Introduction
The transforming growth factor-β (TGF-β) pathway
appears to serve a dual function in tumor development and
progression. It suppresses early tumorigenesis, but also
facilitates malignant transformation and invasion (1). Therefore, the function of TGF-β in
tumorigenesis is controversial (2).
There is growing evidence to suggest that
constitutive and somatically acquired alterations in TGF-β
signaling are associated with an increased risk of colorectal
cancer (3,4). Colorectal cancer cells evade the
antiproliferative effects of TGF-β by acquiring mutations in
components of this signaling pathway. Common mutations of TGF-β
pathway components, including ligands, receptors, Smads and
Smad-interacting transcription factors, increases the risk of
developing colorectal cancer (5–7).
Cross-talk between TGF-β, Smads and other cell signaling pathways
is also activated by the TGF-β receptors, through either
phosphorylation or direct interaction (8). For example, type 1 transforming growth
factor β receptor (TGFBR1) may also participate in the regulation
of other non-Smad signaling pathways, including phosphoinositide
3-kinase (PI3K)/protein kinase B (AKT), p38 mitogen-activated
protein kinase (MAPK) and nuclear factor-κB (9).
TGFBR1*6A, a common allele located at exon 1 of the
TGFBR1 gene, has been reported to act as a low-penetrance
tumor-susceptibility allele in human colorectal cancer cell lines.
It is also less effective at transducing TGF-β signaling compared
with the TGFBR1*9A wild type (10).
Functional studies have demonstrated that the TGFBR1*6A allele is
associated with an increased risk of various different
malignancies, including breast cancer and osteosarcoma (11). A meta-analysis conducted by Wang et
al (12) has also indicated that
the TGFBR1*6A allele increases the risk of colorectal cancer. Zhang
et al (13) also concluded
that TGFBR1*6A may be low-penetrance, but has a statistically
significant increased risk of colorectal cancer. Furthermore,
TGFBR1*6A has been demonstrated to increase the migration and
invasion of MCF-7 breast cancer cells in response to TGF-β1
(14). The results suggest that the
variant TGFBR1*6A may serve an oncogenic function in cancer
development, switching the TGF-β1 growth inhibitory signals into
growth stimulatory signals (15).
Certain studies have confirmed the presence of an
association between TGFBR1*6A and colorectal cancer, but others
have failed to establish any such association (16–18).
Therefore, the molecular mechanism underlying the contribution of
TGFBR1*6A to colorectal cancer development remains under
investigation and the function of this variant in colorectal cancer
remains controversial (19). A
combined analysis of six studies assessing TGFBR1*6A in colon
cancer cases and controls indicated that TGFBR1*6A carriers were at
an increased risk of developing colorectal cancer (20), but a large case control study did not
confirm this association (21). Based
on this controversy and the uncertain association between TGFBR1*6A
and non-Smad pathways in colorectal cancer, the present study was
conducted as a means to assess the effect of TGFBR1*6A polymorphism
on colorectal cancer cells. The present study also evaluated the
association between TGFBR1*6A and the non-Smad pathways in terms of
tumor cell migration and invasion.
Materials and methods
Cell culture and transfection
The human colorectal cancer SW48 and DLD-1 cell
lines were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA) and were cultured according to ATCC
recommendations. The SW48 and DLD-1 cells were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 2 mM glutamax
(Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin, 100
µg/ml streptomycin and 250 ng/ml amphoterycin (all Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified atmosphere
including 5% CO2. Cells were transfected with 0.5 µg/ml
pCMV5-TGFBR1*6A-HA (supplied by Professor Boris Pasche) (22), or with an empty vector alone.
Transfections were performed using Lipofectamine (Gibco; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
protocols. Stably transfected cells were selected in the presence
of 600 g/ml Geneticin reagent (G418) (Sigma-Aldrich; Merck KGaA).
For maintenance and culturing of transfectant clones, 400 g/ml G418
was added to the medium. Single-cell clones were subsequently
maintained in 400 g/ml G418 and clones positive for TGFBR1*6A
expression were identified by screening via reverse
transcription-polymerase chain reaction (RT-PCR).
DNA extraction and polymerase chain
reaction (PCR)
DNA was extracted from the colorectal cancer cells
using proteinase K digestion (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) at 55°C overnight, followed by phenol/chloroform
extraction and ethanol precipitation. Subsequently, 0.5 ml
phenol/chloroform was added, samples were centrifuged at 14,000 × g
for 15 min at room temperature, followed by further centrifugation
at 12,000 × g for 10 min at room temperature. The supernatant was
then to a fresh cuvette, followed by the addition of 0.5 ml
chloroform, and was centrifuged at 14,000 × g for 5 min at room
temperature, followed by further centrifugation at 12,000 × g for
10 min at room temperature. Subsequently, the supernatant was
transferred to a fresh cuvette, prior to the addition of 50 ml 3 M
NaOAc (pH=6.0) and 0.5 ml 100% ethanol. The cuvette was inverted
several times, centrifuged at 12,000 × g for 10 min at room
temperature. The pellet was then washed once with 70% ethanol and
was left to air dry. The optical density (OD) 260/OD 280 of the DNA
used for PCR amplification was ~1.80. The TGFBR1 exon 1 coding
sequence was as previously described, and PCR amplification was
also performed as previously described (23). PCR was performed using Advantage-GC
Genomic Polymerase Mix (Invitrogen; Thermo Fisher Scientific, Inc.)
