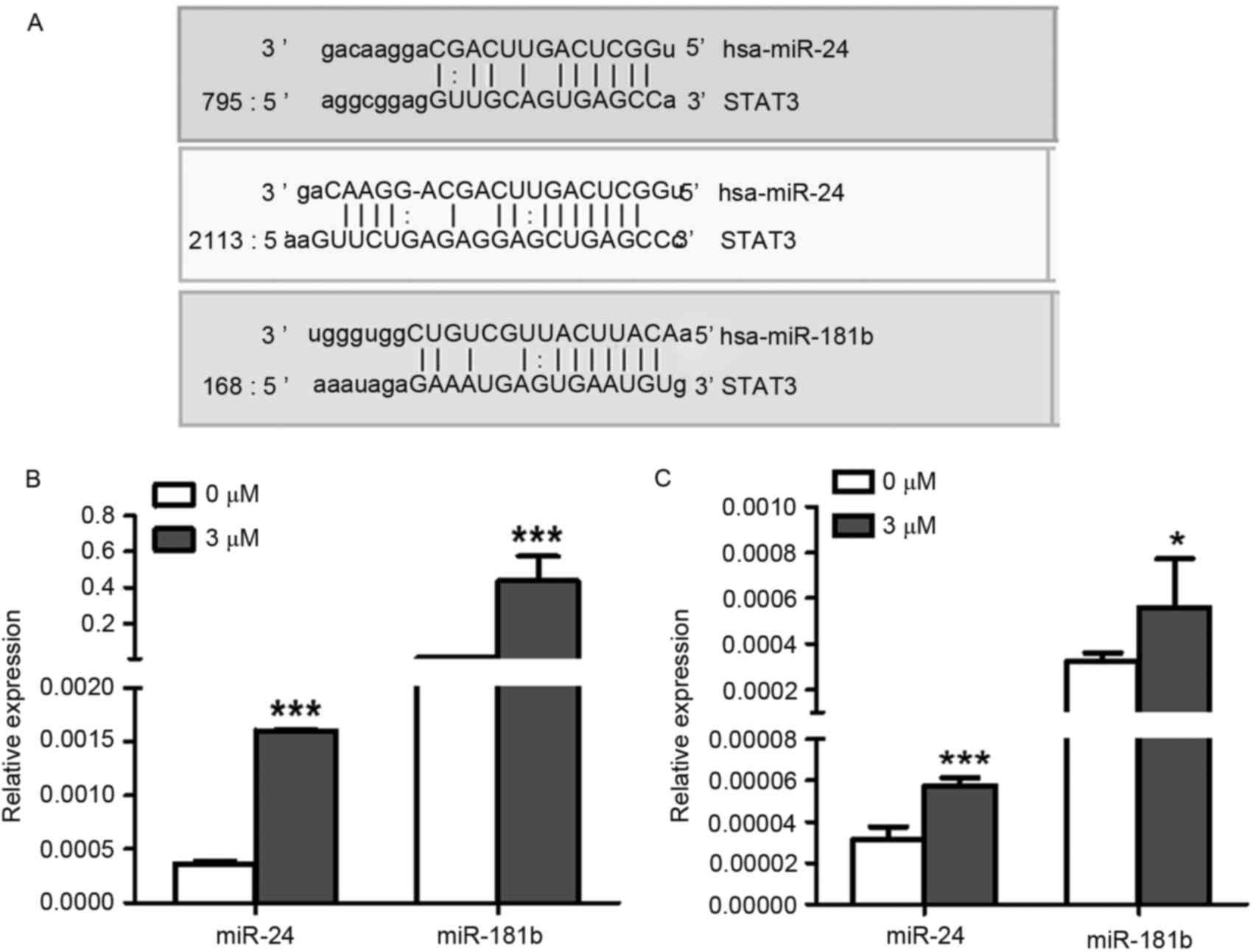
Using microRNA.org
software (http://www.microrna.org/microrna/home.do), the present
study predicted that the 3′ untranslated region (UTR) of STAT3 mRNA
was targeted by miR-24 and miR-181b (Fig.
