Introduction
Large tumor suppressor, homolog 2 (LATS2), is
a tumor suppressor gene implicated in the regulation of cell
proliferation, migration and apoptosis via several cell signaling
pathways, particularly the Hippo, p53 and LATS-LIM domain
kinase-actin pathways (1).
Classically, a tumor suppressor gene is inactivated by a somatic
mutation or promoter hypermethylation. Previous human cancer genome
projects have revealed the presence of LATS2 mutations in
several types of human cancer, including breast cancer, however,
its frequency is low at ~1% (2,3).
Therefore, LATS2 mutations are unlikely to be important in
the pathogenesis of the majority of breast cancer cases. By
contrast, hypermethylation of the LATS2 promoter has been
reported in a higher proportion of breast cancer (50%) (4), acute lymphoblastic leukemia (ALL) (24%)
(5), astrocytoma (71.5%) (6), lung cancer (78.8%) (7), and nasopharyngeal cancer (36.7%)
(8) cases, and the absence of
LATS2 hypermethylation has been confirmed in normal breast
tissues (4) and normal brain tissues
(6). In addition, the association
between LATS2 promoter hypermethylation and reduced
messenger RNA (mRNA) expression levels of LATS2 has been
established in breast cancer (4), ALL
(5) and astrocytoma (6). However, conflicting results have also
been reported, namely, that LATS2 promoter hypermethylation
is rare in head and neck cancer (9),
and that it is more common in normal tissues, compared with tumor
tissues in nasophalyngeal cancer (8)
and gastric cancer (10).
Investigations of the methylation of promoter
regions are usually performed through bisulfite modification to
convert the methylation status into a sequence difference. The
present study carefully examined the LATS2 promoter sequence
following bisulfite modification and noted that several sequences
with a high degree of homology to the LATS2 promoter
sequence were present in intron 2 (pseudo promoter sequence). As
previous methylation studies of the LATS2 promoter have used
a methylation-specific polymerase chain reaction (MSP) assay, it is
possible that the MSP assay may amplify the LATS2 promoter
sequence and also the pseudo promoter sequences in intron 2,
resulting in the conflicting results of LAST2 promoter
methylation previously reported (4–10). This
indicates that a more accurate technique is required for the
investigation of LATS2 promoter methylation. Advancements in
sequence technology with the development of next generation
sequencing (NGS), has made it possible to quantitatively determine,
with high accuracy, the methylation status of a gene. It is
anticipated that bisulfite sequencing with NGS can be used to
evaluate the methlyation status of the LATS2 promoter more
accurately than is possible using a conventional MSP assay.
The aim of the present study was, first, to evaluate
the methylation status of the LATS2 promoter region using
bisulfite sequencing with NGS, and second, to clarify the
correlation between LATS2 promoter methylation and gene
expression in breast cancer, using breast cancer cell (BCC) lines
and breast cancer tissues.
Materials and methods
Patients and breast tumor samples
Pairs of tumor tissue and normal tissue samples were
obtained from 11 patients with primary breast cancer, who underwent
breast conserving surgery or mastectomy between 2001 and 2004 at
Osaka University Hospital (Osaka, Japan). Patients treated with
neoadjuvant chemotherapy and/or hormonal therapy were excluded. The
clinicopathological characteristics of the patients recruited are
summarized in Table I. The tissue
samples were snap frozen in liquid nitrogen and stored at −80°C
until use. The remaining surgical specimens were fixed with 10%
buffered formaldehyde. The present study was approved by the
Institutional Review Board for Clinical Research of Osaka
University Hospital, and informed consent was obtained from each
patient prior to surgery.
 | Table I.Clinicopathological characteristics
of patients/tumors for comparison of the large tumor suppressor,
homolog 2 methylation in paired breast tumor and normal breast
tissues. |
Table I.
Clinicopathological characteristics
of patients/tumors for comparison of the large tumor suppressor,
homolog 2 methylation in paired breast tumor and normal breast
tissues.
| Characteristic | Patients, n
(%) |
|---|
| Age (years) |
|
|
<50 | 4 (36) |
|
≥50 | 7 (64) |
| Menopausal
status |
|
|
Premenopausal | 4 (36) |
|
Postmenopausal | 7 (64) |
| Tumor size
(cm) |
|
| ≤2 | 3 (27) |
|
>2 | 8 (73) |
| Lymph node
metastasis |
|
|
Negative | 8 (73) |
|
Positive | 3 (27) |
| Histological
type |
|
|
Invasive ductal carcinoma | 11 (100) |
|
Other | 0 (0) |
| Histological
grade |
|
| 1 and
2 | 5 (45) |
| 3 | 6 (55) |
| Estrogen
receptor |
|
|
Negative | 6 (45) |
|
Positive | 5 (55) |
| Progesterone
receptor |
|
|
Negative | 6 (45) |
|
Positive | 5 (55) |
| Human epidermal
growth factor receptor 2 |
|
|
Negative | 7 (64) |
|
Positive | 4 (36) |
DNA extraction and sodium bisulfite
treatment
Total DNA from cell lines was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scietnific,
Inc., Waltham, MA, USA), and the DNeasy® Blood and
Tissue kit (Qiagen, Inc., Valencia, CA, USA) was used to extract
total DNA from the breast tissues. The genomic DNA (1 µg) was
treated with sodium bisulfite using an EpiTect®
Bisulfite kit (Qiagen, Inc.).
