Introduction
Colon cancer is a common malignant tumor of the
digestive tract, from which morbidity and mortality have been
increasing in recent years. Tumor invasion and metastasis are the
main causes of colon cancer-associated death, but the precise
mechanism mediating these processes remains unclear. Previous
studies have primarily focused on the characteristics of cancer
cells; however, the effects of the tumor microenvironment on
invasion and metastasis have increasingly been gaining attention in
recent years (1).
The tumor microenvironment has an important role in
tumorigenesis and disease progression. Tumor necrosis factor
(TNF)-α is an inflammatory cytokine frequently present in the tumor
microenvironment. TNF-α is primarily secreted by tumor-associated
macrophages and it initiates chronic inflammation (2). TNF-α has biphasic effects, by which it
induces tumor cell apoptosis at high doses while it accelerates
tumor invasion and metastasis with long-term administration of low
doses (3). Tumor cell invasion is
potentiated when tumor cells are co-cultured with macrophages, an
effect primarily mediated by the secretion of TNF-α by macrophages
(4). In addition, TNF-α induces the
expression of angiogenic factors, promotes tumor angiogenesis, and
accelerates tumor metastasis (5).
However, the specific molecular mechanisms by which TNF-α
facilitates tumor cells migration and invasion are unclear.
Tumor-associated calcium signal transducer protein
(TROP)-2 is a transmembrane glycoprotein expressed at high levels
in a variety of human epithelial tumors, including oral (6), pancreatic (7), gastric (8), ovarian (9), lung (10),
and prostate cancer (11). By
contrast, TROP-2 is barely detectable in non-cancerous tissues.
Furthermore, TROP-2 is strongly associated with tumor invasion and
metastasis, and with disease prognosis (12,13). A
previous study from our group have demonstrated that both TROP-2
and TNF-α levels are elevated in colon cancer cells and that TROP-2
and TNF-α levels are directly associated (12). The aim of the present study was to
explore the effect of TNF-α on the regulation of TROP-2 levels in
the context of colon cancer cell migration and invasion.
Materials and methods
Cells and reagents
The human colon cancer cell line HCT-116 was kindly
provided by Professor Haitao Shen (Department of Pathology, Hebei
Medical University, Hebei, China). The cells were frozen in liquid
nitrogen. Recombinant human TNF-α was purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). The Lipofectamine 2000
transfection kit was purchased from Invitrogen (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). TROP-2 small interfering
(si)RNA (5′-CGTGGACAACGATGGCCTCTA-3′) and the negative control
siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′) were purchased from Biovol
Biotechnology (Shanghai, China). Matrigel, a mixture of basement
membrane matrix proteins, was purchased from BD Biosciences
(Franklin Lakes, NJ, USA). Monoclonal mouse anti-human primary
TROP-2 antibody (cat. no. ab65006; 1:1,000) and GAPDH antibody
(cat. no. ab8245; 1:1,000) were purchased from Abcam (Cambridge,
MA, USA). Monoclonal mouse antibodies against human phosphorylated
(p)-c-Jun N-terminal kinase (JNK; cat. no. 9251; 1:1,000), total
(t)-JNK (cat. no. 9252; 1:1,000), p-p38 (cat. no. 9211; 1:1,000),
t-p38 (cat. no. 9212; 1:1,000), p-extracellular signal-regulated
kinase (p-ERK) 1/2 (cat. no. 9101; 1:1,000), and t-ERK1/2 (cat. no.
9107; 1:1,000) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). The specific p-ERK1/2 inhibitor PD98059 was
purchased from Cayman Chemical Company (Ann Arbor, MI, USA).
Horseradish peroxidase-conjugated secondary antibody was purchased
from Beijing Zhongshan Jinqiao Biotechnology Co., Ltd. (1:4,000;
Beijing, China).
Cell culture
HCT-116 human colon cancer cells were cultured in
McCoy's 5A medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (Invitrogen;
Thermo Fisher Scientific, Inc.) and 1% antibiotics (penicillin and
streptomycin). The cells were incubated at 37°C in a 5%
CO2 atmosphere.
Western blot analysis
HCT-116 cells were seeded in culture dishes
(5×105 cells/dish) and cultured until they reached 80%
confluence. Then, the cells were cultured in serum-free medium for
an additional 24 h. The cells were subsequently incubated with
various concentrations of TNF-α for the indicated period of time.
Once cells were harvested, total proteins (50 µg) were separated
using SDS-PAGE (10% gel) and the separated proteins were
transferred to polyvinylidene fluoride membranes. The membranes
were blocked with 5% skim milk at room temperature for 1 h, and
subsequently incubated at 4°C overnight with the primary antibody
of interest and anti-GAPDH as a loading control. Then, the
membranes were washed three times with TBS/0.1% Tween-20 and
incubated with goat anti-rabbit secondary antibody at room
temperature for 2 h. The protein signals were detected using
enhanced chemiluminescence (Beyotime Institute of Biotechnology,
Haimen, China). Western blot results were analyzed using ImageJ
software (version 1.41; National Institutes of Health, Bethesda,
MD, USA).
Wound healing assay
HCT-116 cells in the logarithmic phase were seeded
in 6-well plates. Once the cells reached 80% confluence, wounds
were generated by scratching the cells with a 100-µl pipette tip.
Then, cells were washed with PBS and treated with TNF-α at a final
concentration of 20 µg/l in serum-free medium for 24 h. Control
cells were treated with PBS in serum-free medium. Images of the
cells were captured at 0 and 24 h after the wound was created. The
width of the wound at 5 randomly selected sites was measured using
a Leica TCS-SP5 confocal microscope (magnification, ×200; Leica
Microsystems, Inc., Buffalo Grove, IL, USA), and the average wound
width was calculated. The wound healing rate was calculated using
ImageJ (version 2.1; National Institutes of Health) (14). The experiment was conducted in
triplicate.