in a total volume of 25 ml containing 50 ng DNA and 1.25 U Platinum
Taq DNA polymerase (Invitrogen; Thermo Fisher Scientific,
Inc.). Following initial denaturation for 10 min at 95°C, 35 cycles
of PCR amplification were performed as follows: 95°C for 1 min,
68°C for 1 min, and 72°C for 1 min followed by a 5-min final
extension at 72°C. For single-strand conformation polymorphism
analysis, PCR products (5 µl) were diluted with 15 µl loading
buffer (10 mM EDTA, 98% deionized formamide and 5 mg/ml Blue
Dextran 2000). Denaturation through heating was performed at 98°C
for 10 min, and then quenched on ice for 2 min. Then, 20 µl of this
solution was added to each lane of an 8% neutral polyacrylamide
gel. Electrophoresis was performed at 300 V in 1X TAE
(Tris-acetate-EDTA) buffer (Sigma-Aldrich; Merck KGaA) at a
temperature of 10°C. The DNA was purified using the QIAquick PCR
purification kit (Qiagen GmbH, Hilden, Germany), according to the
manufacturer's protocol. Finally, purified DNA fragments were
directly sequenced by the same forward or reverse primers utilized
in the original PCR amplification (Thermo Fisher Scientific, Inc.),
performed as previously described (22).
Cell proliferation assay
Cells (1×104) were seeded onto 96-well
plates. The cell growth mediated by TGF-β1 was determined using an
MTT assay. To assess the growth inhibitory effects of TGF-β1, sw48,
control vector-modified sw48 and TGFBR1*6A-modified sw48
(sw48/TGFBR1*6A) cells were seeded, at a density of 10,000
cells/well, onto 96 well plates in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% (v/v)
heat-inactivated fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), and were incubated for 48 h prior to incubation
for 48 h in serum-free Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc.) in the absence or presence of
TGF-β1 (5 ng/ml). The assay was initiated by adding MTT solution at
a final concentration of 100 µg MTT/well. Wells were then
aspirated, 100 µl dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA)
was added to each well to dissolve the purple formazan, and the
plate was agitated for 15 min. Cells were subsequently subjected to
MTT assays at discrete time periods of 24, 48 and 72 h. Plates were
read at 460 nm in a spectrophotometer.
In vitro invasion/migration
assays
BioCoat Matrigel invasion chambers [12-well cell
culture inserts containing an 8.0 µm polyethylene terephthalate
(PET) membrane with a uniform layer of Matrigel matrix; BD
Biosciences, Franklin Lakes, NJ, USA] were used to assess cell
invasion. Cell migration was assessed in BioCoat control cell
culture chambers (12-well cell culture inserts containing an 8.0 µm
PET membrane without a Matrigel layer). The membranes (1.0
ml/chamber) were rehydrated with warm serum-free Dulbecco's
modified Eagle's medium (Sigma-Aldrich; Merck KGaA) for 2 h. In
brief, cells that were pre-incubated for 48 h at room temperature
in minimum essential medium (MEM; Sigma-Aldrich; Merck KGaA), in
the presence or absence of 5 ng/ml TGF-β1, were seeded into the
upper wells at a density of 0.5×105 cells/500 µl MEM.
The lower chambers were filled with MEM (Sigma-Aldrich; Merck KGaA)
containing 10% FBS, which acted as a chemoattractant. The chambers
were incubated for 48 h at 37°C in a 5% CO2 atmosphere.
Cells from the upper surface of the membranes were removed by
scrubbing with a cotton swab. Those on the lower surface of the
membranes were fixed for 5 min at room temperature with 100%
methanol and stained with Wright-Giemsa [0.4 % (w/v) in methanol,
pH 6.8); Sigma-Aldrich; Merck KGaA)] for 2 min at room temperature.
The number of cells that penetrated into each filter was counted in
five random optical microscopic fields, under ×20 magnification, by
a technician unaware of the experimental settings. The percentage
of invading cells was expressed as the ratio of the mean cell
number from the invasion chamber to the mean cell number from the
control chamber, according to the manufacturer's protocols. Each
assay was performed on duplicate filters and the experiments were
repeated twice.