Quantitative LATS2 promoter
methylation analysis using NGS
The GS Junior system (Roche Diagnostics, Basel,
Switzerland) was used for the NGS methylation assay for region X
(Fig. 1A) in accordance with the
manufacturer's protocol, and data were analyzed using GS Amplicon
Variant Analyzer software (version 2.7; Roche Diagnostics)
(11). The LATS2 gene sequence
was obtained from the University of California Santa Cruz browser
(NM_014572, December 2013; GRCh38/hg38; https://genome.ucsc.edu). The MI was calculated by
dividing the number of reads of cytosine by the total number of
reads at each CpG site. Sequence alignment between regions X and
Z1-4 of the LATS2 are shown in Fig. 1B. The NGS primers for region X, used
for the frozen tissues and cell lines (NGS primer X-long; Fig. 1A), were as follows: Forward
5′-TTTTTTAGAAAGAAAATATATATGGGGAGG-3′ and reverse
5′-AAAAAAACCACCAAATTACCAACTAATAAC-3′ (chr13:
21,061,921–21,062,405). For all 16 CpG sites, the average MI of 13
CpG sites was defined as the LATS2 MI (2nd-14th CpG;
Fig. 2A). The NGS primers for region
X, used for the DNA extracted from formalin-fixed paraffin-embedded
(FFPE) tissues (NGS primer X-short, Fig.
1A), were as follows: Forward
3′-TTATTGGGATAGTGGAATTAAATAATTAAG-5′ and reverse
3′-AAAATTTCTCCAAATTAATCAAACTAATCT-5′ (chr13:
21,061,987–21,062,136). The NGS short primer included six CpG sites
corresponding to the 10–15th CpG, and the average MI of five CpG
sites (10–14th CpG) was defined as the LATS2 MI. The NGS
primers for region Y (NGS primer Y; Fig.
1A) were as follows: Forward 5′-GGGTAAATATTTAAGTTTGGGGGTA-3′
and reverse 5′-AACCCCCTCACCTCCAAAAAC-3′ (chr13: 21061335-21061816),
and the NGS primers for region Z1 (NGS primer Z1, Fig. 1A) were as follows: Forward
5′-TTATGTTTGGTTAGAAATTTTTTTT-3′ and reverse
5′-ATCTACCAACACAATACCCAATACATA-3 (chr13:
21,039,732–21,040,116).
 | Figure 1.Schematic representation of the
LATS2 gene and primer design for DNA methylation analysis of
LATS2. (A) Two primer sets were designed for DNA methylation
analysis of LATS2 (region X) using NGS, including NGS
primers for DNA from cell lines and frozen tissues (X-long; red
arrows) and from formalin-fixed paraffin-embedded tissues (X-short;
blue arrows). MSP primers used in previous studies (4–9), and NGS
primers for regions Y and Z1 are also shown. The LATS2
genome sequence was obtained from the University of California
Santa Cruz browser (NM_01,4572, December 2013; GRCh38/hg38). (B)
Sequence alignment between regions X and Z1-4 of the LATS2
gene. Bisulfite converted cytosines are shown as ‘u’. Regions
corresponding to X and Z1-4 are indicated with gray bars. The
X-long and X-short NGS primers were designed for the unmatched
region (red and blue capital letters, respectively). The locations
of MSP primers and Sanger sequence primers used in previous studies
(4,6–8) are
indicated by solid and dotted lines, respectively. LATS2,
large tumor suppressor, homolog 2; NGS, next generation sequencing;
MSP, methylation-specific polymerase chain reaction; F, forward; R,
reverse. |
In situ hybridization (ISH) for LATS2
mRNA
The QuantiGene®ViewRNA ISH Tissue Assay
kit (Affymetrix, Inc., Santa Clara, CA, USA) was used in accordance
with the manufacturer's protocol. FFPE sections (4 µm thick) of the
tumor tissues were incubated for 10 min at 98°C with 200 ml 1X
pretreatment solution (Affymetrix, Inc.), followed by protease
digestion for 10 min. The LATS2 View RNA™ Probe
set (Affymetrix, Inc.) was hybridized for 2 h at 40°C using a
Hybridizer (Dako, Glostrup, Denmark). ISH images were captured
using a fluorescent microscope (BX63; Olympus, Tokyo, Japan).