Transwell invasion assay
The invasive ability of cells was assessed using a
Transwell assay. The invasive potential of HCT-116 cells was
assessed by determining the number of cells that passed through a
polycarbonate membrane (8 µm pore size) in 24-well transwell
chambers. The upper Transwell filters (Corning Inc., Corning, NY,
USA) were coated with Matrigel (BD Biosciences). Single cell
suspensions of HCT-116 cells were cultured until they reached the
logarithmic phase, and the cells (1×105) were
subsequently seeded into the upper chamber of Transwell filters in
serum-free medium supplemented with 20 µg/l TNF-α. Complete medium
was added to the lower chambers. The cells were incubated for 24 h,
and non-migratory cells on the upper side of the filter were
removed using cotton swabs. Cells that had migrated through the
pores of the filter were fixed in methanol at 4°C for ~20–30 min,
stained with 0.1% crystal violet for 30 min at 37°C, and washed
with PBS. The number of migratory cells was determined using an
inverted light microscope at a magnification of ×200. The average
number of cells in five randomly selected fields was calculated,
and the experiment was conducted in triplicate.
siRNA-mediated silencing of
TROP-2
In order to knock-down endogenous TROP-2 expression,
TROP-2-specific siRNA was transfected into cells using
Lipofectamine® 2000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Sequences were as follows: TROP-2 siRNA,
5′-CGTGGACAACGATGGCCTCTA-3′; control siRNA,
5′-UUCUCCGAACGUGUCACGUTT-3′. Cells were transfected with TROP-2
siRNA (50 nM) or negative control siRNA (50 nM) using
Lipofectamine® 2000. Non-transfected cells (blank
control group) were included in the analysis. Two days after
transfection, transfected cells were harvested and TROP-2 mRNA and
protein expression was evaluated by reverse
transcription-quantitative polymerase chain reaction and western
blot analysis.
RT-qPCR
RT-qPCR was used to determine TROP-2 mRNA expression
levels of the human colon cancer HCT-116 cell line. Primers were as
follows: TROP-2 forward, 5′-TCACCAACCGGAGAAAGTCG-3′ and reverse,
5′-AGGAAGCGTGACTCACTTGG-3′; β-actin forward,
5′-CACGAAACTACCTTCAACTCC-3′ and reverse,
5′-CATACTCCTGCTTGCTGATC-3′. Following transfection, HCT-116 cells
were harvested for total RNA extract using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). For reverse
transcription, RNase-free EP tubes containing Mix I (5 µl total
RNA, 0.5 µl of 50 µmol/l oligo(dT), 0.5 µl random primer, 1 µl of
10 mmol/l dNTP solution and 5 µl diethylpyrocarbonate-treated
water) were incubated in a 65°C water bath for 5 min and
immediately transferred to ice water for 1 min. Then, 4 µl of 5X
first-strand buffer, 2 µl of 0.1 mol/l DTT, 1 µl of 40 U/µl
RNaseOUT (Invitrogen; Thermo Fisher Scientific, Inc.), and 1 µl
SuperScript III reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.) were added to Mix I for a total reaction volume
of 20 µl. The reactions were incubated as follows: 5°C for 5 min,
50°C for 60 min, and 70°C for 15 min. The resulting cDNA products
were placed on ice immediately following the reaction. qPCR was
performed using SYBR Premix DimerEraser (Takara Biotechnology Co.,
Ltd., Dalian, China). The conditions for the qPCR were as follows:
50°C for 30 min, a denaturation step at 95°C for 2 min, 40 cycles
of 95°C for 10 sec and 60°C for 30 sec, and an elongation step at
70°C for 30 sec. All experiments were conducted in triplicate.
Relative gene expression levels were calculated using the
2−∆∆Cq method (15).
Statistical analysis
SPSS software (version 17.0; SPSS, Inc., Chicago IL,
USA) was used for statistical analyses. Data with a normal
distribution are presented as the mean ± standard deviation. A
t-test was used to compare differences in means between two groups.
One-way ANOVA was used to compare differences in means among
multiple groups, and the student Newman-Keuls-q method was used for
multiple comparisons between groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effect of TNF-α on TROP-2 levels in
colon cancer cells
Previous studies from our group have demonstrated
that TNF-α and TROP-2 protein levels are abundant in colon cancer,
and higher levels of these proteins are associated with a poor
prognosis (12). To investigate the
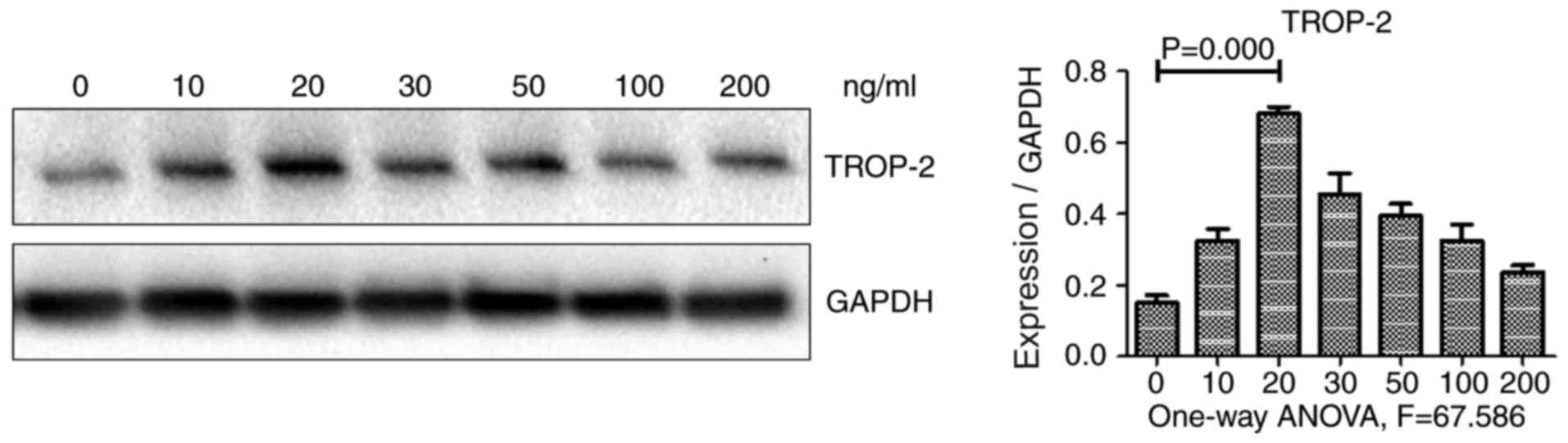
effect of TNF-α on TROP-2 expression, HCT-116 colon cancer cells
were incubated with various concentrations of TNF-α, and TROP-2
protein levels were subsequently examined using western blot
analysis. As illustrated in Fig. 1,
TROP-2 protein expression levels exhibited a biphasic response,
increasing in cells incubated with decreased concentrations of
TNF-α (10 and 20 µg/l), but decreasing in cells incubated with
increased concentrations (30, 50, 100 and 200 µg/l). TROP-2 protein
levels were the highest in cells incubated with 20 µg/l TNF-α.
