Introduction
Gastric cancer is the one of most common causes of
cancer-associated mortality globally and clinical approaches for
treatment of gastric cancer are limited, with the median survival
time for advanced gastric cancer remaining at 8–10 months (1). Dysplasia and Barrett's esophagus have
been reported to be associated with the development of esophageal
adenocarcinoma, while Helicobacter pylori infection,
atrophic gastritis, intestinal metaplasia and dysplasia are
associated with gastric adenocarcinoma (2). In 20–30% of gastric and
gastro-esophageal junction cancer cases, gastric cells overexpress
human epidermal growth factor receptor 2 (HER2), which is
indicative of a poor prognosis (3).
Trastuzumab (Tzb) is a humanized monoclonal antibody
that targets the HER2 gene. Tzb is one of the first
molecular-targeting drugs to be developed and was originally
introduced for the treatment of HER2-positive advanced breast
cancer (4). Tzb has also been widely
used to treat HER2-positive gastric cancer (1). Tzb, induces antibody-dependent cellular
cytotoxicity and confers an overall survival benefit in
HER2-positive advanced gastric cancer (3). However, Tzb treatment remains under
investigation in order to further elucidate its potential
utilization and underlying mechanisms (5). Tzb in combination with chemotherapy may
be considered as a novel standard option for patients with
HER2-positive advanced gastric or gastro-esophageal junction cancer
(6). However, with increased
durations of Tzb treatment, the risk of developing resistance to
the drug is also increased. In addition, details of the mechanisms
underpinning Tzb resistance remain unclear. Therefore, it is
important to explore the mechanisms underlying drug resistance in
order to combat this problem.
Autophagy is the cellular degradation process in
which cellular proteins and organelles are engulfed by
double-membrane autophagosomes and are degraded in lysosomes
(7). Perturbations in autophagy have
been observed in gastric cancer (8,9). In cancer
cells, autophagy has both pro-survival and pro-death functions and,
thus, the action of autophagy in cancer cells remains
controversial. Autophagy may act as a survival mechanism that
provides energy and protects cancer cells from the cell death
induced by multiple antitumor treatments; however, autophagy is
also a cell death mechanism in response to anticancer therapies
(10). Furthermore, autophagy
modulates the development of gastric cancer by affecting a range of
pathological events, including tumor angiogenesis and changes to
the tumor microenvironment (11).
Wu et al (10)
revealed that loss of the autophagy regulator beclin 1 is
significantly correlated with HER2 amplification in patients with
breast cancer. Notably, HER2 signaling and responsiveness to Tzb
appear to dynamically interact with the tumor-suppressive and
tumorigenic functions of autophagy (12). Previously, autophagy has been reported
to protect against Tzb-induced cytotoxicity in HER2-overexpressing
breast tumor spheroids (13). A study
has revealed that the autophagy inhibitor, chloroquine, overcomes
Tzb resistance in HER2-positive breast cancer SK-BR3 cells and have
confirmed that HER2-overexpressing breast cancer cells may require
autophagy in order to maintain the Tzb-resistant phenotype
(14). However, these studies are
focused on breast cancer, with only limited data regarding the
association between autophagy and HER2 expression in gastric
adenocarcinoma being reported. The present study investigated the
function of autophagic flux in a Tzb-resistant gastric cancer cell
line in order to study its mechanism of action.
Materials and methods
Materials
Tzb was provided by Ningbo No. 2 Hospital (Zhejiang,
China), solubilized in water (stock solution at 21 mg/ml), stored
at 4°C and used within 1 month. Dimethylsulfoxide (DMSO),
3-methyladenine (3MA), MTT, crystal violet, hydroxychloroquine
(HCQ) and bafilomycin A1 (BafA1) were purchased from Sigma-Aldrich;
Merck KGaA (Darmstadt, Germany). Everolimus was provided by the
China State Institute of Pharmaceutical Industry (Shanghai, China).
RPMI-1640 medium, 10 U/ml penicillin-streptomycin (P/S), 0.25%
trypsin, fetal bovine serum (FBS) and bovine serum albumin (BSA)
were purchased from Invitrogen (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). Cell Counting Kit-8 (CCK-8) was purchased from
Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). Cell lysis
buffer, polyvinylidene difluoride (PVDF) membranes, and Tween-20
were purchased from Weiao Inc. (Shanghai, China). Glutaraldehyde,
Epon 812, DDSA, NMA and DMP-30 were purchased from Sinopharm Inc.
(Beijing, China).
Cell culture
Human gastric cancer NCI-N87 and SGC 7901 cell
lines, and the human breast cancer SK-BR3 cell line, which was used
as the positive control, were purchased from the Shanghai Institute
of Biochemistry and Cell Biology (Shanghai, China). The cells were
cultured in full medium (P/S and RPMI-1640 medium, supplemented
with 10% FBS) at 37°C in a humidified atmosphere with 5%
CO2.