Western blot analysis
A total of 48 h after transfection, the supernatant
from the cells was transferred to a 10-cm petri dish, prior to
being used for western blot analysis. Cells were washed with cold
phosphate-buffered saline and were lysed in 70 µl lysis buffer [50
mM Tris-Cl (pH 8.1), 10 mM EDTA, 1% SDS, 1% protein inhibitor;
Beyotime Institute of Biotechnology, Haimen, China] for 10 min at
4°C. Cell lysates were centrifuged at 5,000 × g at 4°C for 10 min
to pellet the cell lysates. The concentration of cellular protein
was determined using a BCA Protein Assay kit (Beyotime Institute of
Biotechnology). Total protein (60 µg) was mixed with a 5X loading
buffer, heated at 100°C for 5 min, and separated on 10% sodium
dodecyl sulfate-polyacrylamide gels. Following electrophoresis, the
proteins were transferred onto a Millipore Immobilon-P transfer
membrane (EMD Millipore, Billerica, MA, USA) using a Semi-Dry
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with Tris
buffer (0.025 M Tris-HCl, 0.192 M glycine, and 20% MeOH). The
membrane was blocked for 1 h at room temperature with 5% non-fat
milk in TBS-Tween 20. Subsequently, the membranes were incubated at
4°C overnight with the following antibodies: Mouse monoclonal
antibodies against β-actin (dilution, 1:1,000; cat. no. sc-70319;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and SMAD family
member 2 (Smad2; dilution, 1:500; cat. no. sc133098; Santa Cruz
Biotechnology, Inc.), and rabbit polyclonal antibodies against
phosphorylated (p)-Smad2 (Ser465/467; dilution, 1:800; cat. no.
8828; Cell Signaling Technology, Inc., Danvers, MA, USA), p38 MAPK
(dilution, 1:800; cat. no. 8690; Cell Signaling Technology, Inc.),
p-p38 (Thr180/Tyr182) MAPK (dilution, 1:1,000; cat. no. 9211; Cell
Signaling Technology, Inc.), extracellular-signal-regulated kinases
1/2 (ERK1/2; dilution, 1:800; cat. no. 9102; Cell Signaling
Technology, Inc.) and p-Erk1/2 (Thr202/Tyr204; dilution, 1:800;
cat. no. 9106; Cell Signaling Technology, Inc.). Following washing
in TBS-Tween20 3 times, the membranes were then incubated with a
horseradish peroxidase-conjugated anti-mouse immunoglobulin G (IgG)
(dilution, 1:1,000; cat. no. 14709) and anti-rabbit IgG against
(p)-Smad2 (dilution, 1:1,000; cat. no. 14708; both Cell Signaling
Technology, Inc.) for 1 h at room temperature. The membranes were
subsequently incubated with BeyoECL Plus reagents (Beyotime
Institute of Biotechnology), according to the manufacturer's
protocols. Images were captured using a motored molecular imaging
system (Molecular Imaging Vilber Fusion X7; Vilber Lourmat,