These results demonstrated that low concentrations of TNF-α
upregulated TROP-2 expression in HCT-116 cells. Therefore, TNF-α at
a concentration of 20 µg/l was used in the subsequent
experiments.
Effect of TNF-α on TROP-2
regulation
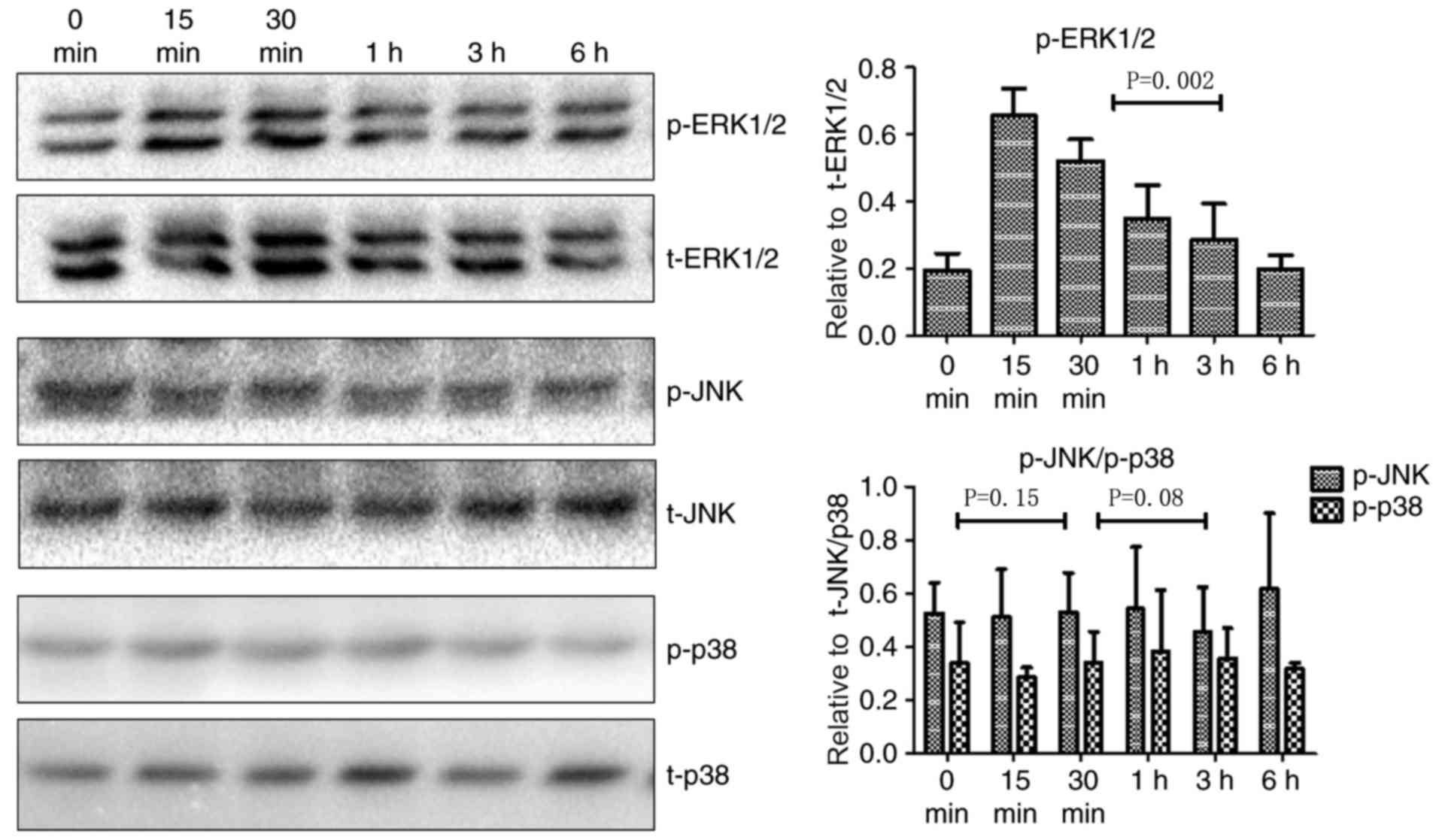
Little is known about the signaling pathways that
mediate TNF-α-induced upregulation of TROP-2. The mitogen-activated
protein kinase (MAPK) signaling pathway is involved in multiple
biological processes in tumors, including cell proliferation, cell
cycle regulation, migration, and invasion. Since the MAPK pathway
can be activated by a variety of inflammatory cytokines and TROP-2
is associated with aggressive tumor cell behaviors, the role of
MAPK signaling in TNF-α-mediated regulation of TROP-2 was
investigated. JNK, p38, and ERK1/2 are key members of the MAPK
signaling pathway. As illustrated in Fig.