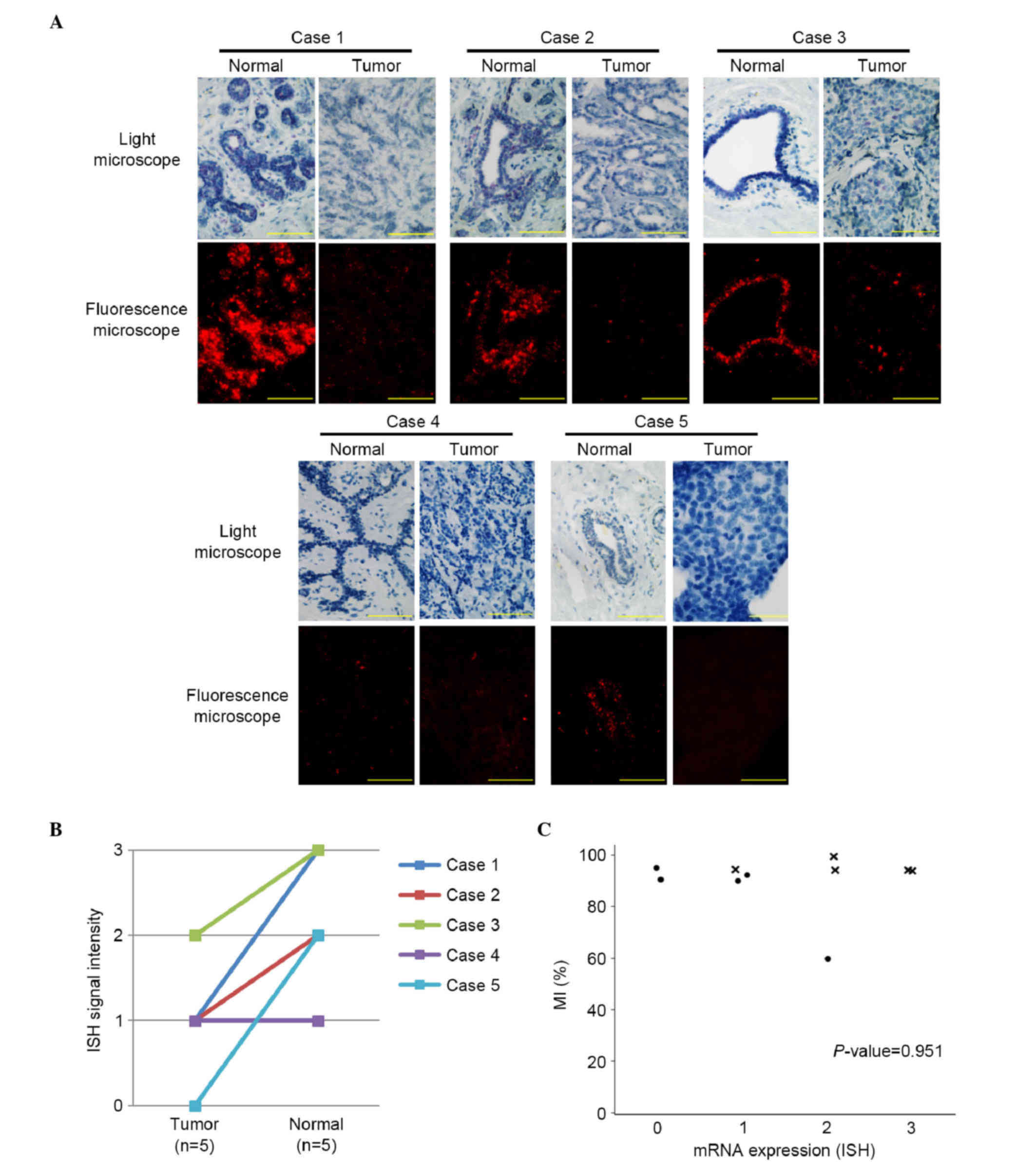
Signal intensity was semi-quantitatively classified into four
categories (0, negative; 1+, weak; 2+, intermediate; and 3+,
strong) based on the number of cytoplasmic fluorescent dots in five
non-overlapping fields at high-power magnification (×400).
Isolation of breast tumor cells using
magnetic-activated cell sorting (MACS)
The breast cancer cells and normal epithelial cells
were isolated from the FFPE tissues using the MACS method with the
EasySep Human EpCAM Positive Selection Cocktail, the EasySep Human
MUC1 Positive Selection Cocktail and EasySep Magnetic Particles
(Stemcell Technologies, Inc., Vancouver, BC, Canada), as previously
described (12). Total DNA was
extracted from these isolated cells using the QIAamp®
DNA FFPE Tissue kit (Qiagen, Inc.).
Cell lines and analysis of
demethylation using 5-aza-2′-deoxycytidine (5-aza)
The 12 BCC lines (MCF7, ZR75-1, T47D, ZR75-30,
MDA-MB-361, BT474, SKBR3, AU565, MDA-MB-453, MDA-MB-231, MDA-MB-468
and BT-20) and two normal breast epithelial cell lines (MCF10A and
HMEC) were cultured according to the culture guides of the American
Type Culture Collection (Manassas, VA, USA). For the investigation
of demethylation, the cultured cells were seeded into a six-well
plate at a density of 1.5×105 cells/well, and
treated with 5 µmol/l 5-aza (Sigma-Aldrich; Merck Millipore) or
with dimethylsulfoxide as a control for 72 h at 37°C, with the
medium replaced every 24 h.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was isolated from the cell lines using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and 1 µg of total RNA was reverse-transcribed into single
strand cDNA using random primers and the ReverTra Ace®
qPCR RT kit (Toyobo Co., Ltd., Osaka, Japan). LATS2 and
GAPDH TaqMan® Gene Expression Assays
(Hs00324396_m1 and Hs02758991_g1; catalog nos., 4351372 and
4331182, respectively; Applied Biosystems; Thermo Fisher
Scientific, Inc.) were used for qPCR on the LightCycler 480
Real-time PCR System (Roche Diagnostics). The qPCR primers and
probes used for the measurement of LATS2 and GAPDH mRNA expression
were designed by the Applied Biosystems (Thermo Fisher Scientific,
Inc.). The cycling conditions were as follows: 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec, 60°C for 60 sec and 50°C
for 10 sec. Absolute mRNA expression levels of LATS2 and
GAPDH were determined by the standard amplification curves
obtained by serially diluted PCR products with known
concentrations. The expression of LATS2 was normalized to
that of GAPDH, and each assay was performed in duplicate.
The result for each of the 5-aza treated BCC lines was normalized
with that of its untreated control.
Statistical analysis
The JMP statistical software package (version 10;
SAS Institute, Cary, NC, USA) was used for statistical analyses.
The association between the expression of LATS2 and MI was
assessed using the Kruskal-Wallis test. The Wilcoxon signed-rank
test was used for comparison of the expression levels of
LATS2 in the cancer and normal tissues. All statistical
analyses were two-sided and P<0.05 was considered to indicate a
statistically significant difference.