Establishment of Tzb-acquired
auto-resistance in HER2-positive NCI-N87 cells
Establishment of NCI-N87/Tzb-resistant pools was
performed as previously described (15), with Tzb-naive NCI-N87 cells being
treated with increasing concentrations of Tzb for 10 months. The
NCI-N87 cells were initially treated with 50 µg/ml Tzb for 2 months
(twice weekly) followed by 100 µg/ml Tzb for 3 months (twice
weekly). Two pools (Tzb POOL1 and Tzb POOL2), which resisted
continuous growth in 250 µg/ml Tzb for 5 months (twice weekly),
were selected for further study. The resistant cells were
maintained in RPMI-1640 medium without Tzb for ≥2 days prior to
further experiments.
MTT assay
The NCI-N87, POOL1 or POOL2 cells were seeded at
4,000 cells/100 µl into 96-well plates, and allowed to attach
overnight. The medium was removed, and 95 µl fresh full medium with
5 µl Tzb (final concentration 1, 3.2, 10, 32, 100 µg/ml), BafA1 (50
nM), 3MA (1 µM), HCQ (50 µM) or everolimus (0.1, 1, 10 µM). The
control cells treated with 5 µl PBS was used as vehicle. For
testing the combination effect of Tzb (10 µg/ml) and autophagy
inhibitors, the concentration of BafA1, 3MA, and HCQ is 25 nM, 500
and 25 µM respectively. After 72 h culture (37°C, 5%
CO2), 10 µl MTT solution (0.5 mg/ml) was added into each
well at 37°C for 4 h and 100 µl DMSO was used to dissolve the
purple formazan. Following agitation, the optical densities were
measured at 595 nm using a microplate reader (BioTek PowerWave XS2;
BioTek Instruments, Inc., Winooski, VT, USA).
CCK-8 assay
The NCI-N87 or POOL2 cells were seeded at 1,000
cells/100 µl into 96 well plates, and allowed to attach overnight.
The medium was removed, and 95 µl fresh full medium with 5 µl Tzb
(final concentration 1, 3.2, 10, 32 or 100 µg/ml), or everolimus
(0.1, 1 or 10 µM). The control cells treated with 5 µl PBS was used
as vehicle. When assessing the combination effect of Tzb (10 µg/ml)
and an autophagy inhibitor, the concentration of BafA1, 3MA, and
HCQ is 25 nM, 500 and 25 µM, respectively. After a 72 h culture
(37°C, 5% CO2), 10 µl CCK-8 solution was added into each
well at 37°C for 2 h and the optical densities were measured at 450
nm using a microplate reader (PowerWave XS2; BioTek Instruments,
Inc.).
Clonogenic assay
Cells were cultured in 6-well plates at a density of
100 cells/well and were incubated (37°C, 5% CO2) for 24
h with full medium to allow for attachment. Following incubation,
cells were treated with 100 µg/ml Tzb for 72 h. The control cells
treated with 5 µl PBS was used as vehicle. Allowed to grow in a
Tzb-free full medium for 14 days (37°C, 5% CO2), fixed
with cold ethanol for 15 min and stained with 0.1% crystal violet
in PBS for 30 min at 25°C. Images from light microscopy
(magnification, ×4; Olympus IX73) were captured using CellSens
Dimension software (version 1.15) (both from Olympus Corporation,
Tokyo, Japan).
Western blot analysis
Protein lysates were obtained with cell lysis buffer
for 30 min at 4°C. The protein (20 µg/lane) was fractionated by 10%
SDS-PAGE and transferred onto PVDF membranes. After 1 h blocking
with 5% BSA at 25°C, the membranes were incubated at 4°C overnight
with different primary antibodies diluted in 3% BSA in 1% Tween-20
in TBS. The membranes were probed with the following antibodies:
HER2 (cat. no. 4290; Cell Signaling Technology, Inc., Danvers, MA,
USA; dilution 1:1,000), light chain 3B (LC3B) (cat. no. L7543;
dilution 1:1,000; Sigma-Aldrich; Merck KGaA), sequestosome 1
(SQSTM1; cat. no. PM045; dilution 1:2,000; MBL International
Corporation, Woburn, MA, USA), phosphorylated (p)-protein kinase B
(Akt; cat. no. 2965; dilution 1:1,000), Akt (cat. no. 2966;
dilution 1:1,000), p-mechanistic target of rapamycin (mTOR; cat.
no. 2983; dilution 1:1,000) and mTOR (cat. no. 5536; dilution
1:1,000) (all from Cell Signaling Technology, Inc.). GAPDH (cat.
no. 5014; dilution 1:2,000; Cell Signaling Technology, Inc.) was
used as the reference protein. Subsequently, the membranes were
incubated with horseradish peroxidase-conjugated secondary
anti-rabbit IgG antibody (cat. no. 7074; dilution 1:2,000; Cell
Signaling Technology, Inc.) for 1 h at room temperature. Protein
bands were visualized using an enhanced chemiluminescence (ECL) kit
using ECL blotting detection reagents (cat. no. 54-61-00; KPL,
Inc., Gaithersburg, MD, USA).