2, p-ERK1/2 levels increased in cells treated with TNF-α at 15
min post-stimulation, whereas there were no significant changes in
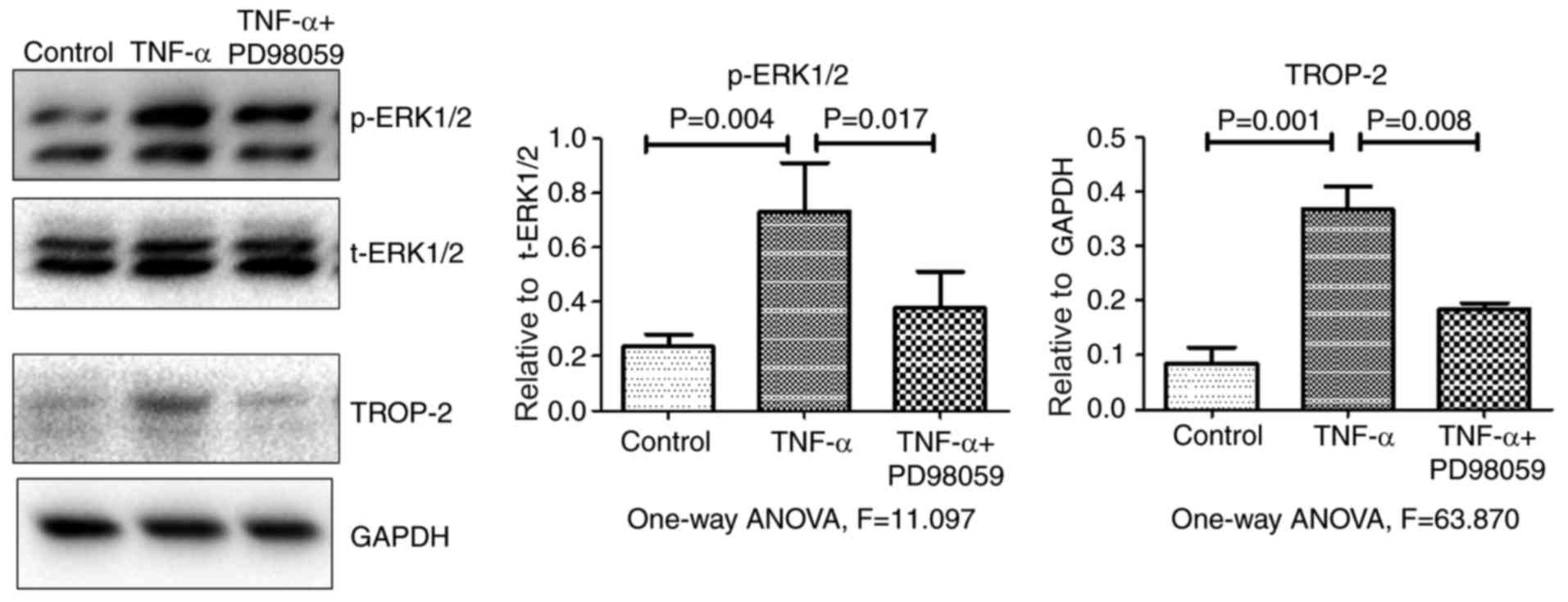
p-JNK and p-p38 levels. Fig. 3
demonstrates that, compared with cells treated with TNF-α alone,
p-ERK1/2 and TROP-2 protein levels significantly decreased in cells
that were pretreated with the specific ERK1/2 inhibitor PD98059 (20
µM) for 30 min. Together, the present data suggested that TNF-α
stimulation upregulated TROP-2 protein expression levels by
activating the ERK1/2 signaling pathway.
TNF-α promotes colon cancer cell
migration and invasion by upregulating TROP-2
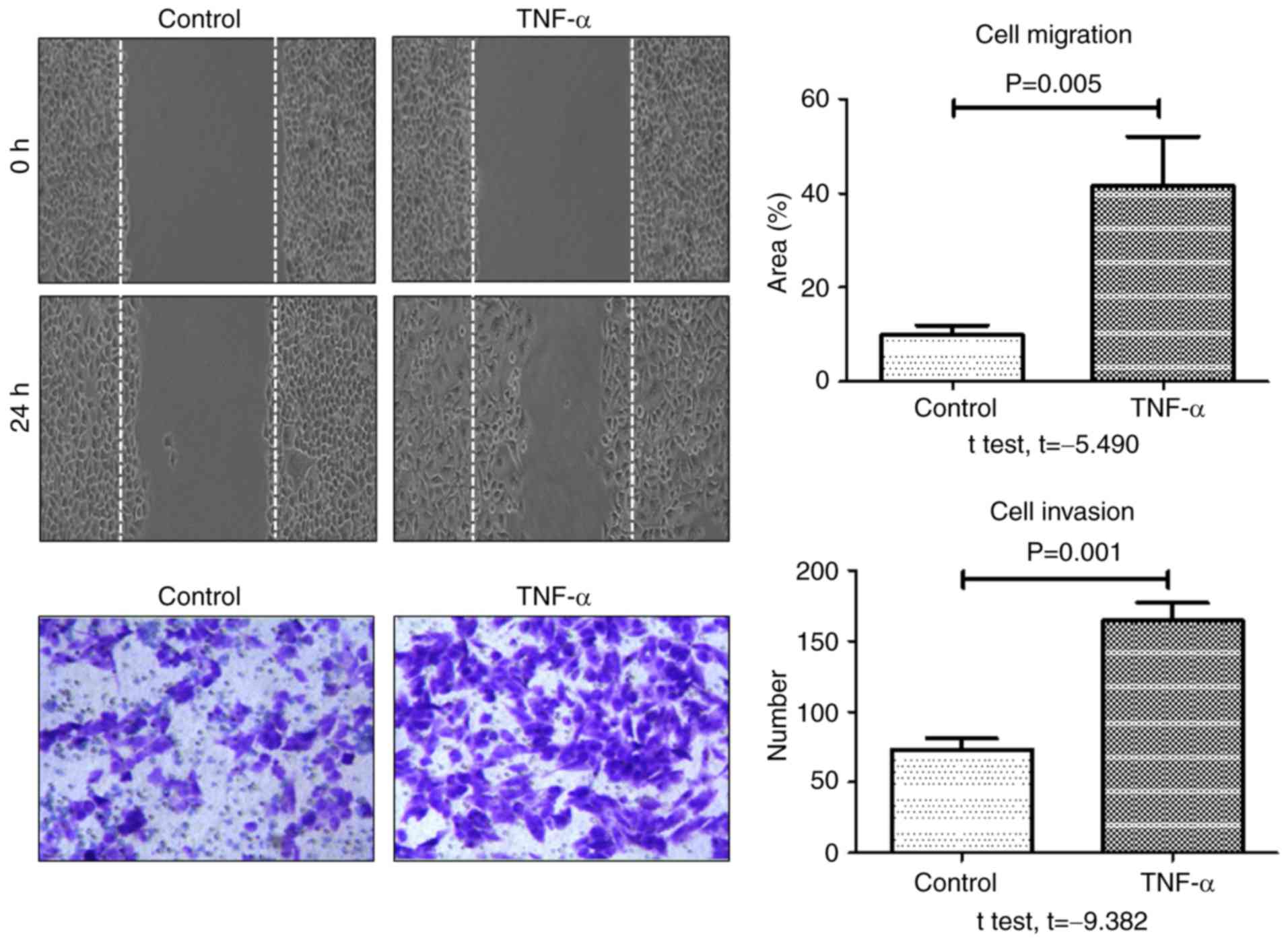
Aggressive cancer cells are characterized by an
increase in cell migration and invasion abilities. To investigate
the role of TNF-α in metastasis, the effect of TNF-α stimulation on
migration and invasion of HCT-116 cells was evaluated using and
wound healing and transwell assays, respectively. As presented in
Fig. 4, the rate of wound healing and
the number of invasive cells at 24 h were significantly increased
in the TNF-α-treated group compared with the control group. These
results indicated that TNF-α significantly enhanced the migratory
and invasive potential of HCT-116 colon cancer cells.