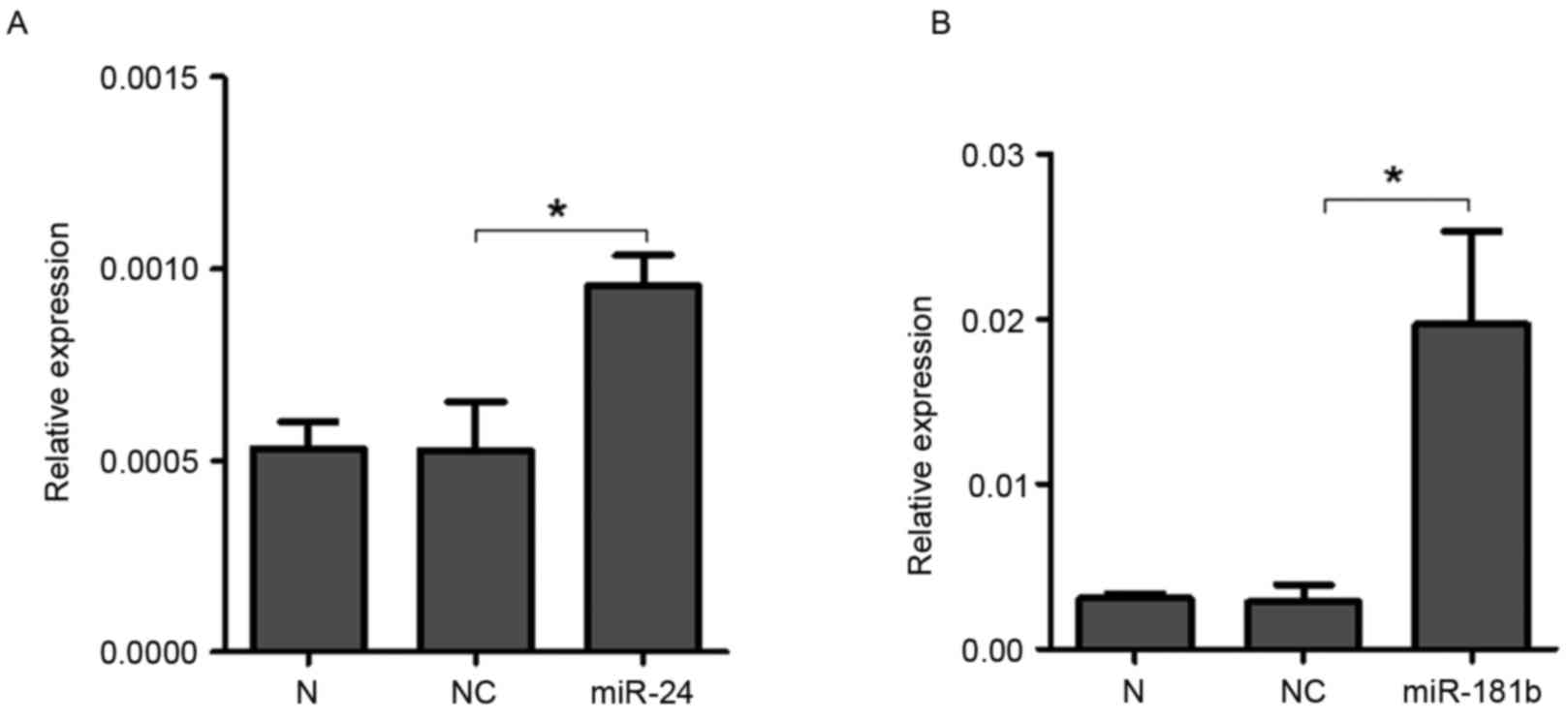
3A). Furthermore, the expression levels of miR-24 (P<0.0001)
and miR-181b (P=0.0006) were both significantly increased in 3 µM
celastrol-treated A549 cells compared with the control treatment
(Fig. 3B), indicating that celastrol
may decrease the expression level of STAT3 via up-regulation of
miR-24 and miR-181b. Additionally, LTEP-a-2 cells were also used to
confirm the effects of celastrol on the expression levels of miR-24
and miR-181b with 3 µM celastrol stimulation. As expected, miR-24
(P=0.0005) and miR-181b (P=0.03) were upregulated in
celastrol-treated LTEP-a-2 cells (Fig.
