|
1
|
Arzumanyan A, Reis HM and Feitelson MA:
Pathogenic mechanisms in HBV-and HCV-associated hepatocellular
carcinoma. Nat Rev Cancer. 13:123–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology. 142:1264.
e1–1273. e1. 2012. View Article : Google Scholar
|
|
3
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
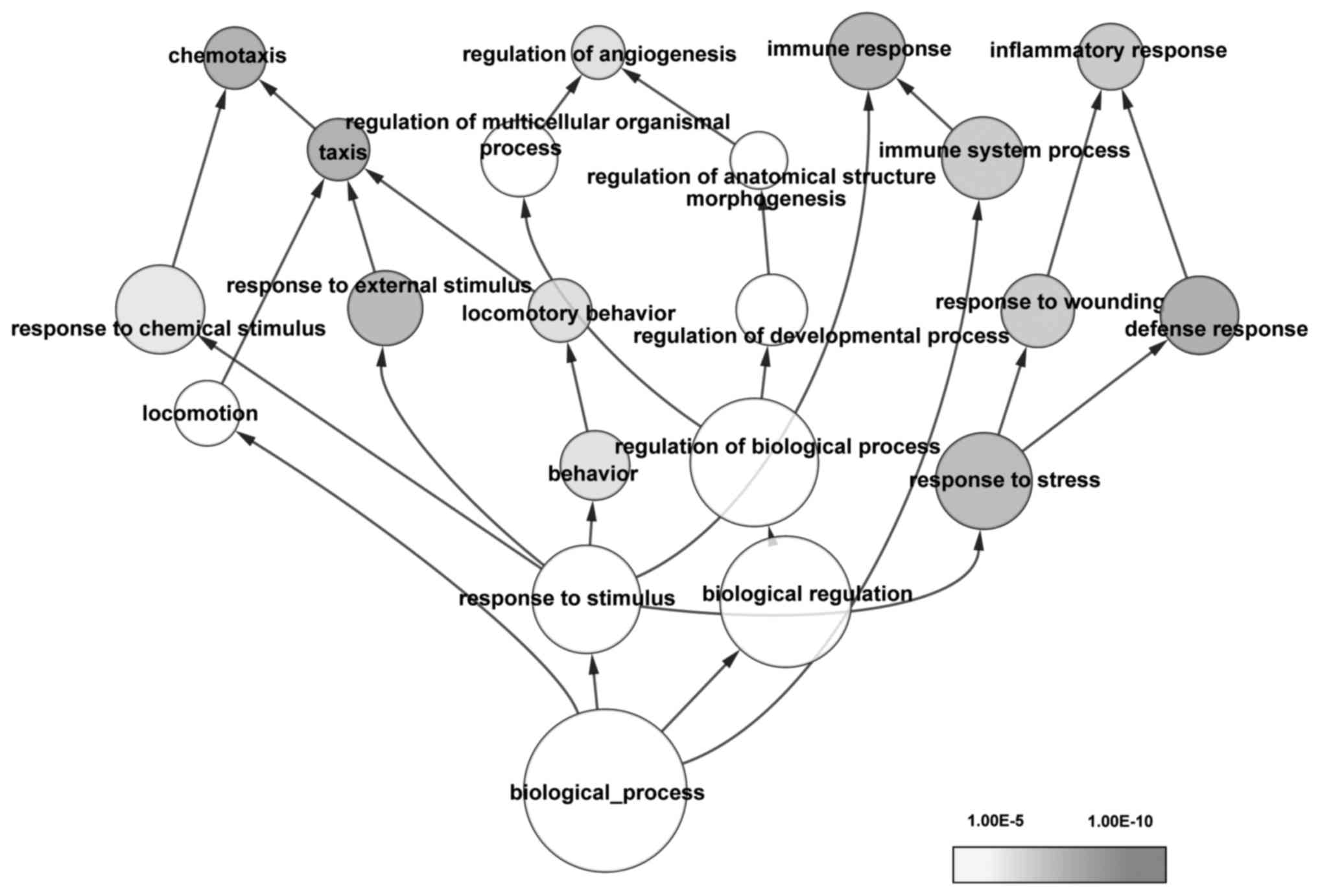
|
Bruix J and Sherman M; American
Association for the Study of Liver Diseases, : Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
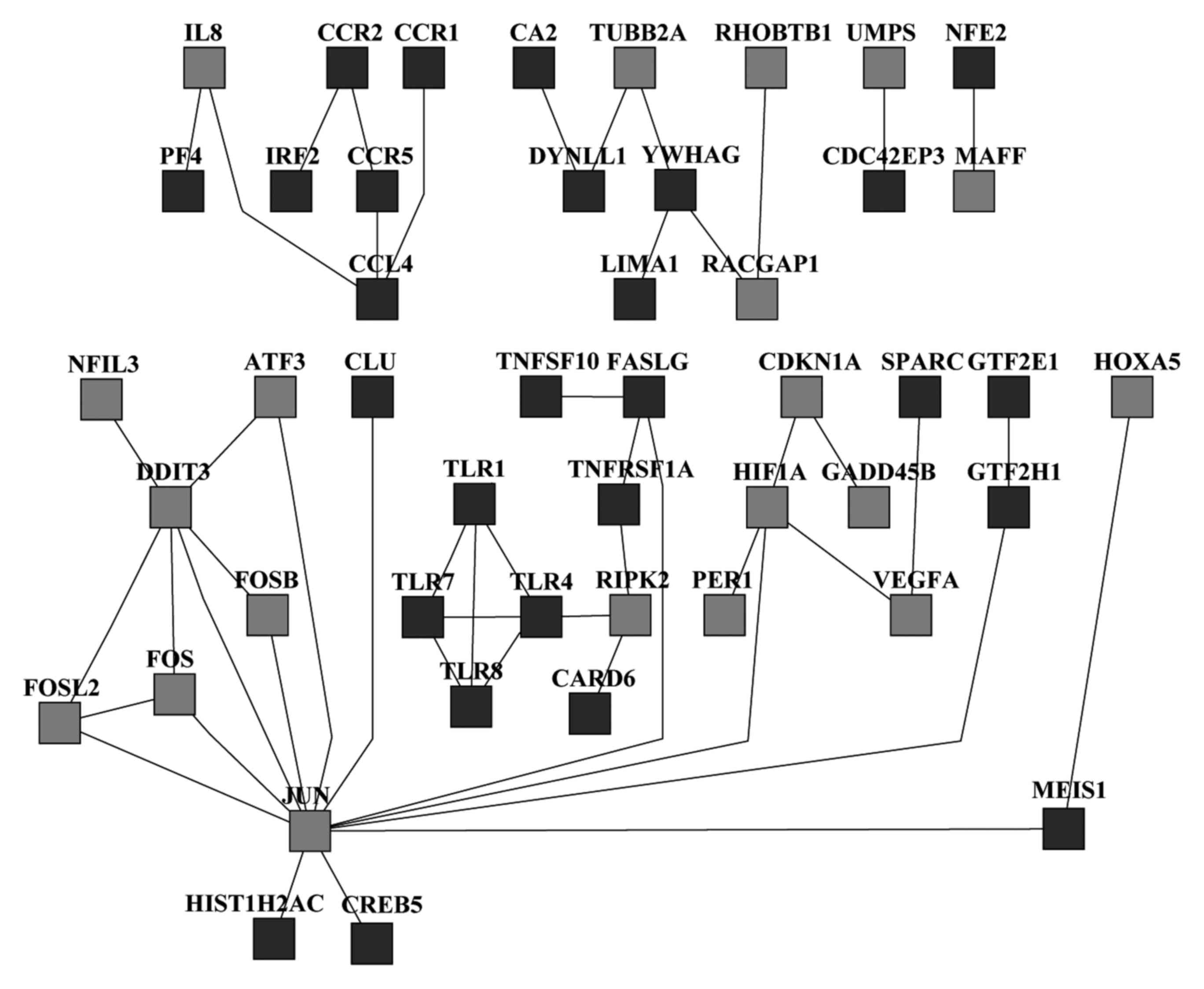
2011. View Article : Google Scholar : PubMed/NCBI
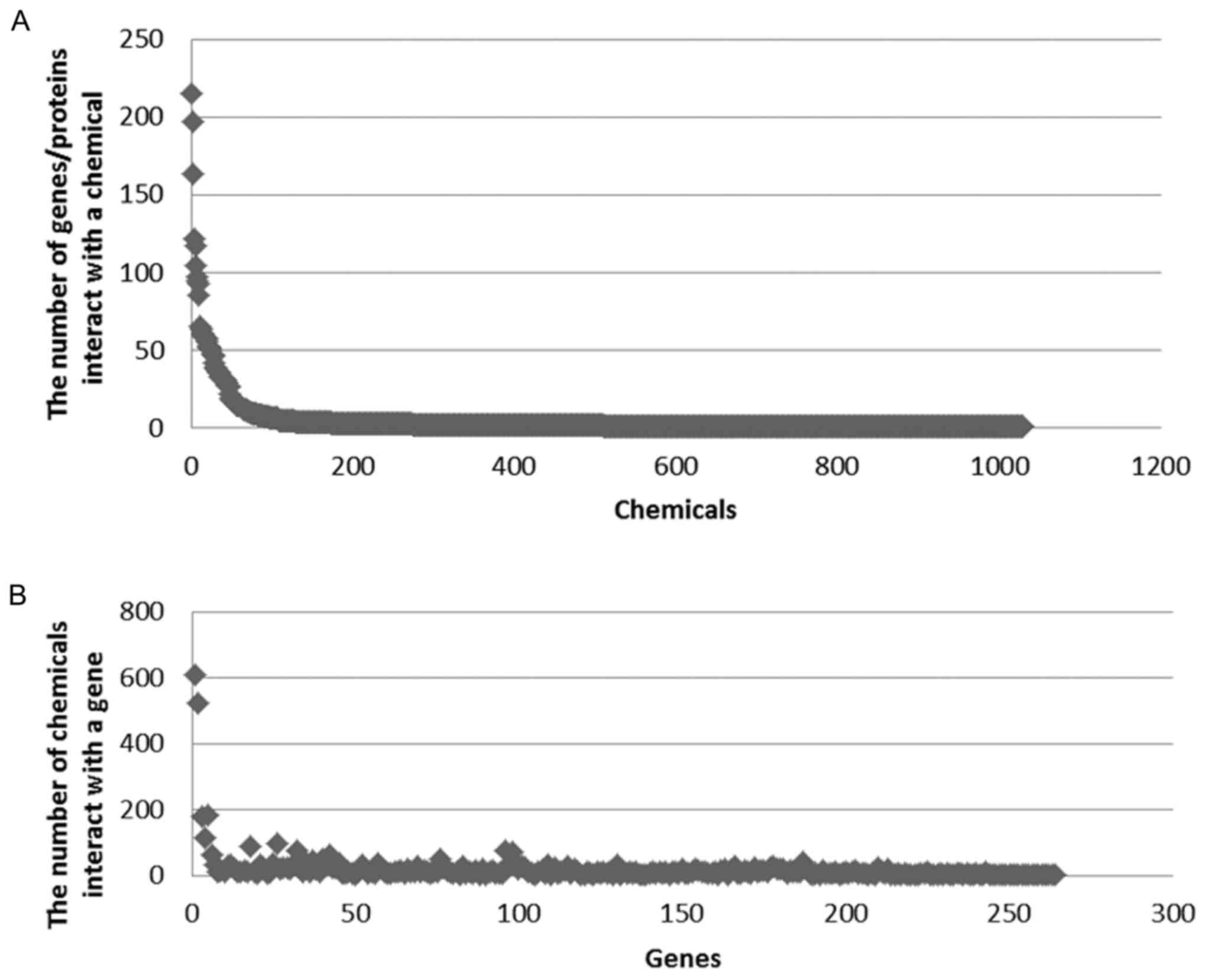
|
|
5
|
European association for the study of the
liver and European organisation for research and treatment of
cancer: EASL-EORTC clinical practice guidelines: Management of
hepatocellular carcinoma. J Hepatol. 56:908–943. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yong KJ, Gao C, Lim JS, Yan B, Yang H,
Dimitrov T, Kawasaki A, Ong CW, Wong KF, Lee S, et al: Oncofetal
gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med.
368:2266–2276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Furuta M, Kozaki K, Tanimoto K, Tanaka S,
Arii S, Shimamura T, Niida A, Miyano S and Inazawa J: The
tumor-suppressive miR-497-195 cluster targets multiple cell-cycle
regulators in hepatocellular carcinoma. PLoS One. 8:e601552013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cillo C, Schiavo G, Cantile M, Bihl MP,
Sorrentino P, Carafa V, D' Armiento M, Roncalli M, Sansano S,
Vecchione R, et al: The HOX gene network in hepatocellular
carcinoma. Int J Cancer. 129:2577–2587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Revill K, Wang T, Lachenmayer A, Kojima K,
Harrington A, Li J, Hoshida Y, Llovet JM and Powers S: Genome-wide
methylation analysis and epigenetic unmasking identify tumor
suppressor genes in hepatocellular carcinoma. Gastroenterology.