Although it was demonstrated that TNF-α upregulated
TROP-2 protein levels and enhanced the invasive potential of colon
cancer cells, it remained unclear if the latter effect was mediated
by the upregulation of TROP-2. Therefore, the role of TROP-2 in the
TNF-α-induced migration and invasion of colon cancer cells was
examined by silencing TROP-2 expression with a specific siRNA. The
negative control group was transfected with a non-specific
scrambled siRNA, and non-transfected cells were used as a blank
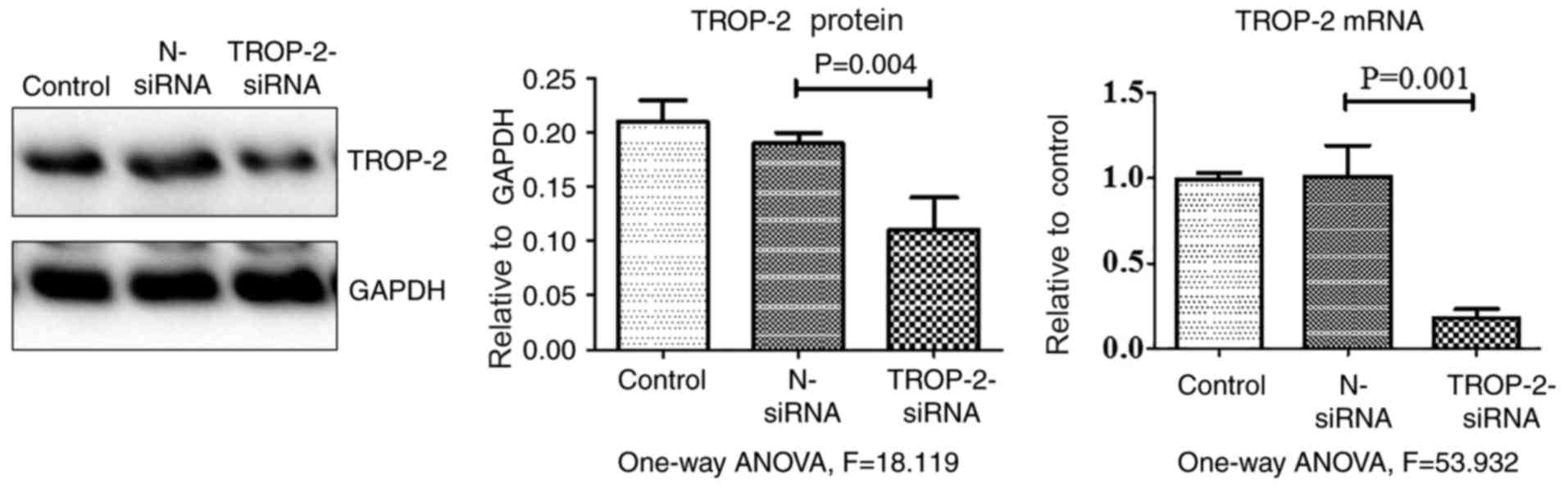
control. As presented in Table I and
Fig. 5, TROP-2 expression levels in
cells transfected with TROP-2 siRNA significantly decreased at both
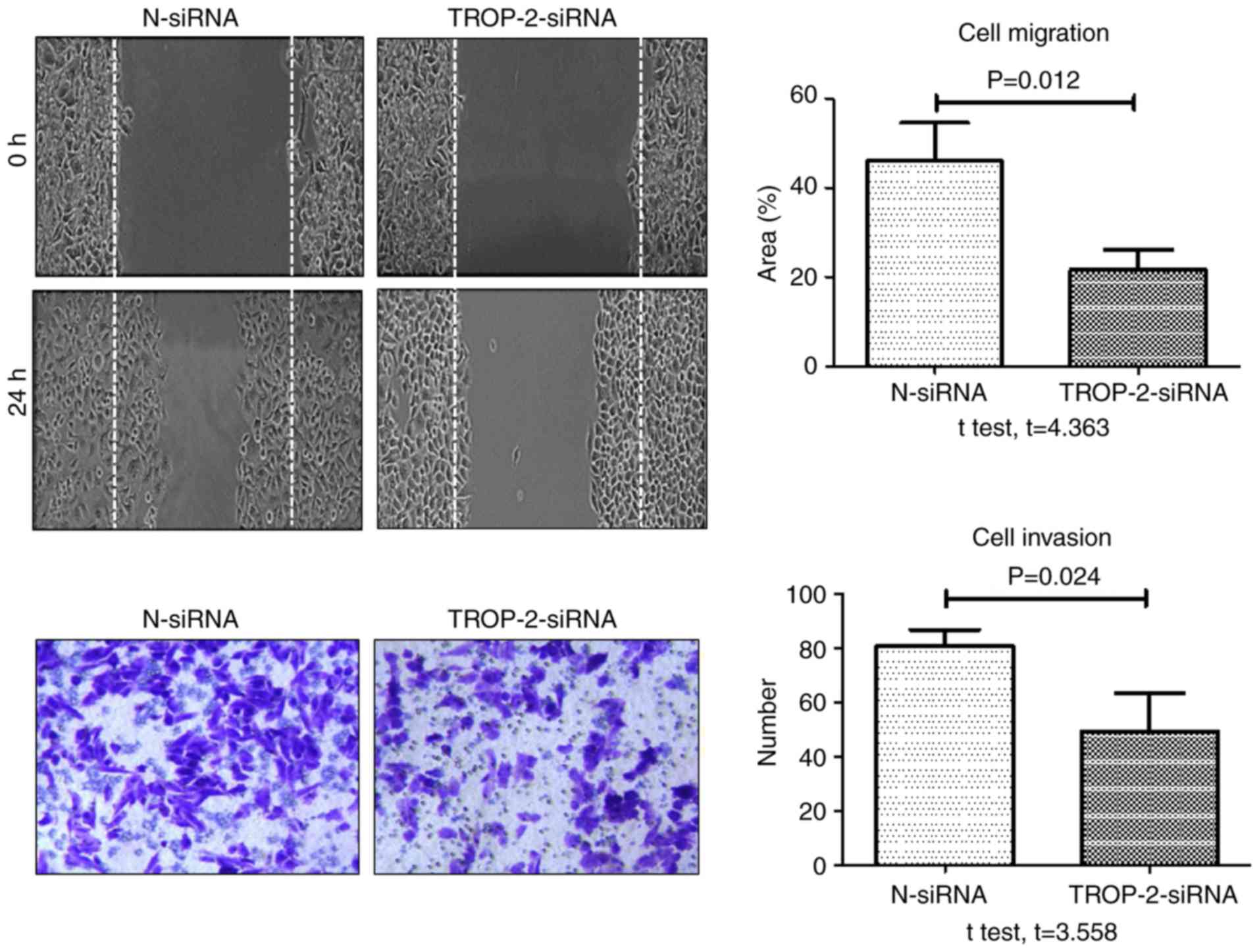