Marne-la-Vallée, France).
Statistical analysis
Results were presented as the mean ± standard
deviation and were analyzed using GraphPad Prism 5 software
(GraphPad Software, Inc., La Jolla, CA, USA). Differences between
groups were assessed using two-way analysis of variance, followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference. All experiments were repeated
at least 3 times.
Results
Analysis of TGFBR1 mutations in
colorectal cancer cell lines
To investigate potential TGFBR1 alterations in
colorectal cancer cell lines, PCR and sequencing was performed on
SW48 and DLD-1 cell lines. SW48 cells were confirmed to carry a
TGFBR1*9A/*9A genotype, and DLD-1 cells carried a *6A/*9A genotype
(Table I). SW48 and DLD-1 cell lines
were then selected for further research.
 | Table I.Analysis of TGFBR1 mutation in
colorectal cancer cell lines. |
Table I.
Analysis of TGFBR1 mutation in
colorectal cancer cell lines.
| Cell line | GCG repeats | TGFBR1 mutant |
|---|
| DLD-1 | CTGGCG GCG GCG GCG GCG GCG
CTGCTCCCGGGGCCACGGGT | *6A/9A |
| SW48 | CTGGCG GCG GCG GCG GCG GCG GCG GCG
GCG CTGCTCCCGGGGGCGACGGGTGAGCGGCGGCGC | *9A/9A |
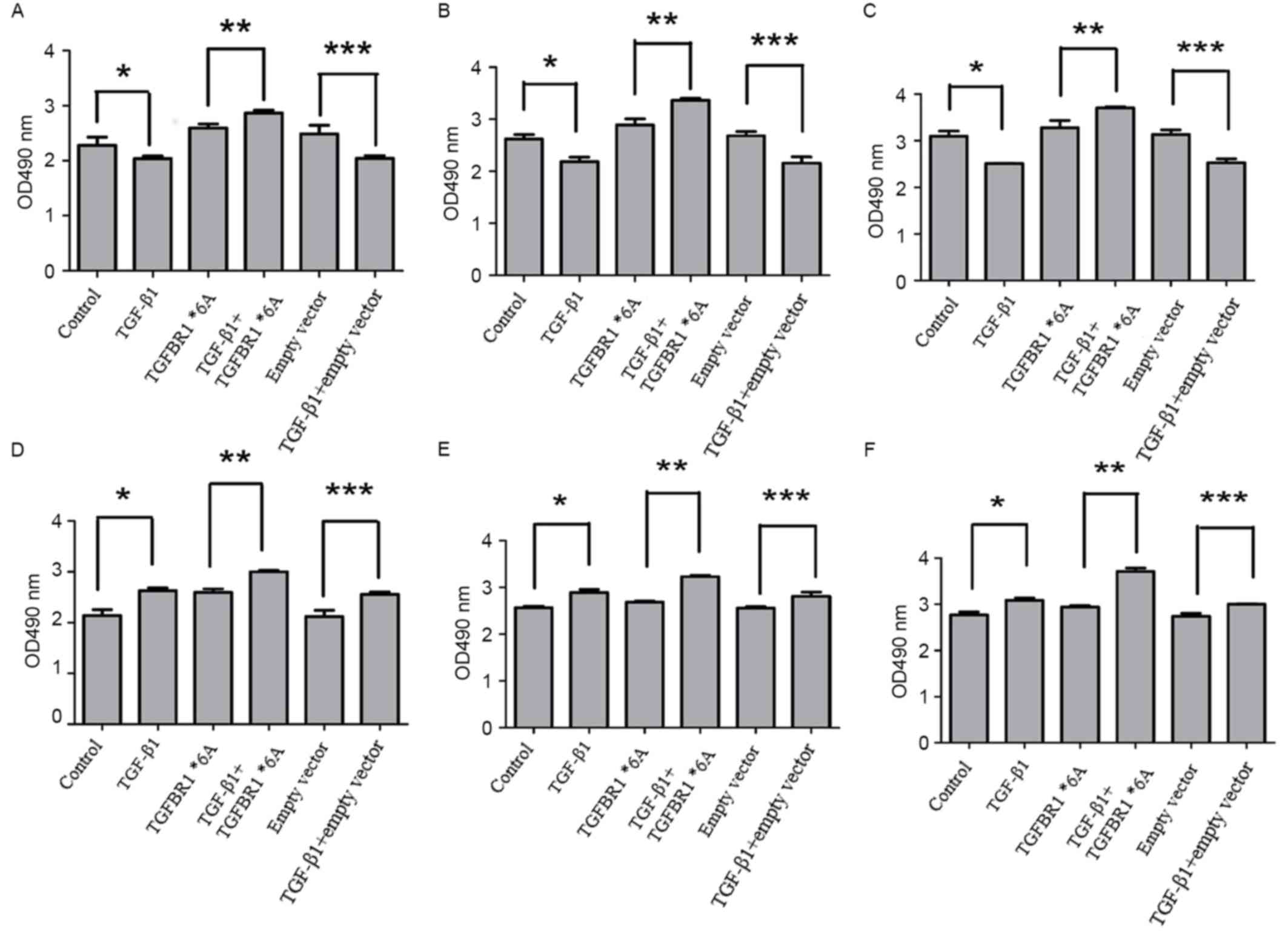
TGFBR1*6A increases colorectal cancer
cell proliferation
To investigate whether the TGFBR1*6A allele
increased colorectal cancer cell proliferation, the TGF-β1-mediated
proliferation of SW48 and DLD-1 cells was assessed following
transfection with TGFBR1*6A plasmids or an empty vector. As
presented in Fig. 1A-C, when compared
with other controls, the proliferation of SW48 cells was inhibited
when exposed to TGF-β1 (5 ng/ml) for 24, 48 and 72 h (P<0.05).
However, when SW48 cells were transfected with TGFBR1*6A plasmids,
the proliferation of these cells increased following exposure to
TGF-β1 (5 ng/ml) for 24, 48 and 72 h, compared with those not
treated with TGF-β1 (Fig. 1A-C;
P<0.05). These data suggested that the overexpression of
TGFBR1*6A may promote cell growth and tumorigenicity in colorectal
cancer cells. Fig. 1D-F demonstrate
that DLD-1 cell proliferation increased following exposure to
TGF-β1 (5 ng/ml) for 24, 48 and 72 h compared with controls
(P<0.05). Furthermore, the proliferation of DLD-1 cells
transfected with TGFBR1*6A plasmids treated with TGF-β1 (5 ng/ml)
was increased compared with control cells that were not treated
with TGF-β1 (Fig. 1D-F; P<0.05).
These phenomena suggested that TGFBR1*6A may switch TGF-β1-mediated
inhibition of proliferation into stimulation of proliferation in
colorectal cancer cells. Each experiment was performed at least 4
times in triplicate.