Transmission electron microscopy
(TEM)
A total of 1×107 cells were digested with
0.25% trypsin at 37°C and washed twice with PBS. The cell
precipitation was fixed with 2.5% glutaraldehyde in PBS for 3 h at
4°C, followed by 1% OsO4 for 2 h at 4°C. After washing with PBS,
the samples were progressively dehydrated in a graduated series of
ethanol solutions (50, 70, 80, 90, 95 and 100%) at 4°C and embedded
in the mixture of Epon 812, DDSA, NMA and DMP-30 at 37°C for 12 h,
and at 60°C for 48 h. Following dehydration, the cells were cut in
to 70 nm sections, and stained with 3% uranyl acetate and lead
citrate for 15 min at 25°C, the ultrastructure of cells were
analyzed with a TEM (magnification, ×16,000; cat. no. JEM-1230;
JEOL, Ltd., Tokyo, Japan).
Statistical analysis
All of the data are presented as the mean ± standard
deviation of three independent experiments. Statistical comparisons
were performed using Student's t-test and repeated-measures one-way
analysis of variance followed by Dunnett's post hoc test using SPSS
software (version 19; IBM Corp., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Tzb induces gastric cancer NCI-N87
cell death
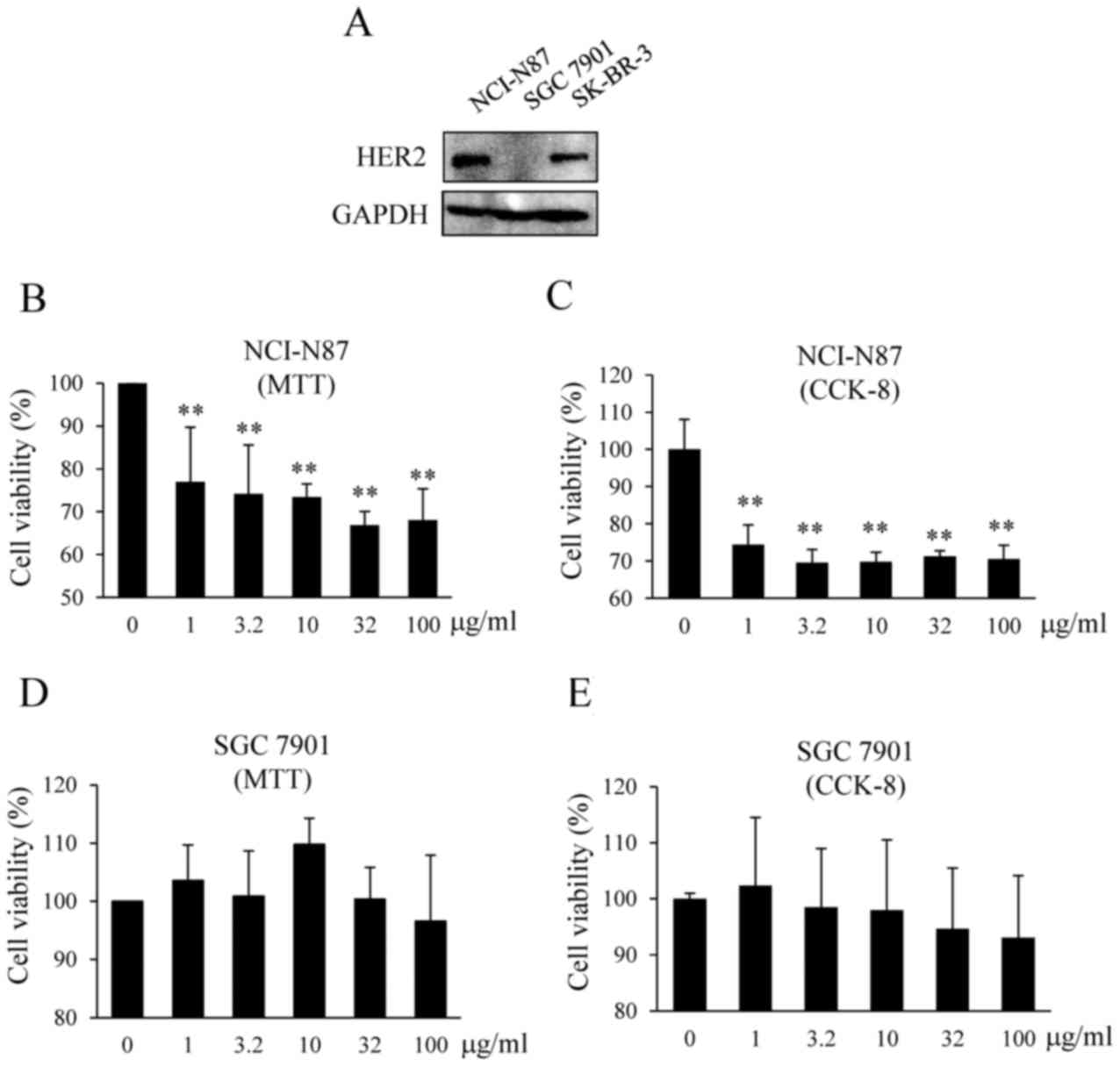
HER2 was expressed in human gastric cancer NCI-N87
cells, but not in SGC 7901 cells (Fig.