the mRNA and protein level compared with control groups. When
migration and invasion were examined, the rate of TNF-α-induced
wound healing and number of invasive cells at 24 h were
significantly decreased in the TROP-2 siRNA group compared with the
negative control siRNA group (Fig.
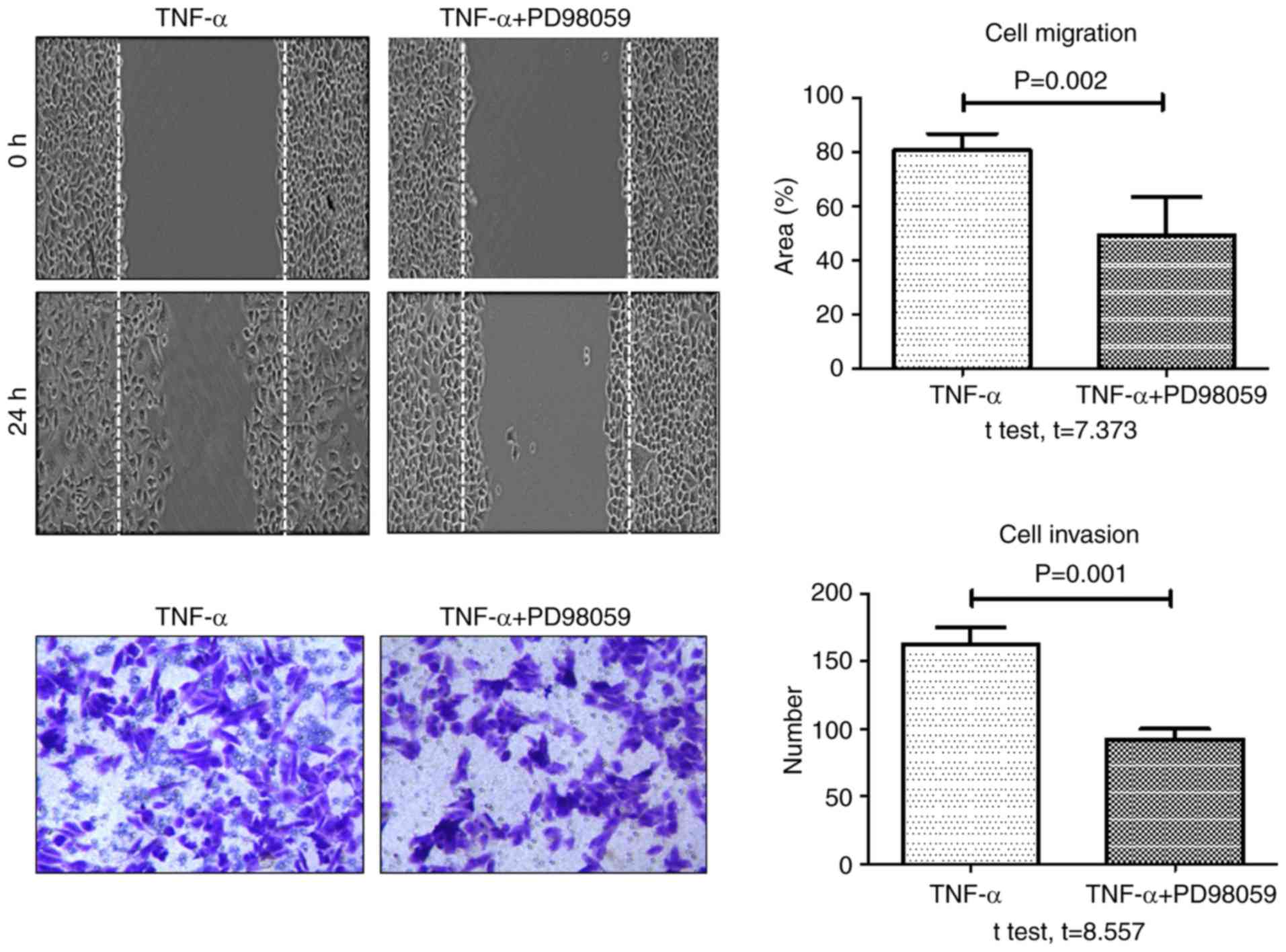
6). In addition, the rate of TNF-α-induced wound healing and
invasion significantly decreased in cells pretreated with PD98059
(20 µM) for 30 min, compared with cells treated with TNF-α alone
(Fig. 7). These data demonstrated
that TROP-2 knockdown inhibited TNF-α-induced invasion and
migration in colon cancer cells, and that TNF-α promoted invasion
and migration in colon cancer cells by upregulating TROP-2 protein
expression via the ERK1/2 signaling pathway.