3C).
STAT3 phosphorylation and Bcl-2/Bax
ratio is suppressed by miR-24 and miR-181b
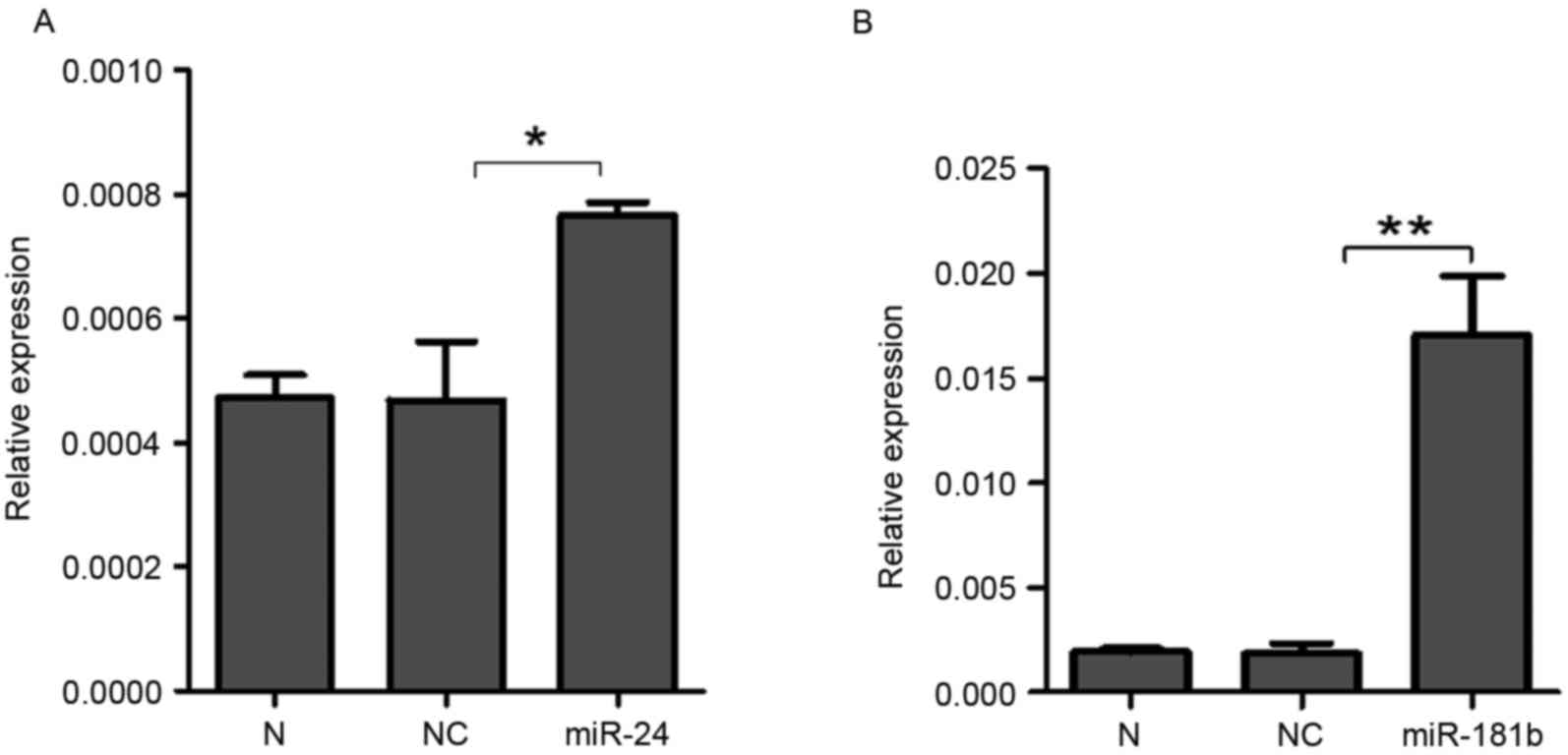
To further investigate the effects of miR-24 and
miR-181b on STAT3, synthesized miR-24 and miR-181b was transfected
into A549 cells. RT-qPCR demonstrated that the expression levels of
miR-24 (P=0.04, Fig. 4A) and miR-181b
(P=0.006, Fig. 4B) significantly
increased in the A549 cells treated with miR-24 and miR-181b
compared with the negative control group, which indicated that
miR-24 and miR-181b were successfully transfected into A549 cells.