1A). Breast cancer SKBR 3 cells were used as a positive
control. The viability of NCI-N87 and SGC 7901 cells treated with
Tzb was examined using MTT and CCK-8 assays. Tzb (1–100 µg/ml) was
revealed to significantly induce NCI-N87 cell death (Fig. 1B and C), but did not induce the death
of SGC 7901 cells (Fig. 1D and E),
suggesting that NCI-N87 cells are more sensitive to Tzb.
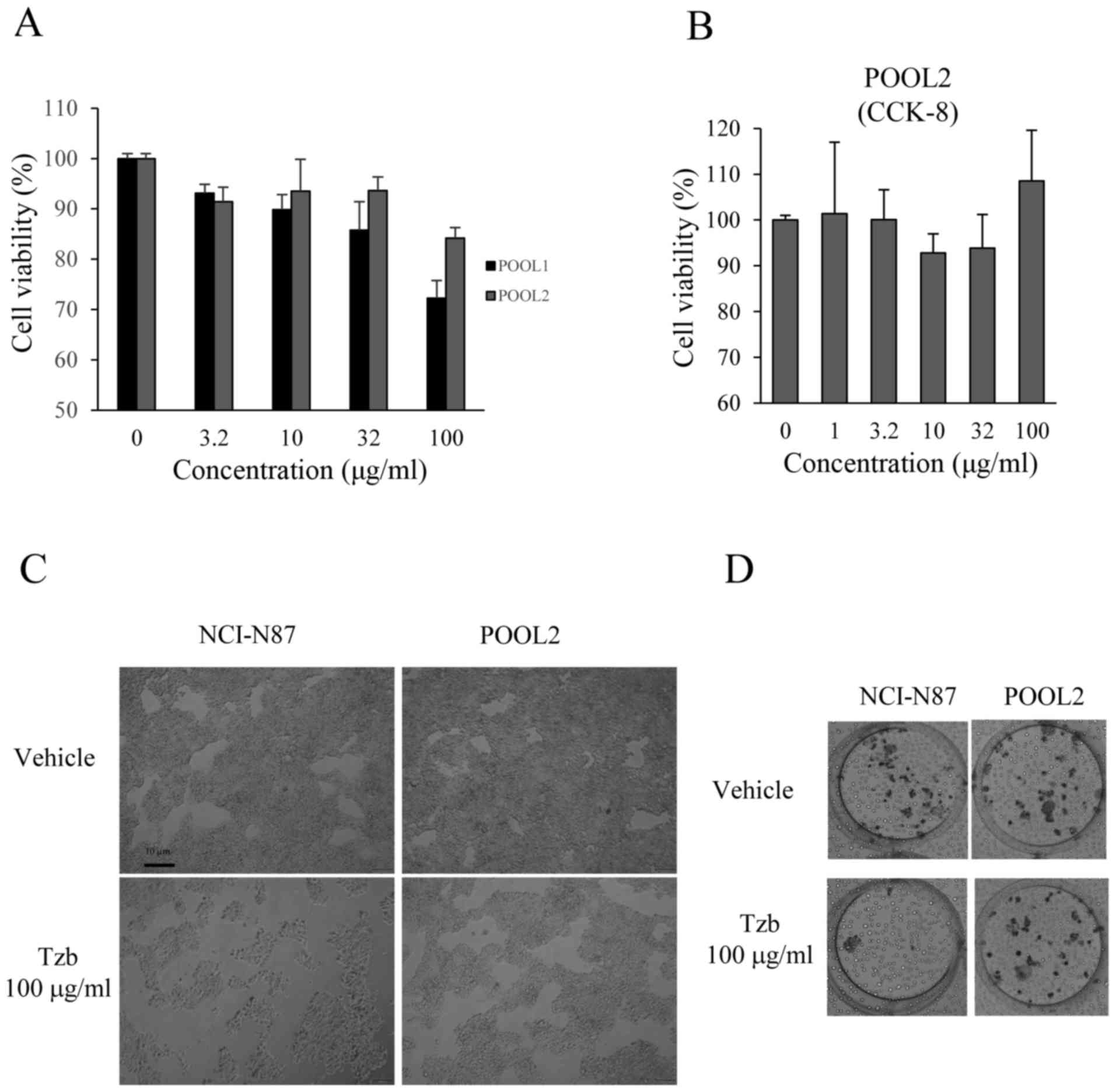
HER2-positive Tzb-resistant gastric cancer cells
were established by treating NCI-N87 parental cells with increasing
concentrations of Tzb. After 10 months of being maintained with
Tzb, two pools of Tzb-refractory cells (Tzb POOL1 and Tzb POOL2)
were obtained. Resistance to Tzb was examined using an MTT assay. A
concentration of 100 µg/ml Tzb did not significantly decrease cell
viability in POOL1 or POOL2 cells compared with the parental cells
(Fig. 2A), and as the resistance of
POOL2 was stronger than that of POOL1. The data obtained from the
CCK-8 assay demonstrated that the viability of POOL2 cells did not
significantly alter with Tzb treatment (Fig. 2B). The NCI-N87 parental cells and the
Tzb-refractory cells (Tzb-resistant POOL2) were incubated with 100
µg/ml Tzb for 72 h and were observed under a light microscope. The
number of parental cells was visibly decreased following Tzb
treatment (Fig. 2C), while the effect
of Tzb was less notable in the POOL2 cells. Additionally, as
demonstrated in Fig. 2D, NCI-N87
cells treated with 100 µg/ml Tzb for 72 h did not form colonies,
indicating that Tzb may inhibit the proliferation of parental