 | Table I.Relative expression levels and mean
band intensities of TROP-2 mRNA and protein levels in cells
transfected with TROP-2 siRNA. |
Table I.
Relative expression levels and mean
band intensities of TROP-2 mRNA and protein levels in cells
transfected with TROP-2 siRNA.
| Group | TROP-2 mRNA | TROP-2 protein |
|---|
| siRNA |
0.02±0.01a |
0.10±0.04a |
| Negative control |
1.02±0.02a |
0.19±0.01a |
| Blank control |
1.00±0.01a |
0.21±0.02a |
| P-value | 0.001 | 0.004 |
Discussion
The tumor microenvironment is a popular topic in
cancer research, and scholars consider tumor-associated
inflammation as the seventh hallmark of cancer (16). The inflammatory microenvironment is
strongly associated with tumorigenesis and cancer progression;
however, the mechanism by which it promotes invasion and metastasis
remains unclear. TNF-α is a key inflammatory cytokine present in
the tumor microenvironment. Although TNF-α has been linked to tumor
invasion and metastasis, the specific mechanisms underlying this
association are unclear. Previous studies have demonstrated that
TNF-α can activate the MAPK/ERK pathway, upregulate matrix
metallopeptidase (MMP)-9, CD26 and fibroblast activation protein
(FAP)-α levels, and enhance tumor cell invasion and metastasis in
breast cancer (17). TNF-α may also
upregulate MMPs in oral cancer cells, thereby promoting invasion
and metastasis (18). In addition,
TNF-α promotes lymphangiogenesis and lymphatic metastasis in
gallbladder carcinoma by activating the ERK1/2 or the activator
protein-1/vascular endothelial growth factor (VEGF)-D pathway
(19). However, there are no previous
reports describing the role of TNF-α in colon cancer invasion and
metastasis.
TROP-2 is a transmembrane glycoprotein expressed at
high levels in a variety of human epithelial tumors. TROP-2
is strongly associated with tumor invasion and metastasis, and high
levels of TROP-2 are associated with a poor prognosis in gastric
carcinoma (8). However, the mechanism
underlying TROP-2 overexpression in tumors remains unclear. The
mechanism by which TROP-2 is upregulated in tumors may provide
insights that could facilitate the development of approaches for
early diagnosis and treatment.
In previous studies, both TROP-2 and TNF-α were
markedly expressed at the protein level in a large number of colon
cancer tissue samples (12).
Therefore, it was speculated that TNF-α may upregulate TROP-2
protein levels in colon cancer cells, thereby promoting tumor cell
migration and invasion. The findings of the present study validate
this hypothesis.
In the present study, the effect of various
concentrations of TNF-α on TROP-2 protein levels was examined in
colon cancer cells. TROP-2 protein levels increased in cells
incubated with low concentrations of TNF-α and decreased in cells
incubated with higher concentrations. Maximum TROP-2 levels were
observed in cells treated with 20 µg/l TNF-α. It is hypothesized
that TROP-2 levels are decreased in cells treated with high
concentrations of TNF-α potentially due to increased cytotoxicity.
Wound healing and cell invasion were enhanced in cells treated with
20 µg/l TNF-α compared with control untreated cells. Thus, the
present data demonstrated that low concentrations of TNF-α
upregulated TROP-2 protein expression and promoted colon cancer
cell migration and invasion.
To further confirm the hypothesis that low
concentrations of TNF-α promote invasion in colon cancer cells by
upregulating TROP-2, expression of TROP-2 was silenced using siRNA
technology. Cell migration and invasion were significantly
inhibited in TROP-2 siRNA-transfected HCT-116 cells treated with
TNF-α, compared with control siRNA-transfected cells. These data
demonstrate that the inflammatory cytokine TNF-α promotes migration
and invasion in colon cancer cells by upregulating TROP-2.
To date, there have been no reports describing the
mechanism by which inflammatory cytokines, such as TNF-α,
upregulate TROP-2. Choo et al (20) demonstrated that TNF-α may induce colon
cancer cells to metastasize to the lungs by activating the ERK
signaling pathway. Hagemann et al (21) reported that TNF-α secretion by
tumor-associated macrophages promotes the invasion and metastasis
of breast cancer cells by activating the JNK pathway and
upregulating MMPs and VEGF. Muthukumaran et al reported that
TNF-α enhances the metastatic potential of SKA and SKOV-3 ovarian
cancer cells by activating the JNK signaling pathway and modulating
CD44 levels (22). Therefore, it was
hypothesized in the present study that TNF-α may upregulate TROP-2
in HCT-116 cells by activating the MAPK pathway. The results
demonstrated that p-ERK1/2 levels significantly increased in cells
treated with TNF-α and this effect was significantly inhibited in
cells pretreated with PD98059, a specific ERK1/2 inhibitor. The
effect of PD98059 on wound healing and invasion assays suggested
that the TNF-α-induced tumor migration and invasion may be mediated
by ERK1/2. By contrast, the phosphorylation levels of the MAPK
proteins p38 and JNK did not significantly change in cells treated
with TNF-α, suggesting that these proteins do not serve a role in
the TNF-α-induced upregulation of TROP-2.