Results
Methylation status of the LATS2
promoter in breast tumor and normal tissues
In the present study, 11 pairs of frozen breast
tumor and normal tissues were analyzed using an NGS-based
methylation assay which involved bisulfite sequencing with NGS.
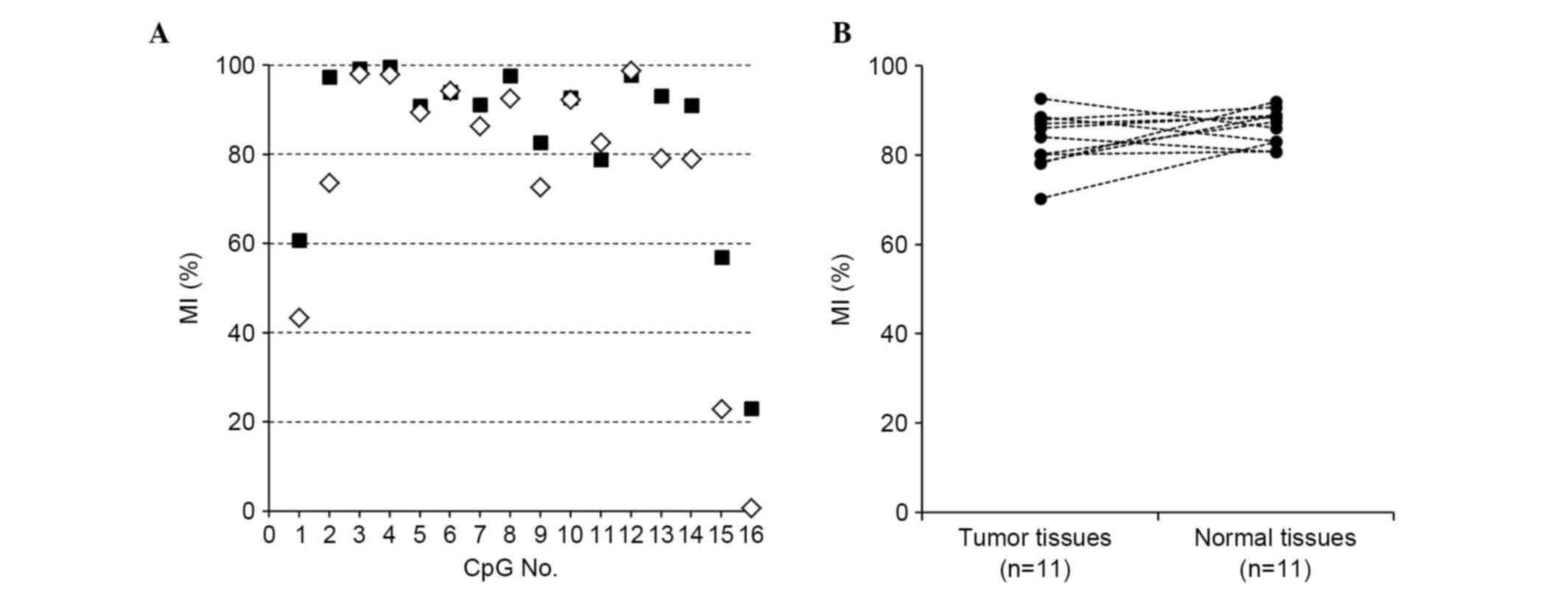
Representative results for the LATS2 MI at each CpG site of
region X are shown in Fig. 2A,
demonstrating that its methylation was uniformly high in the tumor
and normal tissues (median 84.0 and 87.4%, respectively; Fig. 2B). The tumor cells and normal
epithelial cells were then isolated using the MACS method from five
pairs of FFPE tumor and normal tissues, respectively, and subjected
to the NGS-based methylation assay. The isolated tumor cells
exhibited high MI (median, 90.6%; range, 59.9–95.1%), as did the
normal epithelial cells (median, 94.4%; range, 94.1–99.7%).
mRNA expression of LATS2 and its
correlation with LATS2 promoter methylation in breast tumor and
normal tissues
ISH for LATS2 mRNA was used to examine the
five pairs of FFPE breast tumor and normal tissues (Fig. 3A). In four of the five pairs, the mRNA
expression of LATS2 was higher in the normal breast
epithelial cells, compared with the tumor cells (Fig. 3B). No significant correlation was
observed between the mRNA expression of LATS2 and the
LATS2 MI (P=0.951; Fig.
3C).
Promoter methylation of LATS2 and its
effect on gene expression in BCC lines
To examine the methylation status of LATS2
region X in BCC lines, NGS-based methylation assays were performed
using 12 BCC lines and two normal breast cell lines. Similar to the
findings for the breast tumor and normal tissues, the LATS2
promoter was found to be uniformly hypermethylated in all BCC lines
and HMEC cells (Fig. 4A).
Subsequently, the correlation between LATS2 promoter
methylation and the mRNA expression of LATS2 was
investigated using RT-qPCR analysis. No inverse correlation was
observed between the mRNA expression of LATS2 and
LATS2 MI (Pearson's correlation coefficient −0.051; Fig. 4A). Treatment of these BCC lines with a
demethylating reagent (5 µM 5-aza) had minimal effect on the mRNA
expression of LATS2 in any of the cell lines, suggesting
that the mRNA expression of LATS2 was not directly regulated
by methylation of its promoter (Fig.
4B). NGS-based methylation assays using eight BCC lines and two
normal breast cell lines were then performed for region Y in the
CG-rich region located 333 bp downstream of region X, and for
region Z1, which was most homologous to region X in intron 2
(Fig. 1A). Region Y was completely
unmethylated and region Z1 showed a high level of methylation in
all the cell lines. Neither the methylation status of regions Y or
Z1 was correlated with the mRNA expression of LATS2
(Fig. 5A and B).