145:1424–1435. e1-25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris CC and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Zhu X, Wu L, Yang R, Yang Z, Wang Q
and Wu F: MicroRNA-122 suppresses cell proliferation and induces
cell apoptosis in hepatocellular carcinoma by directly targeting
Wnt/β-catenin pathway. Liver Int. 32:752–760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Dong Z, Liang J, Cao C, Sun J, Ding
Y and Wu D: As an independent prognostic factor, FAT10 promotes
hepatitis B virus-related hepatocellular carcinoma progression via
Akt/GSK3β pathway. Oncogene. 33:909–920. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goyal L, Muzumdar MD and Zhu AX: Targeting
the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res.
19:2310–2318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Han C, Lu L, Magliato S and Wu T:
Hedgehog signaling pathway regulates autophagy in human
hepatocellular carcinoma cells. Hepatology. 58:995–1010. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma W, Wong CC, Tung EK, Wong CM and Ng IO:
RhoE is frequently down-regulated in hepatocellular carcinoma (HCC)
and suppresses HCC invasion through antagonizing the
Rho/Rho-Kinase/Myosin phosphatase target pathway. Hepatology.
57:152–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi M, Chen MS, Sekar K, Tan CK, Ooi LL
and Hui KM: A blood-based three-gene signature for the non-invasive
detection of early human hepatocellular carcinoma. Eur J Cancer.
50:928–936. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang JX, Yu C, Li ZP, Xiao J, Zhang H,
Chen MY and Sun CY: Insights into significant pathways and gene
interaction networks in peripheral blood mononuclear cells for
early diagnosis of hepatocellular carcinoma. J Cancer Res Ther.
12:981–989. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smyth GK: Limma: Linear models for
microarray dataBioinformatics and computational biology solutions
using R and Bioconductor. Springer; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
21
|
Smyth GK: Linear models and empirical
Bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article32004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ferreira JA: The Benjamini-Hochberg method
in the case of discrete test statistics. Int J Biostat. 3:Article
112007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Altermann E and Klaenhammer TR:
PathwayVoyager: Pathway mapping using the Kyoto encyclopedia of
genes and genomes (KEGG) database. BMC Genomics. 6:602005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao J, Ade AS, Tarcea VG, Weymouth TE,
Mirel BR, Jagadish HV and States DJ: Integrating and annotating the
interactome using the MiMI plugin for cytoscape. Bioinformatics.
25:137–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mattingly CJ, Colby GT, Forrest JN and
Boyer JL: The comparative toxicogenomics database (CTD). Environ
Health Perspect. 111:793–795. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wiegers TC, Davis AP, Cohen KB, Hirschman
L and Mattingly CJ: Text mining and manual curation of
chemical-gene-disease networks for the comparative toxicogenomics
database (CTD). BMC Bioinformatics. 10:3262009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davis AP, Wiegers TC, Roberts PM, King BL,
Lay JM, Lennon-Hopkins K, Sciaky D, Johnson R, Keating H, Greene N,
et al: A CTD-Pfizer collaboration: Manual curation of 88,000
scientific articles text mined for drug-disease and drug-phenotype
interactions. Database (Oxford). 2013:bat0802013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moghaddam SJ, Haghighi EN, Samiee S,
Shahid N, Keramati AR, Dadgar S and Zali MR: Immunohistochemical
analysis of p53, cyclinD1, RB1, c-fos and N-ras gene expression in
hepatocellular carcinoma in Iran. World J Gastroenterol.
13:588–593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Watanabe T, Hiasa Y, Tokumoto Y, Hirooka
M, Abe M, Ikeda Y, Matsuura B, Chung RT and Onji M: Protein kinase
R modulates c-Fos and c-Jun signaling to promote proliferation of
hepatocellular carcinoma with hepatitis C virus infection. PLoS
One. 8:e677502013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan Q, He M, Deng X, Wu WK, Zhao L, Tang
J, Wen G, Sun X and Liu Y: Derepression of c-Fos caused by
MicroRNA-139 down-regulation contributes to the metastasis of human
hepatocellular carcinoma. Cell Biochem Funct. 31:319–324. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li S, Fu H, Wang Y, Tie Y, Xing R, Zhu J,
Sun Z, Wei L and Zheng X: MicroRNA-101 regulates expression of the
v-fos FBJ murine osteosarcoma viral oncogene homolog (FOS) oncogene
in human hepatocellular carcinoma. Hepatology. 49:1194–1202. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen Z, Yao C, Wang Z, Yue L, Fang Z, Yao
H, Lin F, Zhao H, Sun YJ, Bian XW, et al: Vastatin, an Endogenous
Antiangiogenesis Polypeptide That Is Lost in Hepatocellular
Carcinoma, Effectively Inhibits Tumor Metastasis. Mol Ther.