In conclusion, TNF-α is a key inflammatory cytokine
in the tumor microenvironment, and the present results suggest that
it may promote colon cancer invasion by upregulating TROP-2
expression via the ERK1/2 signaling pathway. Therefore, the ERK1/2
pathway may represent a potential therapeutic target for the
treatment of colon cancer.
Acknowledgements
The present study was partly supported by the
National Natural Science Foundation of China (grant nos. 81-172317,
30972887, and 81572856) and Beijing Municipal Administration of
Hospitals Clinical Medicine Development of Special Funding Support
(grant no. ZYLX201504). The authors would like to thank the members
of the Beijing Friendship Hospital laboratory for providing
technical assistance and our colleagues for help during the course
of this study.
Glossary
Abbreviations
Abbreviations:
|
TNF-α
|
tumor necrosis factor-α
|
|
TROP-2
|
tumor-associated calcium signal
transducer protein-2
|
References
|
1
|
Eiró N and Vizoso FJ: Inflammation and
cancer. World J Gastrointest Surg. 27:62–72. 2012. View Article : Google Scholar
|
|
2
|
Calcinotto A, Grioni M, Jachetti E, Curnis
F, Mondino A, Parmiani G, Corti A and Bellone M: Targeting TNF-α to
neoangiogenic vessels enhances lymphocyte infiltration in tumors
and increases the therapeutic potential of immunotherapy. J
Immunol. 188:2687–2694. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ikemoto S, Sugimura K, Yoshida N, Wada S,
Yamamoto K and Kishimoto T: TNF alpha, IL-1 beta and IL-6
production by peripheral blood monocytes in patients with renal
cell carcinoma. Anticancer Res. 20:317–321. 2000.PubMed/NCBI
|
|
4
|
Wu Y, Deng J, Rychahou PQ, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsushita S, Nitanda T, Furukawa T,
Sumizawa T, Tani A, Nishimoto K, Akiba S, Miyadera K, Fukushima M,
Yamada Y, et al: The effect of a thymidine phosphorylase inhibitor
on angiogenesis and apoptosis in tumors. Cancer Res. 59:1911–1916.
1999.PubMed/NCBI
|
|
6
|
Fong D, Spizzo G, Gostner JM, Gastl G,
Moser P, Krammel C, Gerhard S, Rasse M and Laimer K: TROP2: A novel
prognostic marker in squamous cell carcinoma of the oral cavity.
Mod Pathol. 21:186–191. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fong D, Moser P, Krammel C, Gostner JM,
Margreiter R, Mitterer M, Gastl G and Spizzo G: High expression of
TROP2 correlates with poor prognosis in pancreatic cancer. Br J
Cancer. 99:1290–1295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mühlmann G, Spizzo G, Gostner J, Zitt M,
Maier H, Moser P, Gastl G, Zitt M, Müller HM, Margreiter R, et al:
TROP2 expression as prognostic marker for gastric carcinoma. J Clin
Pathol. 62:152–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bignotti E, Todeschini P, Calza S,
Falchetti M, Ravanini M, Tassi RA, Ravaggi A, Bandiera E, Romani C,
Zanotti L, et al: Trop-2 overexpression as an independent marker
for poor overall survival in ovarian carcinoma patients. Eur J
Cancer. 46:944–953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pak MG, Shin DH, Lee CH and Lee MK:
Significance of EpCAM and TROP2 expression in non-small cell lung
cancer. World J Surg Oncol. 10:532012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trerotola M, Rathore S, Goel HL, Li J,
Alberti S, Piantelli M, Adams D, Jiang Z and Languino LR: Trop-2
and alpha-2beta1 integrin surface receptors as markers of putative
human prostate cancer stem cells. Am J Transl Res. 2:135–144.
2010.PubMed/NCBI
|
|
12
|
Zhao P, Yu HZ and Cai JH: Clinical
investigation of TROP-2 as an independent biomarker and potential
therapeutic target in colon cancer. Mol Med Rep. 12:4364–4369.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stepan LP, Trueblood ES, Hale K, Babcook
J, Borges L and Sutherland CL: Expression of Trop2 cell surface
glycoprotein in normal and tumor tissues: Potential implications as
a cancer therapeutic target. J Histochem Cytochem. 59:701–710.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schnider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Guo Q, Zhao Y, Chen J, Wang S, Hu
J and Sun Y: BRAF-activated long non-coding RNA contributes to cell
proliferation and activates autophagy in papillary thyroid
carcinoma. Oncol Lett. 8:1947–1952. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer related inflammation, the seventh hallmark
of cancer: Links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wolczyk D, Zaremba-Czogalla M,
Hryniewicz-Jankowska A, Tabola R, Grabowski K, Sikorski AF and
Augoff K: TNF-α promotes breast cancer cell migration and enhances
the concentration of membrane-associated proteases in lipid rafts.
Cell Oncol (Dordr). 39:353–363. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao ZL, Yang CC, Xu XH, et al:
Experimental study on TNF-α promoting invasion and metastasis of
oral cancer cells by regulating MMPs. Stomatology. 34:9–12.
2014.
|
|
19
|
Hong H, Jiang L, Lin Y, He C, Zhu G, Du Q,
Wang X, She F and Chen Y: TNF-alpha promotes lymphangiogenesis and
lymphatic metastasis of gallbladder cancer through the
ERK1/2/AP-1/VEGF-D pathway. BMC Cancer. 16:2402016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choo MK, Sakurai H, Koizumi K and Saiki I:
Stimulation of cultured colon 26 cells with TNF-alpha promotes lung
metastasis through the extracellular signal-regulated kinase
pathway. Cancer Lett. 230:47–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hagemann T, Wilson J, Kulbe H, Li NF,
Leinster DA, Charles K, Klemm F, Pukrop T, Binder C and Balkwill
FR: Macrophages induce invasiveness of epithelial cancer cells via
NF-kappaB and JNK. J Immunol. 175:1197–1205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Muthukumaran N, Miletti-González KE,
Ravindranath AK and Rodríguez-Rodríguez L: Tumor necrosis
factor-alpha differentially modulates CD44 expression in ovarian
cancer cells. Mol Cancer Res. 4:511–520. 2006. View Article : Google Scholar : PubMed/NCBI
|