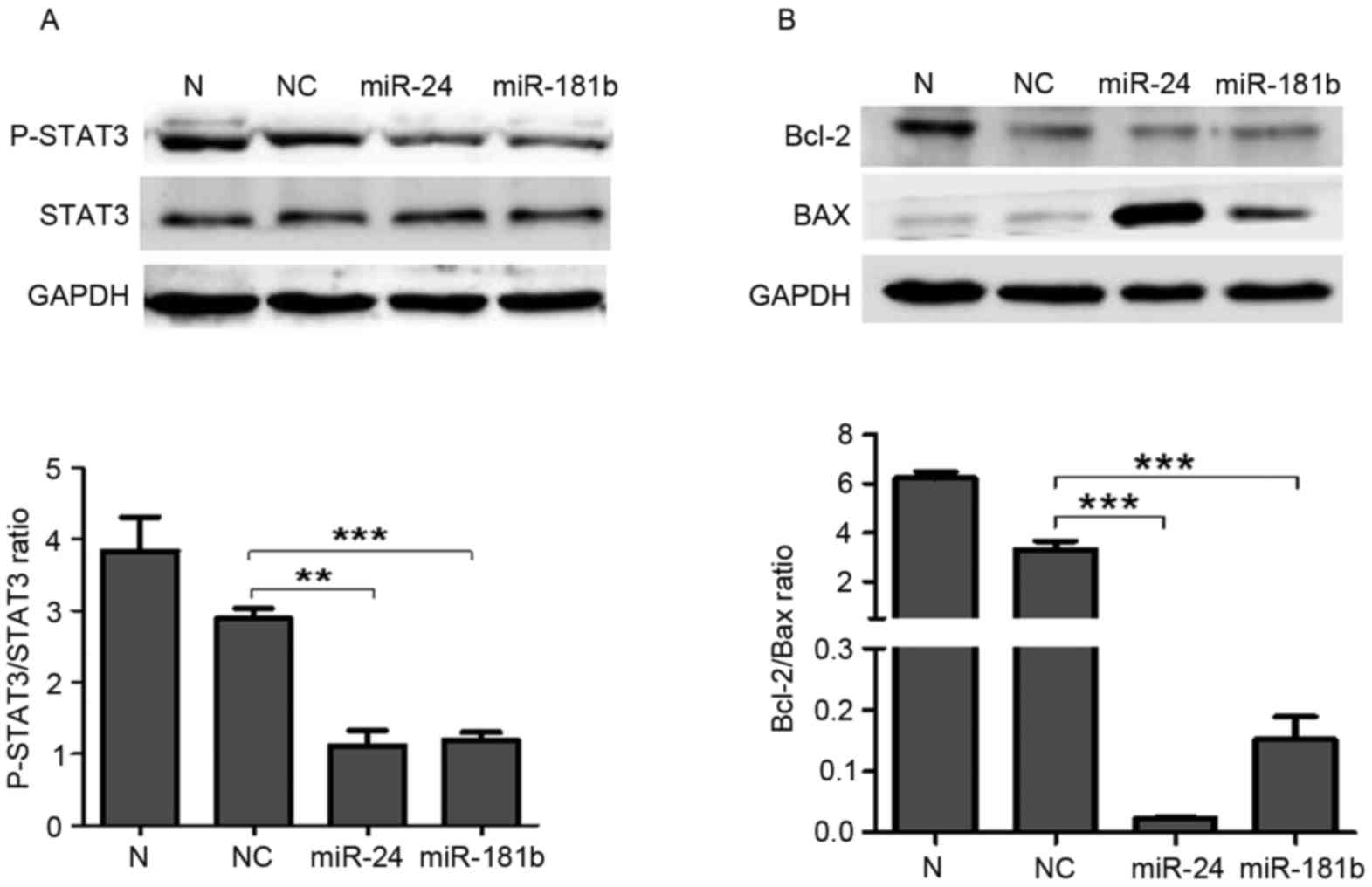
Western blotting revealed that miR-24 (P=0.002) and miR-181b
(P=0.0007) treatment significantly suppressed the p-STAT3 levels
compared with the negative control group (Fig. 5A). Furthermore, miR-24 (P=0.0008) and
miR-181b (P=0.0009) significantly decreased the expression ratio of
Bcl-2 and Bax compared with the negative control group (P<0.001;
Fig. 5B).
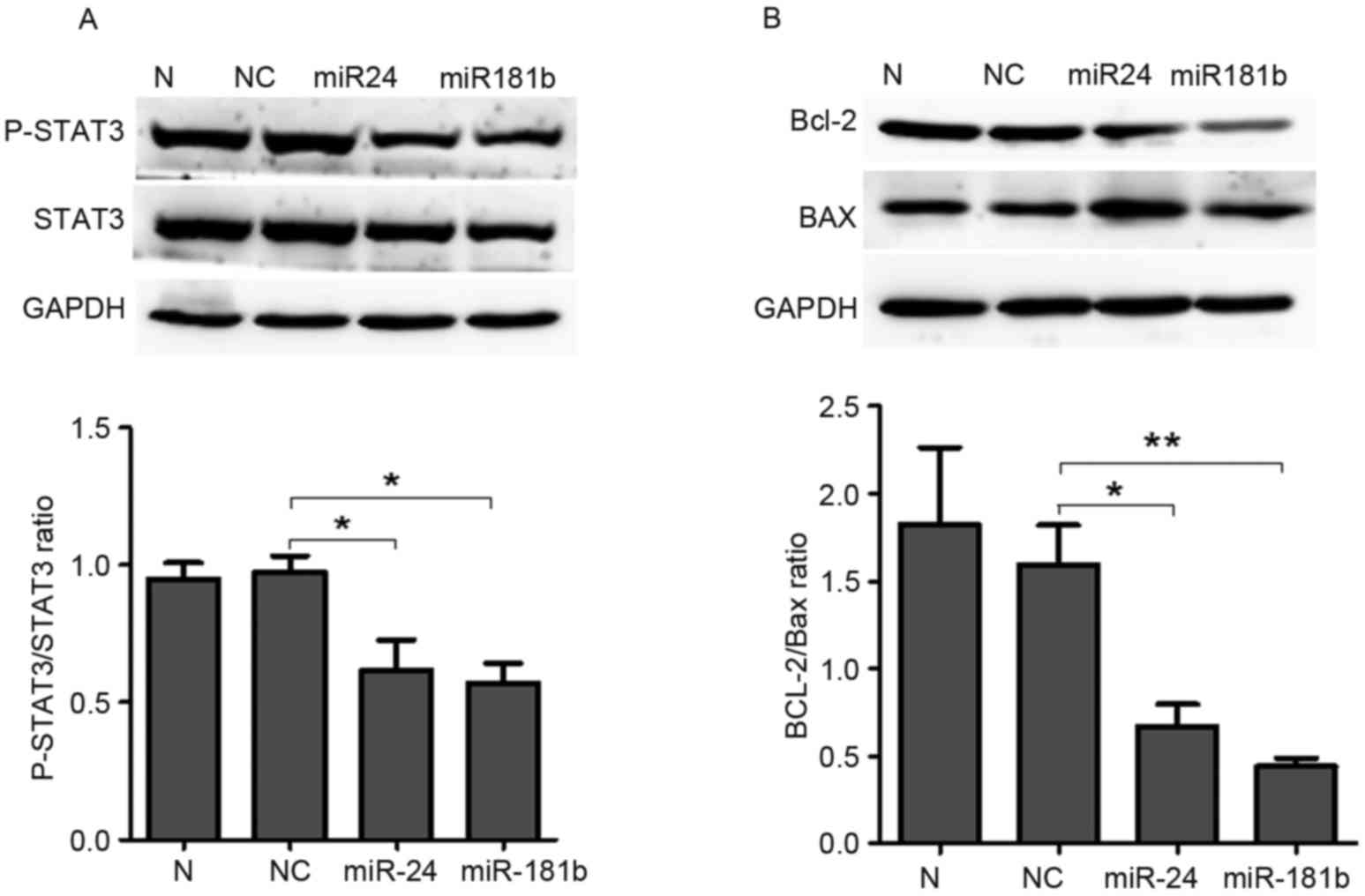
LTEP-a-2 cells were used to confirm these results.
MiR-24 and miR-181b treatment significantly increased the level of
miR-24 (P=0.04, Fig. 6A) and miR-181b
(P=0.04, Fig. 6B) compared with the
negative control group and significantly decreased p-STAT3 compared
with the negative control group (P=0.04 for miR-24 and P=0.04 for
miR-181b; Fig. 7A). In addition,
miR-24 (P=0.03, Fig. 7B) and miR-181b
(P=0.006, Fig. 7B) treatment
significantly decreased the Bcl-2/Bax ratio compared with the
control group in LTEP-a-2 cells. These results suggested that
miR-24 and miR-181b mediated the effects of celastrol on the