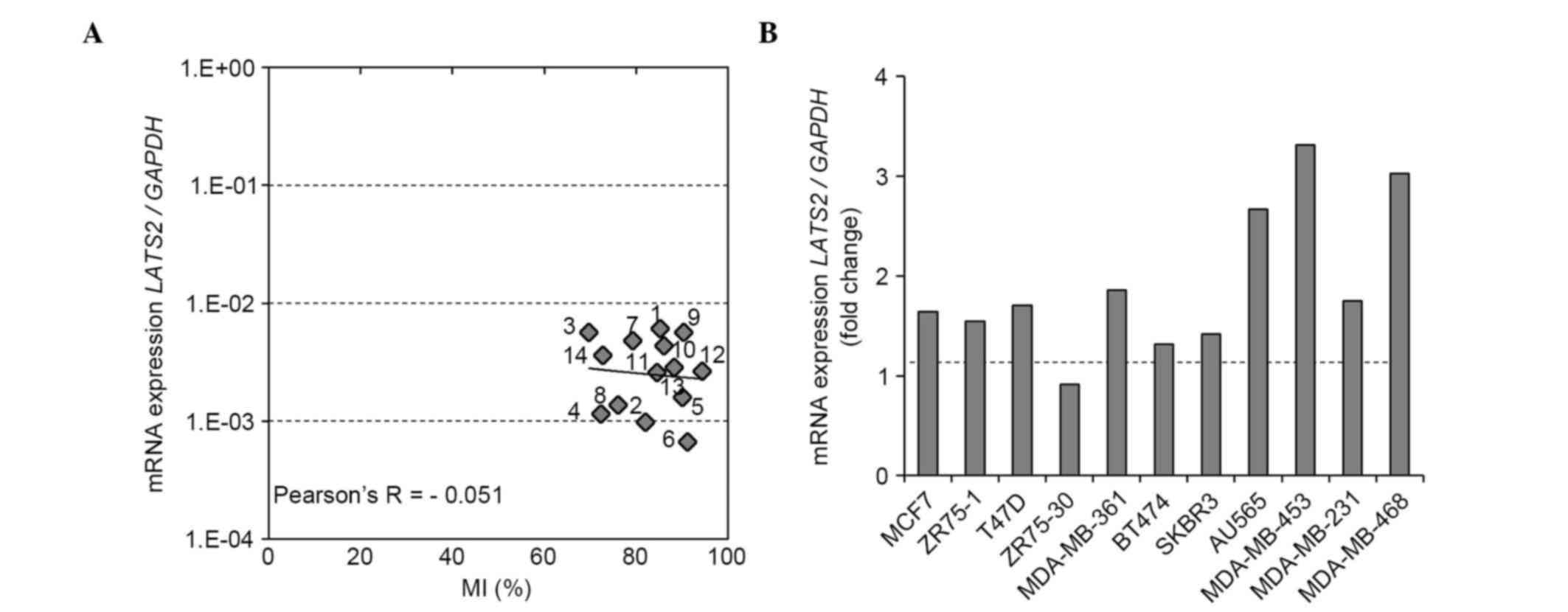 | Figure 4.LATS2 methylation status of
region X, the mRNA expression of LATS2 in breast cancer cell
lines and the effect of demethylating reagent on mRNA expression.
(A) Correlation between LATS2 MI and mRNA expression. The MI
of LATS2 (region X) was determined using a next generation
sequencing-based methylation assay and was compared with the mRNA
expression of LATS2 in 12 BCC lines and two normal breast
epithelial cell lines (MCF10A and HMEC). (B) Effect of 5-aza
treatment on the mRNA expression of LATS2. The mRNA
expression of LATS2 was evaluated following treatment with
5-aza or vehicle (dimethyl sulfoxide) in 11 BCC lines. The fold
changes in the mRNA expression of LATS2 following 5-aza
treatment are shown. LATS2, large tumor suppressor, homolog
2; BCC, breast cancer cell; MI, methylation index; 5-aza,
5-aza-2′-deoxycytidine; 1, MCF7; 2, ZR75-1; 3, T47D; 4, ZR75-30; 5,
MDA-MB-361; 6, BT474; 7, SKBR3; 8, AU565; 9, MDA-MB-453; 10,
MDA-MB-231; 11, MDA-MB-468; 12, BT-20; 13, MCF10A; 14, HMEC. |
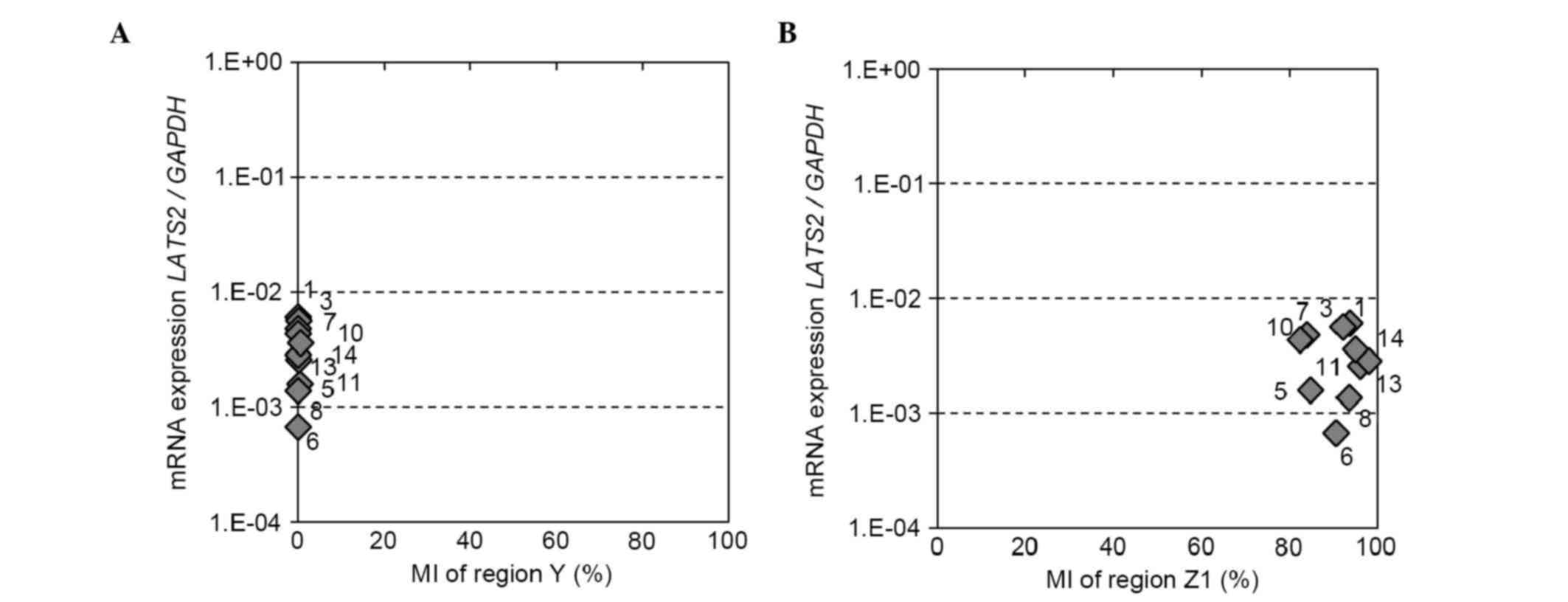 | Figure 5.LATS2 methylation status of
regions Y and Z1, and mRNA expression of LATS2 in breast
cancer cell lines. Correlation between the LATS2 MI of (A)
region Y or (B) region Z1 and mRNA expression. LATS2 MI was
determined using a next generation sequencing-based methylation
assay and was compared with the mRNA expression of LATS2 in
eight breast cancer cell lines and two normal breast epithelial
cell lines (MCF10A and HMEC). LATS2, large tumor suppressor,
homolog 2; MI, methylation index; 1, MCF7; 3, T47D; 5, MDA-MB-361;
6, BT474; 7, SKBR3; 8, AU565; 10, MDA-MB-231; 11, MDA-MB-468; 13,
MCF10A; 14, HMEC. |
Discussion
The present study investigated LATS2 promoter
methylation (region X; Fig. 1A) using
NGS-based methylation assays, a more accurate method, compared with
MSP assays. It was found that the LATS2 promoter exhibited a
high level of methylation in breast tumor cells and normal breast
epithelial cells, and that the mRNA expression of LATS2,
determined using ISH, did not correlate with LATS2 promoter
methylation. In addition, the LATS2 promoter exhibited high
levels of methylation in all 12 BCC lines, regardless of their
subtypes, and in two normal breast cell lines. Furthermore, it was
established that treatment of BCC lines with a demethylating