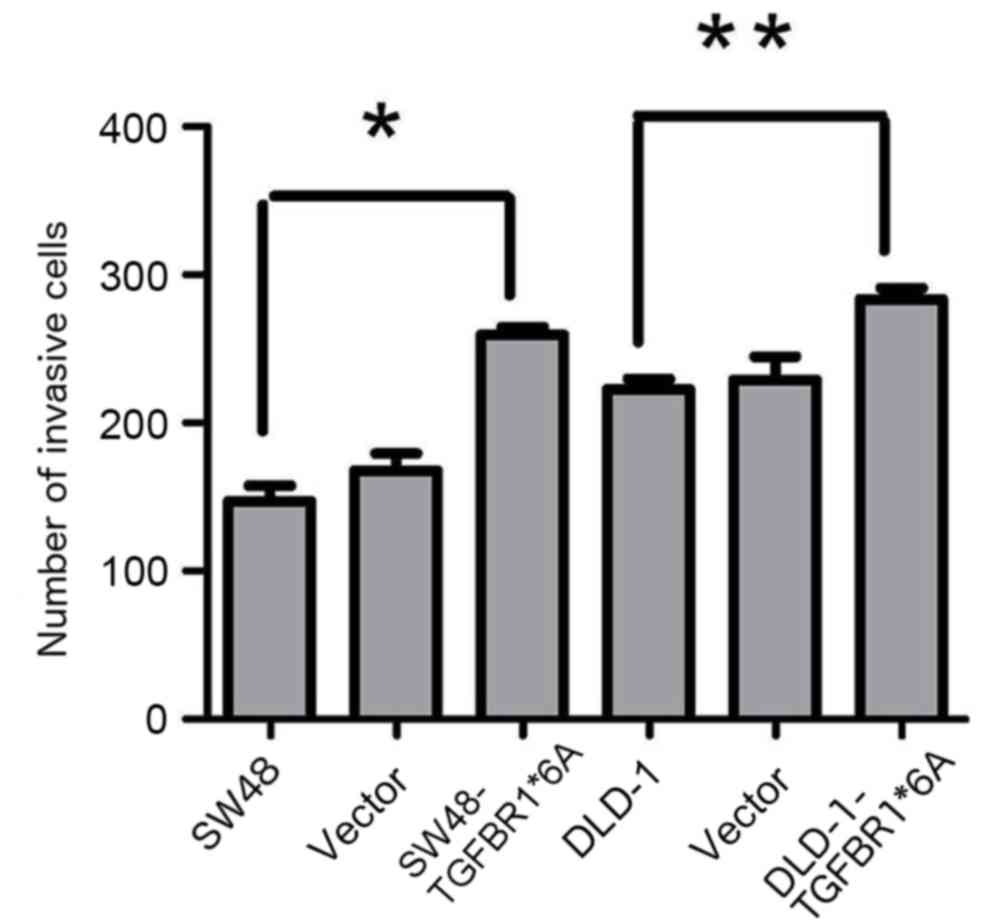
TGFBR1*6A increases invasion in
colorectal cancer cells
To test the hypothesis that TGFBR1*6A is involved in
colorectal cancer development and progression, its ability to
modify migration and invasion was assessed in SW48 and DLD-1 cells
transfected with TGFBR1*6A plasmids or empty vectors. In the
presence of TGF-β1 (5 ng/ml), overexpression of TGFBR1*6A in DLD-1
cells significantly increased invasion compared with the controls
(Fig. 2). Similarly, overexpression
of TGFBR1*6A in SW48 cells resulted in increased invasion compared
with the controls (Fig. 2). These
data indicated that TGFBR1*6A increased invasion in colorectal
cancer cells.
TGFBR1*6A promotes the development and
progression of colorectal cancer via p38 and ERK MAPK
signaling
To investigate the potential mechanisms underlying
the TGFBR1*6A-induced switch from TGF-β1-mediated inhibition
of proliferation to stimulation of proliferation in colorectal
cancer cells, a signaling test was performed using western
blotting. Increased expression of p-p38 and p-ERK1/2 was detected
within 15–30 min of stimulation with exogenous TGF-β1 (5 ng/ml) in
SW48 cells that were transfected with TGFBR1*6A plasmids, compared
with those transfected with the empty vector (Fig. 3). Following treatment with TGF-β1 (5
ng/ml), the empty vector and wild type SW48 (TGFBR1*9A)
cells exhibited activated p-Smad2 signaling, compared with those
transfected with TGFBR1*6A, in which the protein expression of
p-Smad2 was decreased, but the protein expression of p-p38 was
markedly increased and that of p-ERK was slightly increased
(Fig. 3). Therefore, when treated
with TGF-β (5 ng/ml), the wild type SW48 (TGFBR1*9A) and
empty vector control cells activated p-Smad2 signaling, but only
induced little activation of p-p38 and p-ERK signaling (Fig. 3). These results indicated that the
TGFBR1*6A allele may cause increased activity of the p38 and ERK1/2
MAPK signaling pathways rather than the TGF-β1/Smad signaling
pathway, compared with wild type and control cells. This may
facilitate the switch in TGF-β1-mediated signaling to result in
proliferation and invasion in colorectal cancer cells.