phosphorylation levels of STAT3 and the Bcl-2/Bax ratio.
Discussion
Previously, natural preparation represented a
significant portion of the pharmaceutical market compared with
synthesized compounds (2–4). Celastrol is extracted from Thunder God
Vine, which contains a number of therapeutic active compounds
(2). Since the 1970s, celastrol has
been identified to possess anti-inflammatory, anti-oxidative and
tumor cell-inducing apoptotic properties. Furthermore, compared
with other anticancer drugs, celastrol selectively targets tumor
cells and causes no obvious damage to normal hematopoietic cells,
the heart, liver, kidney or other organs (20–22).
Therefore, celastrol is considered to be a relatively safe and
effective anticancer drug. Cumulative studies have identified
numerous molecular targets of celastrol; however, studies
investigating how celastrol affects these targets are rare. The
present study treated the A549 lung adenocarcinoma cell line with
various concentrations of celastrol to observe these effects. The
results suggested that celastrol suppresses the proliferation and
induces apoptosis of A549 cells in a dose-dependent manner. Of
note, it was revealed that STAT3-associated miRNAs participated in
the process of celastrol-mediated induction of A549 cell
apoptosis.
STAT3 is a transcription factor that is
phosphorylated by Janus kinases in response to cytokine activation
(23). Subsequently, it dimerizes and
translocates to the nucleus to activate transcription of
cytokine-responsive genes (24). In
tumorigenesis, STAT3 activation induces tumor progression by
promoting the cell cycle and preventing apoptosis (12,23). The
present study demonstrated that celastrol induces cell apoptosis by
decreasing the levels of STAT3 phosphorylation.
Furthermore, Bcl-2 and Bax have been reported to be
downstream factors of STAT3, and they serve important roles in the
mitochondrial-mediated apoptosis pathway (18,20). Bcl-2
is an anti-apoptotic protein and Bax is a pro-apoptotic protein.