reagent had minimal effect on reactivation of the mRNA expression
of LATS2. Taken together, these results demonstrated that
LATS2 promoter hypermethylation was not implicated in the
silencing of LATS2 in breast cancer and is unlikely to be
important in the pathogenesis of breast cancer.
The present study showed that the mRNA expression of
LATS2, determined using ISH, was lower in tumor cells,
compared with normal breast epithelial cells, suggesting the
possible involvement of the downregulation of LATS2 mRNA in
the pathogenesis of breast cancer. Such a reduction in the mRNA
expression of LATS2 is likely to be induced by mechanisms
other than promoter hypermethylation. Previously, several micoRNAs,
including microRNA (miR)93 in breast cancer (13), miR25 in ovarian cancer (14) and miR31 in lung (15) and endometrial cancer (16), have been reported to directly target
the LATS2 gene and reduce its anti-oncogenic effect. A
previous study identified a LATS2 mutation, which can reduce
the expression of the gene in lung cancer, however, such a mutation
in breast cancer remains to be elucidated (2,3).
Furthermore, the mechanism underlying the downregulated mRNA
expression of LATS2 in tumor cells requires more detailed
investigation.
Until now, reports regarding LATS2 promoter
hypermethylation have been inconsistent in their support for
tumor-specific LATS2 hypermethylation in various types of
human cancer (4–9; Table II). Such
inconsistency is most likely attributable to the low fidelity of
the MSP assay used in these previous studies. A schematic
representation of the LATS2 genome is shown in Fig. 1A. A careful survey of the LATS2
promoter sequence following bisulfite modification revealed the
presence of several sequences (region Z1-Z4 in Fig. 1A) in intron 2, which exhibit a level
of homology with the LATS2 promoter sequence. The most
homologous sequence was region Z1, which was 92% identical to
region X (Fig. 1A), and located
22,024 bp downstream of region X. The alignment of the region X and
Z1-Z4 sequences following bisulfite modification is shown in
Fig. 1B. The MSP primers used in our
previous study (4) were designed on
the basis of Sanger bisulfite sequencing. The primers used for
bidirectional Sanger sequencing hybridize to regions X and Z, and
the sizes of the amplicons from the two regions were almost
identical at 212 bp in region X and 211 or 212 bp in region Z.