cells. However, 100 µg/ml Tzb did not affect the colony-forming
ability of POOL2, suggesting that POOL2 is a Tzb-refractory cell
line.
Tzb-refractory cells exhibit
autophagic flux inhibition
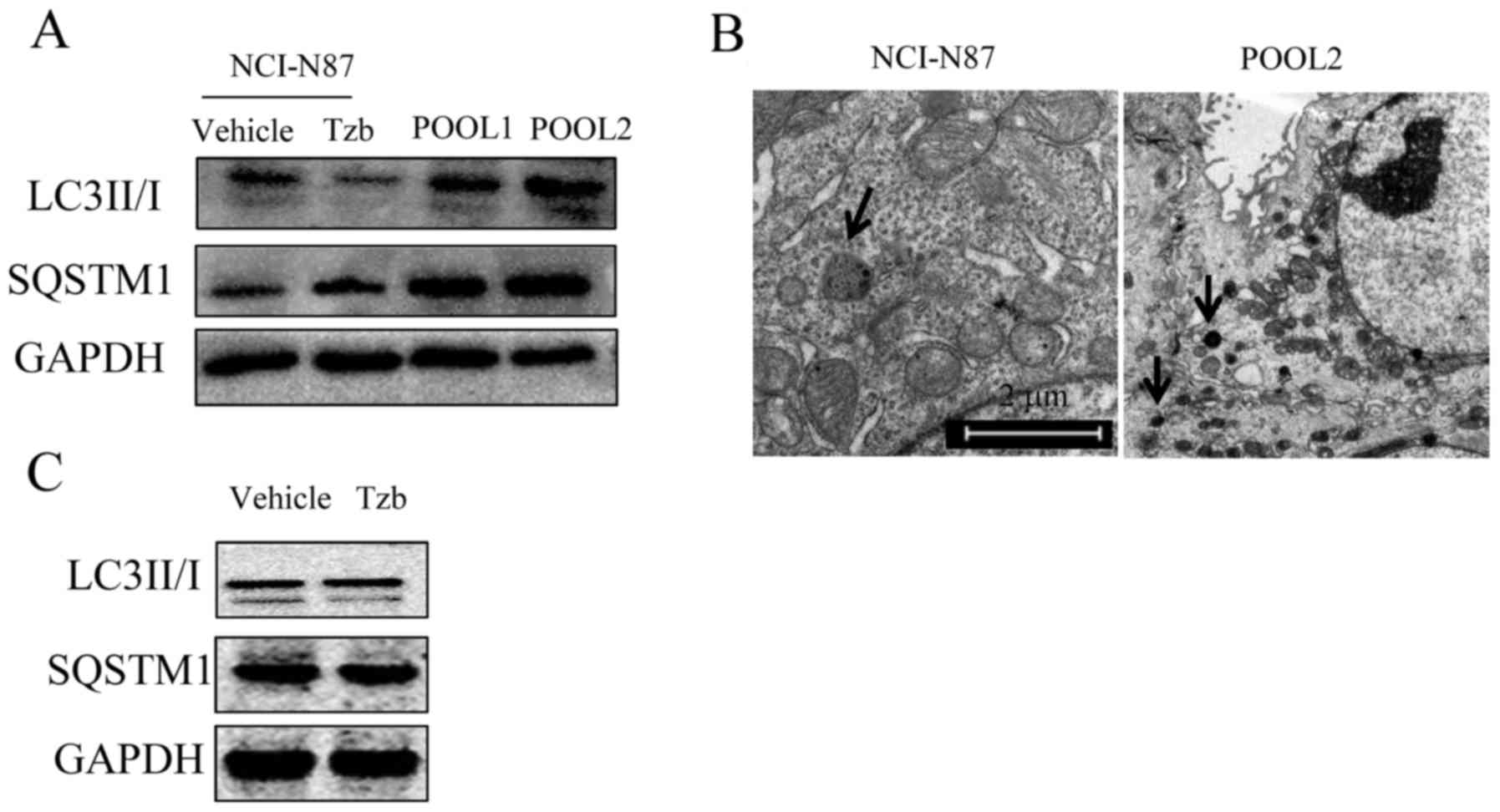
Western blot analysis revealed that the top band (18
kDa) expressed LC3-I and the bottom band (16 kDa) expressed LC3-II,
which is a typical autophagosome marker (Fig. 3A). Furthermore, Tzb reduced the
expression of LC3 I, and the Tzb-resistant POOL1 and POOL2 cells
exhibited an increased expression of LC3-II, suggesting that HER2
may be associated with autophagy. POOL2 demonstrated increased
resistance compared with POOL1, therefore POOL2 was selected for
further study. SQSTM1 is a link between LC3 and ubiquitinated
substrates. The inhibition of autophagic flux is correlated with
increased expression of SQSTM1 (16).
In the present study, SQSTM1 degradation was observed and Tzb
increased the expression of SQSTM1, suggesting that Tzb may inhibit
autophagic flux. POOL1 and POOL2 cells exhibited an elevated SQSTM1
expression compared with the parental cells, suggesting that the
Tzb-refractory cells exhibited autophagic flux inhibition. The TEM
data (Fig. 3B) demonstrated that the
autolysosome in the POOL2 cells was smaller than that in the
parental cells. As demonstrated in Fig.
3C, 100 µg/ml Tzb did not alter the autophagic flux in POOL2.
These results indicated that Tzb-refractory cells exhibited less
autophagic flux and improved viability compared with parental
cells, suggesting that autophagy may induce the cell death of