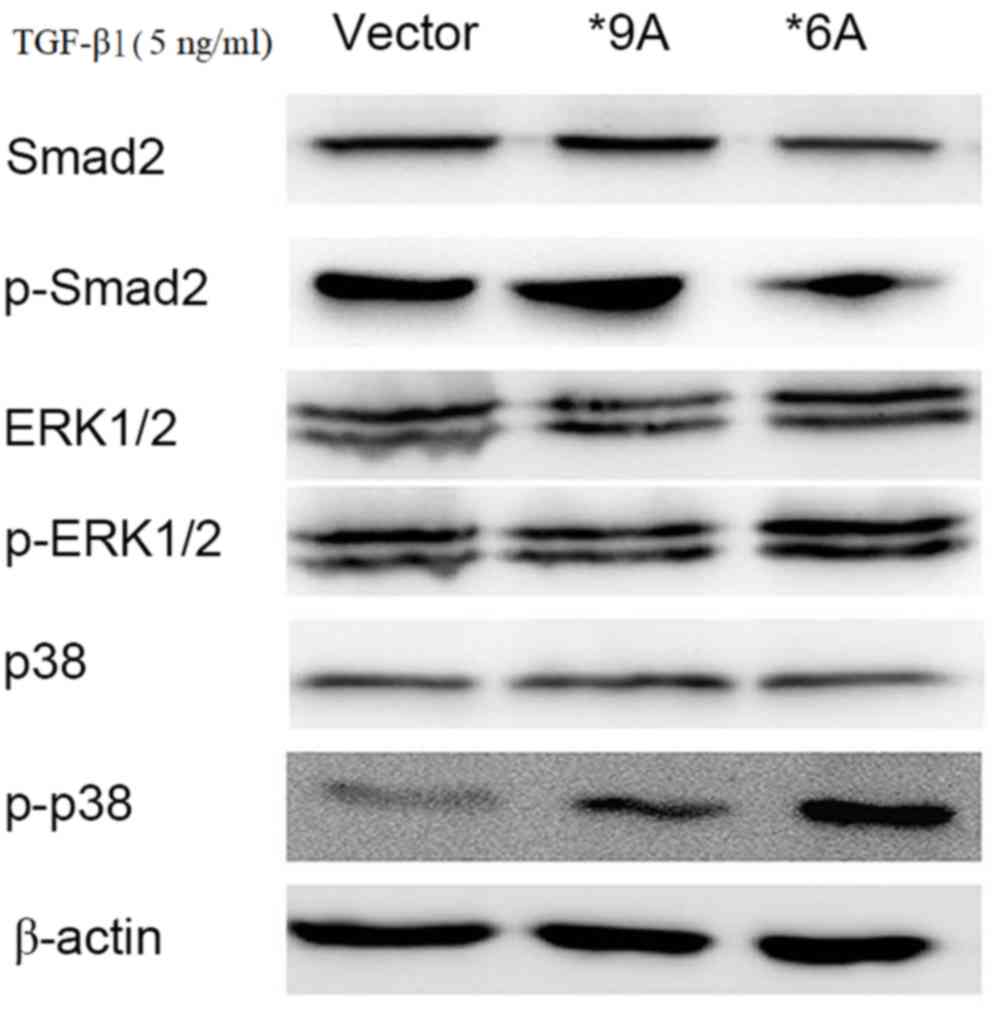 | Figure 3.To investigate the potential
mechanisms underlying the TGFBR1*6A-induced switch from
TGF-β1-mediated growth inhibition to growth stimulation in SW48
colorectal cancer cells, western blotting was performed. Western
blotting demonstrating that increased expression of p-p38 and
p-ERK1/2 were detected within 15–30 min of stimulation with
exogenous TGF-β1 (5 ng/ml) in SW48 cells transfected with a *6A
plasmid. When treated with TGF-β1 (5 ng/ml), the wild type SW48
(TGFBR1*9A) and controls cells activated p-Smad2 signaling, but
only induced little activation of p-p38 and p-ERK signaling.
However, under the same conditions in SW48-*6A cells, TGF-β1
activated both p-p38 and p-ERK signaling, while the expression of
p-Smad2 was decreased. TGFBR1*6A, type 1 transforming growth factor
β receptor; TGF-β1, transforming growth factor-β1; ERK,
extracellular-signal-regulated kinase; MAPK, mitogen-activated
protein kinase; p-, phosphorylated; Smad2, SMAD family member
2. |
Discussion
In normal epithelial cells, TGF-β predominantly
inhibits growth and serves as a tumor suppressor. However, during
the development and progression of malignancies, TGF-β is
transformed into a tumor promoter. Loss of TGF-β-mediated
inhibition of growth appears to be a common, important event that
occurs in colorectal cancer (24).
Multiple colorectal cancer cell lines escape from the
tumor-suppressive effect of TGF-β, becoming resistant to
TGF-β-induced growth inhibition.
There is a growing body of evidence to demonstrated
that TGF-β signaling alterations mediated by mutations or
polymorphisms of TGF-β receptors contribute to the development and
progression of colon cancer. TGFBR1*6A is a common polymorphic
variant of the TGF-β receptor I gene, and an association between
TGFBR1*6A and human colorectal cancer has previously been reported
(23). Studies conducted by Pasche
et al (23) revealed that
there is a significantly higher TGFBR1*6A allelic frequency in
patients with colorectal cancer than in healthy controls.