Therefore, Sanger sequencing may produce varied sequence results
for regions X and Z, leading to the faulty design of MSP primers
(Table III). With such MSP primers,
it is possible that the two regions are amplified at varying ratios
depending on the PCR conditions, which raising concerns as to
whether the assessment of LATS2 promoter methylation is
accurate. By contrast, with NGS, accurate sequencing can be
performed with a single base resolution, which avoids the possible
misreading of similar sequences. In addition, the NGS primers were
designed outside region X to avoid illegitimate hybridization
(Fig. 1A). Therefore, the NGS-based
methylation assay was considered sufficiently accurate to provide a
true assessment of the methylation status of the LATS2
promoter.
| Table II.Summary of previous studies examining
LATS2 methylation in human cancer. |
Table II.
Summary of previous studies examining
LATS2 methylation in human cancer.
| Author (year) | Type of Cancer | Tissue | n | LATS2
methylation positive rate (%)a | Refs. |
|---|
| Visser and Yang
(2010) | Breast | T | 30 | 50 | (1) |
|
|
| N | 6 | 0 |
|
| Steinmann et
al (2009) | Head and neck | T | 54 | 80 | (9) |
|
|
| N | 23 | 90 |
|
| Zhang et al
(2010) | Nasopharyngeal | T | 30 | 37 | (8) |
|
|
| N | 23 | 100 |
|
| Jiang et al
(2006) | Astrocytoma | T | 88 | 72 | (6) |
|
|
| N | 10 | 0 |
|
| Sasakiet al
(2010) | Lung | T | 203 | 79 | (7) |
|
|
| N | – | – |
|
| Jiménez-Velasco
et al (2005) | ALL | T | 66 | 24 | (5) |
|
|
| N | – | – |
|
| Table III.Primer sequences used in previous
studies. |
Table III.
Primer sequences used in previous
studies.
| Author (year) | Primer | Concordance rate
(%) | Refs. |
|---|
| Sanger sequencing
primers |
|
|
|
| Takahashi et
al (2005) |
|
| (4) |
|
| F
5′-TTTTGAGATGGAGTTTTGTT-3′ | – |
|
|
| R
5′-AATTCAAAACCAACCTAACC-3′ | – |
|
|
Methylation-specific primers |
|
|
|
| Takahashi et
al (2005); |
|
| (4) |
| Jiang et al,
(2006); |
|
| (6) |
| Sasaki et
al, (2010); |
|
| (7) |
| Zhang et al,
(2010) |
|
| (8) |
|
| M-F
5′-ATTTCGGTTTATTGTAATTTTC-3′ |
|
|
|
| Region -X:
21,062,261–21,062,240 | 82 |
|
|
| Region -Z1:
21,040,026–21,040,005 | 95 |
|
|
| Region -Z2:
21,029,744–21,029,723 | 91 |
|
|
| Region -Z3:
21,023,926–21,023,905 | 82 |
|
|
| Region -Z4:
21,019,927–21,019,906 | 73 |
|
|
| M-R
5′-AACCAACATAATAAAACCCCG-3′ |
|
|
|
| Region -X:
21,062,138–21,062,117 | 85 |
|
|
| Region -Z1:
21,039,903–21,039,883 | 92 |
|
|
| Region -Z2:
21,029,620–21,029,599 | 90 |
|
|
| Region -Z3:
21,023,804–21,023,783 | 86 |
|
|
| Region -Z4:
21,019,804–21,019,783 | 86 |
|
|
| UM-F
5′-TTTGTTTTTTGGGTTTAAGT-3′ |
|
|
|
| Region -X:
21,062,242–21,062,223 | 95 |
|
|
| Region -Z1:
21,040,007–21,039,988 | 95 |
|
|
| Region -Z2:
21,029,725–21,029,706 | 100 |
|
|
| Region -Z3:
21,023,907–21,023,888 | 100 |
|
|
| Region -Z4:
21,019,908–21,019,889 | 95 |
|
|
|
UM-R5′-CCAACATAATAAAACCCCA-3′ |
|
|
|
| Region -X:
21,062,138–21,062,119 | 84 |
|
|
| Region -Z1:
21,039,903–21,039,885 | 95 |
|
|
| Region -Z2:
21,029,620–21,029,602 | 95 |
|
|
| Region -Z3:
21,023,803–21,023,785 | 89 |
|
|
| Region -Z4:
21,019,803–21,019,785 | 95 |
|
| Jiménez-Velasco
et al (2005) |
|
| (5) |
|
| M-F
5′-GTTTTAGATTCGAAAGGTCGTAGC-3′ | – |
|
|
| M-R
5′-AAAACTAATTAACCCGTAAAACGAT-3′ | – |
|
|
| UM-F
5′-GGTGTTTTAGATTTGAAAGGTTGTAGT-3′ | – |
|
|
| UM-R
5′-AAAAAACTAATTAACCCATAAAACAAT-3′ | – |
|
| Steinmann et
al (2009) |
|
| (9) |
|
| M-F
5′-TTCGTTCGGATTGGTATGCGGTC-3′ | – |
|
|
| M-R
5′-CCATCTTCCCGAAACGCTCACG-3′ | – |
|
|
| UM-F
5′-GGTGTTTTGTTTGGATTGGTATGTGGTT-3′ | – |
|
|
| UM-R
5′-CATCTTCCCAAAACACTCACACCACA-3′ | – |
|
Another CpG island, region Y, located 333 bp
downstream from the sequencing site, was also analyzed using the
NGS-based methylation assay in eight BCC lines and two normal
breast cell lines (Fig. 5A). No
methylation was found in any of these cell lines, a finding which
is consistent with previous reports (4,9).