Apoptosis is largely controlled by the balance between
anti-apoptotic and pro-apoptotic proteins, including the Bcl-2/Bax
ratio. In the present study, celastrol significantly decreased
STAT3 phosphorylation and the Bcl-2/Bax ratio, which supported the
hypothesis that celastrol induces apoptosis by affecting the
expression of STAT3 phosphorylation and the Bcl-2/Bax ratio.
Certain studies have demonstrated that celastrol may
influence the expression levels of numerous miRNAs and achieve the
therapeutic effects via these miRNAs (15,16), which
regulate a number of gene expression levels by targeting the 3′-UTR
of mRNA molecules. To further investigate whether celastrol
affected the expression of STAT3-associated miRNAs, miRNA analysis
software was used to predict that the 3′-UTR of STAT3 was targeted
by miR-24 and miR-181b. Previous studies indicated that miR-24 may
regulate liver inflammation in rats and that miR-181b functions as
a tumor suppressor in glioma (25,26). Of
note, the present study revealed that p-STAT3 was significantly
reduced by miR-24 and miR-181b. In addition, the Bcl-2/Bax ratio
also decreased in miR-24 and miR-181b-treated cells. These results
confirmed that miR-24 and miR-181b may influence STAT3 activation
(27). Furthermore, celastrol
treatment increased the expression levels of miR-24 and miR-181b,
and decreased STAT3 phosphorylation and the Bcl-2/Bax ratio. These
results indicated that celastrol may decrease p-STAT3 levels by
regulating miR-24 and miR-181b.
In conclusion, the present study investigated the
molecular mechanism underlying celastrol in lung cancer therapy. It
was demonstrated that celastrol induced apoptosis of lung cancer
cells by affecting the expression levels of miR-24 and miR-181b,
which further regulated the activation of STAT3. Identification of
the function of miR-24 and miR-181b provides useful information for
the safe and effective application of celastrol in tumor
therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation (grant nos. 31371321, 31440061 and
81530060), and the Shandong Science and Technology Committee (grant
nos. ZR2014HP004, 2015GSF118073, ZR2014HL055 and ZR2013HL003), the
Health and Family Planning Commission of Shandong Province (grant
no. 2014WS0185) and the Shandong Education Department (grant no.
J13LE11).
References
|
1
|
Salminen A, Lehtonen M, Paimela T and
Kaarniranta K: Celastrol: Molecular targets of thunder god vine.
Biochem Biophys Res Commun. 394:439–442. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li-Weber M: Targeting apoptosis pathways
in cancer by Chinese medicine. Cancer Lett. 332:304–312. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Corson TW and Crews CM: Molecular
understanding and modern application of traditional medicines:
Triumphs and trials. Cell. 130:769–774. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patwardhan B and Mashelkar RA: Traditional
medicine-inspired approaches to drug discovery: Can Ayurveda show
the way forward? Drug Discov Today. 14:804–811. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koehn FE: High impact technologies for
natural products screening. Prog Drug Res. 65:175, 177–210.
2008.
|
|
6
|
Lipsky PE and Tao XL: A potential new
treatment for rheumatoid arthritis: Thunder god vine. Semin
Arthritis Rheum. 26:713–723. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Canter PH, Lee HS and Ernst E: A
systematic review of randomised clinical trials of Tripterygium
wilfordii for rheumatoid arthritis. Phytomedicine. 13:371–377.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petronelli A, Pannitteri G and Testa U:
Triterpenoids as new promising anticancer drugs. Anticancer Drugs.