gastric cancer cells.
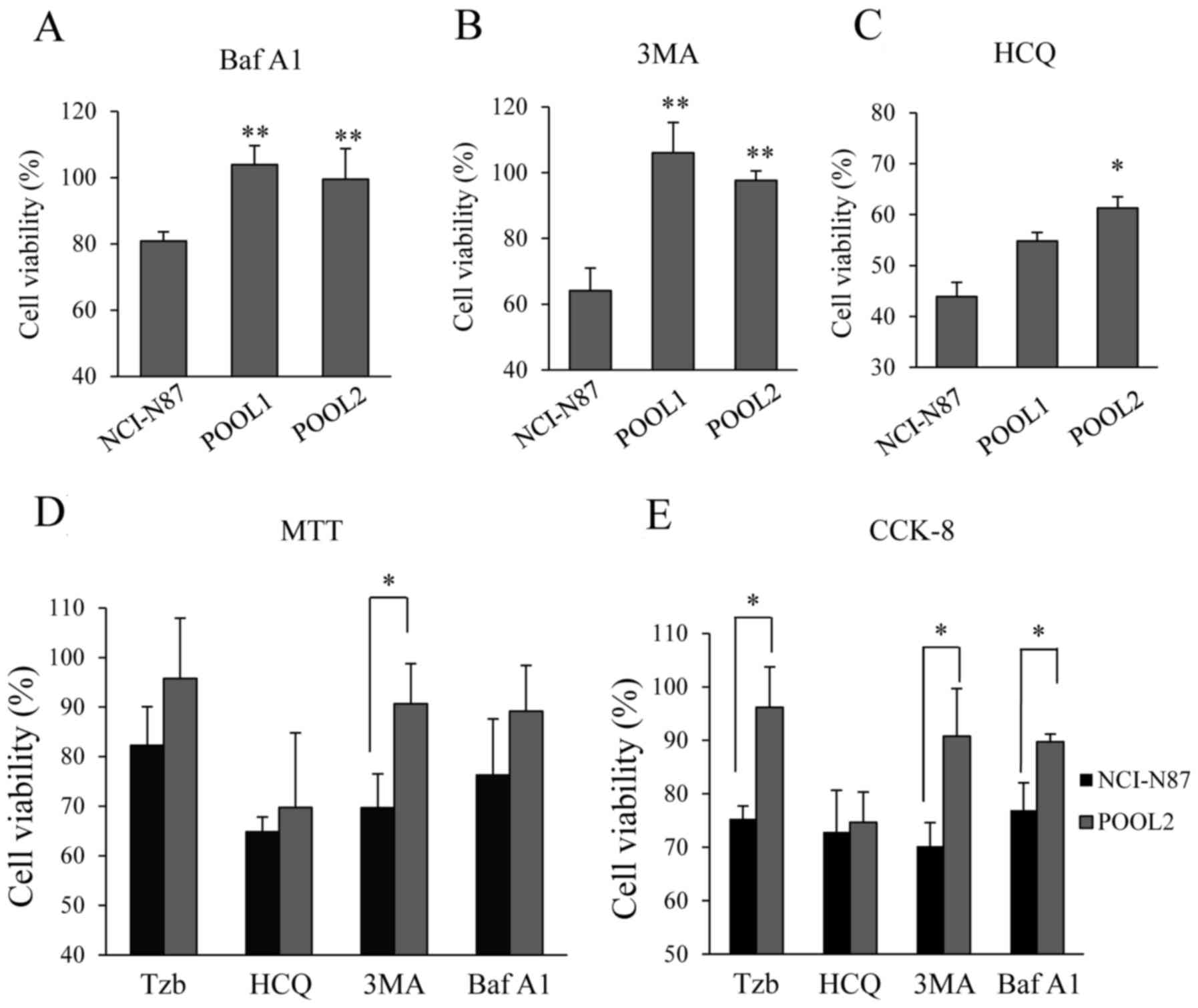
BafA1 is a vacuolar H+-ATPase inhibitor
known to block autophagic flux to prevent fusion between
autophagosomes and lysosomes, 3MA is an inhibitor of the formation
of the pre-autophagosomal structures and HCQ is an autophagic
inhibitor that prevents endosomal acidification and blocks
autophagosome-lysosome fusion (13,17,18). In
the present study, cell viability was studied in both parental
NCI-87 and Tzb-refractory cells following blocking of autophagy
with BafA1 (50 nM), 3MA (1 mM) or HCQ (50 µM). The three autophagy
inhibitors, BafA1, 3MA and HCQ, were revealed to induce parental
NCI-N87 cell death (Fig. 4A-C).
However, no change in the viability of POOL1 or POOL2 cells treated
with BafA1 or 3MA was observed, suggesting that these autophagy
inhibitors did not alter the viability of Tzb-refractory cells. HCQ
overcame Trz resistance, but its effect was less notable than that
in the parental NCI-N87 cells. In addition, the results of MTT and
CCK-8 assays revealed that the combination of Tzb (10 µg/ml) with
an autophagy inhibitor (BafA1, 3MA or HCQ) may inhibit the growth
of parental cells, but neither BafA1 nor 3MA induced Tzb-refractory
cell death (Fig. 4D and E). Although
HCQ may induce Tzb-refractory cell death, this effect was
significantly lower than in the parental cells. These results
indicated that cell Tzb resistance may be due to autophagic flux
inhibition.
Tzb induces resistance via the
Akt/mTOR pathway
The Akt/mTOR signaling pathway is a key negative