Therefore, this CpG island does not appear to be involved in the
epigenetic regulation of the LATS2 gene. In addition, an
NGS-based methylation assay was performed for region Z1, which
contained the most CpG sites among regions Z1-4, in eight BCC lines
and two normal breast cell lines, and found that the methylation
status of region Z1 was uniformly high in all cell lines, and its
methylation status was not associated with the mRNA expression of
LATS2 (Fig. 5B). These results
indicated that region Z1 was unlikely to be involved in the
epigenetic regulation of the mRNA expression of LATS2.
In conclusion, the results obtained in the present
study using NGS-based methylation assays demonstrated that
LATS2 hypermethylation was not involved in the silencing of
the mRNA expression of LATS2. The mRNA expression of
LATS2 was lower in tumor cells, compared with normal
epithelial cells, which suggested the possible involvement of the
downregulation of LATS2 mRNA in the pathogenesis of breast
cancer. However, the mechanism underlying this downregulation
remains to be elucidated. The conflicting results previously
reported for LATS2 promoter methylation obtained using MSP
assays are considered to be attributable to the low fidelity of the
MSP assay.
References
|
1
|
Visser S and Yang X: LATS tumor
suppressor: A new governor of cellular homeostasis. Cell Cycle.
9:3892–3903. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu T, Bachman J and Lai ZC: Evidence for a
tumor suppressor role for the large tumor suppressor genes LATS1
and LATS2 in human cancer. Genetics. 195:1193–1196. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu T, Bachman J and Lai ZC: Mutation
analysis of large tumor suppressor genes LATS1 and LATS2 supports a
tumor suppressor role in human cancer. Protein Cell. 6:6–11. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takahashi Y, Miyoshi Y, Takahata C,
Irahara N, Taguchi T, Tamaki Y and Noguchi S: Down-regulation of
LATS1 and LATS2 mRNA expression by promoter hypermethylation and
its association with biologically aggressive phenotype in human
breast cancers. Clin Cancer Res. 11:1380–1385. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiménez-Velasco A, Román-Gómez J, Agirre
X, Barrios M, Navarro G, Vázquez I, Prósper F, Torres A and
Heiniger A: Downregulation of the large tumor suppressor 2
(LATS2/KPM) gene is associated with poor prognosis in acute
lymphoblastic leukemia. Leukemia. 19:2347–2350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang Z, Li X, Hu J, Zhou W, Jiang Y, Li G
and Lu D: Promoter hypermethylation-mediated down-regulation of
LATS1 and LATS2 in human astrocytoma. Neurosci Res. 56:450–458.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sasaki H, Hikosaka Y, Kawano O, Yano M and
Fujii Y: Hypermethylation of the large tumor suppressor genes in
Japanese lung cancer. Oncol Lett. 1:303–307. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Hu CF, Chen J, Yan LX, Zeng YX
and Shao JY: LATS2 is de-methylated and overexpressed in
nasopharyngeal carcinoma and predicts poor prognosis. BMC Cancer.
10:5382010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Steinmann K, Sandner A, Schagdarsurengin U
and Dammann RH: Frequent promoter hypermethylation of tumor-related
genes in head and neck squamous cell carcinoma. Oncol Rep.
22:1519–1526. 2009.PubMed/NCBI
|
|
10
|
Lim B, Park JL, Kim HJ, Park YK, Kim JH,
Sohn HA, Noh SM, Song KS, Kim WH, Kim YS and Kim SY: Integrative
genomics analysis reveals the multilevel dysregulation and
oncogenic characteristics of TEAD4 in gastric cancer.
Carcinogenesis. 35:1020–1027. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Uji K, Naoi Y, Kagara N, Shimoda M,
Shimomura A, Maruyama N, Shimazu K, Kim SJ and Noguchi S:
Significance of TP53 mutations determined by next-generation ‘deep’
sequencing in prognosis of estrogen receptor-positive breast
cancer. Cancer Lett. 342:19–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Otani Y, Miyake T, Kagara N, Shimoda M,
Naoi Y, Maruyama N, Shimomura A, Shimazu K, Kim SJ and Noguchi S:
BRCA1 promoter methylation of normal breast epithelial cells as a
possible precursor for BRCA1-methylated breast cancer. Cancer Sci.
105:1369–1376. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fang L, Du WW, Yang W, Rutnam ZJ, Peng C,
Li H, O'Malley YQ, Askeland RW, Sugg S, Liu M, et al: MiR-93
enhances angiogenesis and metastasis by targeting LATS2. Cell
Cycle. 11:4352–4365. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Feng S, Pan W, Jin Y and Zheng J: MiR-25
promotes ovarian cancer proliferation and motility by targeting
LATS2. Tumour Biol. 35:12339–12344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Sempere LF, Ouyang H, Memoli VA,
Andrew AS, Luo Y, Demidenko E, Korc M, Shi W, Preis M, et al:
MicroRNA-31 functions as an oncogenic microRNA in mouse and human
lung cancer cells by repressing specific tumor suppressors. J Clin
Invest. 120:1298–1309. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mitamura T, Watari H, Wang L, Kanno H,
Kitagawa M, Hassan MK, Kimura T, Tanino M, Nishihara H, Tanaka S
and Sakuragi N: microRNA 31 functions as an endometrial cancer
oncogene by suppressing Hippo tumor suppressor pathway. Mol Cancer.
13:972014. View Article : Google Scholar : PubMed/NCBI
|