Furthermore, TGFBR1*6A was somatically acquired during colorectal
cancer tumorigenesis and liver metastasis (15). In the present study, SW48 and DLD1
cells were transfected with pCMV5-TGFBR1*6A-HA plasmids or with the
empty vector. Our group observed that TGFBR1*6A-mediated growth
inhibition was weaker than TGFBR1*9A-mediated growth inhibition
when exposed to 5 ng/ml TGF-β1. Transfection of TGFBR1*6A into the
colorectal cancer cells resulted in a significant increase in
cellular invasion. However, the difference between the TGFBR1*6A
cells and TGFBR1*9A cells was independent of TGF-β1/Smad signaling,
suggesting that TGFBR1*6A may switch TGF-β1 growth
inhibitory signals into growth stimulatory signals via
Smad-independent pathways. A previous study has demonstrated that
the biological effects of TGFBR1*6A are mediated by the
signal sequence rather than by the mature receptor, TGFBR1
(14). Following cleavage, the signal
sequence remains in the cytoplasm, and may modulate specific gene
expression or other cellular functions. Therefore, the observed
effects are likely due to secondary signaling events triggered by
the TGFBR1*6A signal sequence. TGFBR1*6A may drive the
proliferation of colorectal cancer cells in conjunction with other
oncogenic pathways, including the Ras/MAPK, c-Jun N-terminal kinase
(JNK) or PI3K/AKT pathways.
The MAPK pathways transduce a large variety of
external signals and lead to a wide range of cellular responses,
including growth, differentiation, inflammation and apoptosis.
Three distinct MAPK pathways have been described in mammalian
cells, including the ERK pathway, the JNK pathway, and the p38 MAPK
pathway (25). The present study
demonstrated that transfection of SW48 cells with the TGFBR1*6A
plasmid resulted in the upregulation of p-p38 and p-ERK protein
expression. These results have led us to hypothesize that TGFBR1*6A
may facilitate SW48 cell metastasis and invasion by increasing the
activation of the p38 and ERK1/2 MAKP pathways.
In summary, the TGFBR1*6A allele increases SW48
colorectal cancer cell invasion and results in the activation of
the p38 MAPK and ERK1/2 MAPK pathways. In the present study, these
effects were observed in the absence of exogenously added TGF-β1.
Furthermore, the TGFBR1*6A phenotype may be a mediator that
switches TGF-β1 growth inhibitory signals into growth stimulatory
signals. As a result of the dual role of TGF-β1 in tumorigenesis, a
comprehensive understanding of TGFBR1*6A biology is required in
order to design successful therapeutics. It is important to
discover novel drugs that mimic the interactions between TGF-β and
its receptors and mechanistically inhibit transduction of TGF-β
signaling and, in turn, eliminate the tumor-promoting activities of
TGF-β.
Glossary
Abbreviations
Abbreviations:
|
TGF-β1
|
transforming growth factor-β1
|
|
TGFBR1
|
type 1 transforming growth factor β
receptor
|
|
ERK1/2
|
extracellular-signal-regulated kinases
1/2
|
References
|
1
|
David CJ, Huang YH, Chen M, Su J, Zou Y,
Bardeesy N, Iacobuzio-Donahue CA and Massagué J: TGF-β tumor
suppression through a lethal EMT. Cell. 164:1015–1030. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Principe DR, Doll JA, Bauer J, Jung B,
Munshi HG, Bartholin L, Pasche B, Lee C and Grippo PJ: TGF-β:
Duality of function between tumor prevention and carcinogenesis. J
Natl Cancer Inst. 106:djt3692014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mehrvarz Sarshekeh A, Advani S, Overman
MJ, Manyam G, Kee BK, Fogelman DR, Dasari A, Raghav K, Vilar E,
Manuel S, et al: Association of SMAD4 mutation with patient
demographics, tumor characteristics and clinical outcomes in
colorectal cancer. PLoS One. 12:e01733452017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ramamoorthi G and Sivalingam N: Molecular
mechanism of TGF-β signaling pathway in colon carcinogenesis and
status of curcumin as chemopreventive strategy. Tumour Biol.
35:7295–7305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
PLOS ONE Staff: Correction: Association of
SMAD4 mutation with patient demographics, tumor characteristics and
clinical outcomes in colorectal cancer. PLoS One. 12:e01782752017.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fleming NI, Jorissen RN, Mouradov D,
Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C,
Lipton L, et al: SMAD2, SMAD3 and SMAD4 mutations in colorectal
cancer. Cancer Res. 73:725–735. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sarshekeh AM, Overman MJ, Kee BK, Fogelman
DR, Dasari A and Singh Raghav KP: Demographic, tumor
characteristics and outcomes associated with SMAD4 mutation in
colorectal cancer. J Clin Oncol. 34:5652016. View Article : Google Scholar
|
|
8
|
Sun XF, Sun XH, Cheng SF, Wang JJ, Feng
YN, Zhao Y, Yin S, Hou ZM, Shen W and Zhang XF: Interaction of the
transforming growth factor-β and Notch signaling pathways in the
regulation of granulosa cell proliferation. Reprod Fertil Dev.