regulator of autophagy in gastric cancer cells (8). Therefore, the present study investigated
whether the Akt/mTOR pathway is involved in the Tzb resistance of
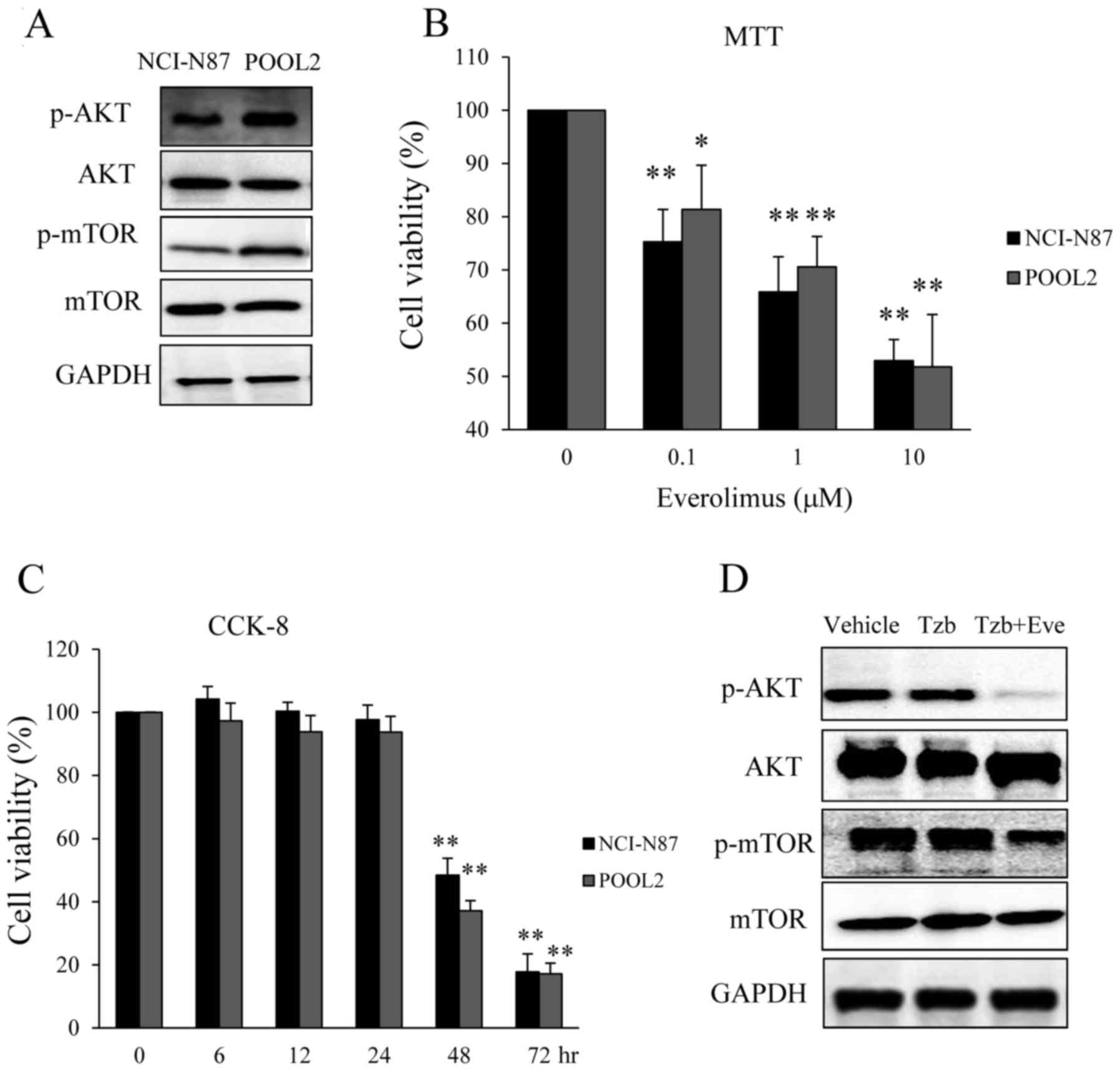
HER2-positive cancer cells. As demonstrated in Fig. 5A, the phosphorylation of Akt and mTOR
was increased in Tzb-resistant HER2 positive cancer cells compared
with the parental cells, suggesting that Tzb resistance is
partially due to alterations in the Akt/mTOR pathway. Notably,
everolimus, a derivative of rapamycin that is functionally similar
to rapamycin and is an allosteric inhibitor of mTOR, is also able
to induce autophagy (19,20), and significantly induced cell death in
parental and Tzb-refractory gastric cancer cells in a dose- and
time-dependent manner (Fig. 5B and
C). As demonstrated in Fig. 5D,
Tzb had little effect on the phosphorylation of Akt and mTOR in
POOL2, however, combining everolimus with Tzb reduced the
activation of Akt and mTOR. Taken together, these results suggest
that Tzb-induction in cells may alter the balance of cellular
autophagic flux, via autophagic flux inhibition or the use of an
autophagy inducer, such as everolimus.
Discussion
Although a number of studies have reported that
autophagy induction is associated with the viability of
HER2-amplified human breast cancer cells in response to the
anti-HER2 monoclonal antibody Tzb, few studies have investigated
the association between autophagy and HER2 in gastric cancer. To
the best of our knowledge, the present study is the first to