20:880–892. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sethi G, Ahn KS, Pandey MK and Aggarwal
BB: Celastrol, a novel triterpene, potentiates TNF-induced
apoptosis and suppresses invasion of tumor cells by inhibiting
NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB
activation. Blood. 109:2727–2735. 2007.PubMed/NCBI
|
|
10
|
Yang H, Chen D, Cui QC, Yuan X and Dou QP:
Celastrol, a triterpene extracted from the Chinese ‘Thunder of God
Vine,’ is a potent proteasome inhibitor and suppresses human
prostate cancer growth in nude mice. Cancer Res. 66:4758–4765.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen M, Rose AE, Doudican N, Osman I and
Orlow SJ: Celastrol synergistically enhances temozolomide
cytotoxicity in melanoma cells. Mol Cancer Res. 7:1946–1953. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dai Y, DeSano JT, Meng Y, Ji Q, Ljungman
M, Lawrence TS and Xu L: Celastrol potentiates radiotherapy by
impairment of DNA damage processing in human prostate cancer. Int J
Radiat Oncol Biol Phys. 74:1217–1225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rajendran P, Li F, Shanmugam MK, Kannaiyan
R, Goh JN, Wong KF, Wang W, Khin E, Tergaonkar V, Kumar AP, et al:
Celastrol suppresses growth and induces apoptosis of human
hepatocellular carcinoma through the modulation of STAT3/JAK2
signaling cascade in vitro and in vivo. Cancer Prev Res (Phila).
5:631–643. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Costantino L and Barlocco D: STAT 3 as a
target for cancer drug discovery. Curr Med Chem. 15:834–843. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sha M, Ye J, Zhang LX, Luan ZY, Chen YB
and Huang JX: Celastrol induces apoptosis of gastric cancer cells
by miR-21 inhibiting PI3K/Akt-NF-κB signaling pathway.
Pharmacology. 93:39–46. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Li Y, Liu D, Sun H and Liu J:
MiR-224 is critical for celastrol-induced inhibition of migration
and invasion of hepatocellular carcinoma cells. Cell Physiol
Biochem. 32:448–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang PY, Sun YX, Zhang S, Pang M, Zhang
HH, Gao SY, Zhang C, Lv CJ and Xie SY: Let-7c inhibits A549 cell
proliferation through oncogenic TRIB2 related factors. FEBS Lett.
587:2675–26812013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
You S, Li R, Park D, Xie M, Sica GL, Cao
Y, Xiao ZQ and Deng X: Disruption of STAT3 by niclosamide reverses
radioresistance of human lung cancer. Mol Cancer Ther. 13:606–616.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Real PJ, Sierra A, De Juan A, Segovia JC,
Lopez-Vega JM and Fernandez-Luna JL: Resistance to chemotherapy via
Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer
cells. Oncogene. 21:7611–7618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang C, Chi YL, Wang PY, Wang YQ, Zhang
YX, Deng J, Lv CJ and Xie SY: miR-511 and miR-1297 inhibit human
lung adenocarcinoma cell proliferation by targeting oncogene TRIB2.
PLoS One. 7:e460902012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li YJ, Zhang YX, Wang PY, Chi YL, Zhang C,
Ma Y, Lv CJ and Xie SY: Regression of A549 lung cancer tumors by
anti-miR-150 vector. Oncol Rep. 27:129–134. 2012.PubMed/NCBI
|
|
23
|
Ji N, Li J, Wei ZKong F, Jin H, Chen X, Li
Y and Deng Y: Effect of celastrol on growth inhibition of prostate
cancer cells through the regulation of hERG channel in vitro.
Biomed Res Int. 2015:3084752015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
DiSanto JP: Cytokines: Shared receptors,
distinct functions. Curr Biol. 7:R424–R426. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dogan I, Cumaoglu A, Aricioglu A and
Ekmekci A: Inhibition of ErbB2 by herceptin reduces viability and
survival, induces apoptosis and oxidative stress in Calu-3 cell
line. Mol Cell Biochem. 347:41–51. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian SS, Lamb P, Seidel HM and RBRosen J
Stein: Rapid activation of the STAT3 transcription factor by
granulocyte colony-stimulating factor. Blood. 84:1760–1764.
1994.PubMed/NCBI
|
|
27
|
Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu
X, Shen Y and Huang TT: Interplay between microRNAs and the STAT3
signaling pathway in human cancers. Physiol Genomics. 45:1206–1214.
2013. View Article : Google Scholar : PubMed/NCBI
|