28:1873–1881. 2015. View
Article : Google Scholar
|
|
9
|
Liu WT, Huang KY, Lu MC, Huang HL, Chen
CY, Cheng YL, Yu HC, Liu SQ, Lai NS and Huang HB: TGF-β upregulates
the translation of USP15 via the PI3K/AKT pathway to promote p53
stability. Oncogene. 36:2715–2723. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jung B, Staudacher JJ and Beauchamp D:
Transforming growth factor βsuper family signaling in development
of colorectal cancer. Gastroenterology. 152:36–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu YS, Pan Y, Li WH, Zhang Y, Li J and Ma
BA: Association between TGFBR1*6A and osteosarcoma: A Chinese
case-control study. BMC Cancer. 10:1692010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Y, Qi X, Wang F, Jiang J and Guo QN:
Association between TGFBR1 polymorphisms and cancer risk: A
meta-analysis of 35 case-control studies. PLoS One. 7:e428992012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang X, Wu L, Sheng Y, Zhou W, Huang Z,
Qu J, Gao G, Cai D and Zhang M: The association of polymorphisms on
TGFBR1 and colorectal cancer risk: A meta-analysis. Mol Biol Rep.
39:2567–2574. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rosman DS, Phukan S, Huang CC and Pasche
B: TGFBR1*6A enhances the migration and invasion of MCF-7 breast
cancer cells through rhoa activation. Cancer Res. 68:1319–1328.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pasche B, Pennison MJ, Jimenez H and Wang
M: TGFBR1 and cancer susceptibility. Trans Am Clin Climatol Assoc.
125:300–312. 2014.PubMed/NCBI
|
|
16
|
Castillejo A, Mata-Balaguer T, Montenegro
P, Ochoa E, Lázaro R, Martínez-Cantó A, Castillejo MI, Guarinos C,
Barberá VM, Guillén-Ponce C, et al: The TGFBR1*6A, allele is not
associated with susceptibility to colorectal cancer in a Spanish
population: A case-control study. BMC Cancer. 9:1932009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ross JP, Lockett LJ, Tabor B, Saunders IW,
Young GP, Macrae F, Blanco I, Capella G, Brown GS, Lockett TJ and
Hannan GN: Little evidence for association between the TGFBR1*6A
variant and colorectal cancer: A family-based association study on
non-syndromic family members from Australia and Spain. BMC Cancer.
14:4752014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McGuire JL, Mcphail M and Rajendran A: The
association of tgfβ signalling pathway gene polymorphisms with
colorectal cancer risk: A meta-analysis. Gastroenterology.
146:S868–S8683. 2014. View Article : Google Scholar
|
|
19
|
Ibrahim T, Yazbeck C, Maalouly G, Haddad
F, Sabbagh C and Chahine G: TGFBR1*6A polymorphism in sporadic and
familial colorectal Carcinoma: A case-control study and systematic
literature review. J Gastrointestinal Cancer. 45:441–447. 2014.
View Article : Google Scholar
|
|
20
|
Pasche B, Kaklamani V, Hou N, Young T,
Rademaker A, Peterlongo P, Ellis N, Offit K, Caldes T, Reiss M and
Zheng T: TGFBR1*6A and cancer: A meta-analysis of 12 case-control
studies. J Clin Oncol. 22:756–758. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ross JP, Lockett LJ, Tabor B, Saunders IW,
Young GP, Macrae F, Blanco I, Capella G, Brown GS, Lockett TJ and
Hannan GN: Little evidence for association between the TGFBR1*6A
variant and colorectal cancer: A family-based association study on
non-syndromic family members from Australia and Spain. BMC Cancer.
14:4752014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pasche B, Luo Y, Rao PH, Nimer SD,
Dmitrovsky E, Caron P, Luzzatto L, Offit K, Cordon-Cardo C and
Renault B: Type I transforming growth factor beta receptor maps to
9q22 and exhibits a polymorphism and a rare variant within a
polyalanine tract. Cancer Res. 58:2727–2732. 1998.PubMed/NCBI
|
|
23
|
Pasche B, Knobloch TJ, Bian Y, Liu J,
Phukan S, Rosman D, Kaklamani V, Baddi L, Siddiqui FS, Frankel W,
et al: Somatic acquisition and signaling of TGFBR1*6A in cancer.
JAMA. 294:1634–1646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu WK, Wang XJ, Cheng AS, Luo MX, Ng SS,
To KF, Chan FK, Cho CH, Sung JJ and Yu J: Dysregulation and
crosstalk of cellular signaling pathways in colon carcinogenesis.
Crit Rev Oncol Hematol. 86:251–277. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Taves S, Berta T, Liu DL, Gan S, Chen G,
Kim YH, Van de Ven T, Laufer S and Ji RR: Spinal inhibition of p38
MAP kinase reduces inflammatory and neuropathic pain in male but
not female mice: Sex-dependent microglial signaling in the spinal
cord. Brain Behav Immun. 55:70–81. 2016. View Article : Google Scholar : PubMed/NCBI
|