24:1358–1368. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Engström K, Willén H, Kåbjörn-Gustafsson
C, Andersson C, Olsson M, Göransson M, Järnum S, Olofsson A,
Warnhammar E and Aman P: The myxoid/round cell liposarcoma fusion
oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma
phenotype in transfected human fibrosarcoma cells. Am J Pathol.
168:1642–1653. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kåbjörn Gustafsson C, Engström K and Åman
P: DDIT3 expression in liposarcoma development. Sarcoma.
2014:9546712014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li T, Su L, Lei Y and Liu X, Zhang Y and
Liu X: DDIT3 and KAT2A proteins regulate TNFRSF10A and TNFRSF10B
expression in endoplasmic reticulum stress-mediated apoptosis in
human lung cancer cells. J Biol Chem. 290:11108–11118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xie M, Sun M, Zhu YN, Xia R, Liu YW, Ding
J, Ma HW, He XZ, Zhang ZH, Liu ZJ, et al: Long noncoding RNA
HOXA-AS2 promotes gastric cancer proliferation by epigenetically
silencing P21/PLK3/DDIT3 expression. Oncotarget. 6:33587–33601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Narendra S, Valente A, Tull J and Zhang S:
DDIT3 gene break-apart as a molecular marker for diagnosis of
myxoid liposarcoma-assay validation and clinical experience. Diagn
Mol Pathol. 20:218–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang Y, Chuang AY, Romano RA, Liégeois
NJ, Sinha S, Trink B, Ratovitski E and Sidransky D: Phospho-∆
Np63α/NF-Y protein complex transcriptionally regulates DDIt3
expression in squamous cell carcinoma cells upon cisplatin
exposure. Cell Cycle. 9:328–338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Riehle KJ, Campbell JS, McMahan RS,
Johnson MM, Beyer RP, Bammler TK and Fausto N: Regulation of liver
regeneration and hepatocarcinogenesis by suppressor of cytokine
signaling 3. J Exp Med. 205:91–103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Taub R: Liver regeneration: From myth to
mechanism. Nat Rev Mol Cell Biol. 5:836–847. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Conner EA, Lee JS, Factor VM and Thorgeirsson SS: Ubiquitous
activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Niwa Y, Kanda H, Shikauchi Y, Saiura A,
Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STAT and FAK signalings in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hsu CN, Lai JM, Liu CH, Tseng HH, Lin CY,
Lin KT, Yeh HH, Sung TY, Hsu WL, Su LJ, et al: Detection of the
inferred interaction network in hepatocellular carcinoma from EHCO
(Encyclopedia of hepatocellular carcinoma genes online). BMC
Bioinformatics. 8:662007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ryschich E, Lizdenis P, Ittrich C, Benner
A, Stahl S, Hamann A, Schmidt J, Knolle P, Arnold B, Hämmerling GJ,
et al: Molecular fingerprinting and autocrine growth regulation of
endothelial cells in a murine model of hepatocellular carcinoma.
Cancer Res. 66:198–211. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Clemens MW: Reply: Association between
agent orange exposure and nonmelanotic invasive skin cancer: A
pilot study. Plast Reconstr Surg. 135:234e–235e. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kennedy GD, Nukaya M, Moran SM, Glover E,
Weinberg S, Balbo S, Hecht SS, Pitot HC, Drinkwater NR and
Bradfield CA: Liver tumor promotion by
2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl
hydrocarbon receptor and TNF/IL-1 receptors. Toxicol Sci.
140:135–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ba Q, Li J, Huang C, Qiu H, Li J, Chu R,
Zhang W, Xie D, Wu Y and Wang H: Effects of benzo [a] pyrene
exposure on human hepatocellular carcinoma cell angiogenesis,
metastasis, and NF-κB signaling. Environ Health Perspect.
123:246–254. 2015.PubMed/NCBI
|
|
53
|
Su Y, Zhao B, Guo F, Bin Z, Yang Y, Liu S,
Han Y, Niu J, Ke X, Wang N, et al: Interaction of benzo [a] pyrene
with other risk factors in hepatocellular carcinoma: A case-control
study in Xiamen, China. Ann Epidemiol. 24:98–103. 2014. View Article : Google Scholar : PubMed/NCBI
|