investigate the function of autophagy in HER2-refractory gastric
cancer.
The present study revealed that Tzb reduced the
expression of LC3 I and increased the expression of SQSTM1 in
NCI-N87 cells, suggesting that Tzb may inhibit autophagic flux.
Tzb-refractory cells exhibited less autophagic flux and increased
viability compared with the parental cancer cells, suggesting that
autophagy may induce cell death in gastric cancer cells. The
involvement of autophagy in tumorigenesis and cancer therapy is
complex. In the present study, the autophagy inhibitors, BafA1 and
3MA, induced NCI-N87 cell death, but did not induce the death of
Tzb-refractory cells. -HCQ was able to overcome Tzb resistance, but
had a weaker effect than that observed in the parental NCI-N87
cells. These results suggest that, in Tzb-refractory NCI-N87 cells,
cell apoptosis is prevented via autophagy inhibition and not
autophagy activation. Certain studies have also reported that the
autophagy inhibitor, chloroquine, is an effective treatment for
Tzb-refractory HER2 gene-amplified metastatic breast cancer
(14), which is in line with the
results of the present study in NCI-N87 cells. As HCQ is not only
an autophagy inhibitor, having other functions that include
Toll-like receptor regulation (21)
and AMP-activated protein kinase induction (22), it may induce cancer cell death via
various pathways not associated with autophagic flux.
However, certain aspects of the results obtained in
NCI-N87 cells in the present study are not consistent with the
previous results associated with breast cancer (13,15). One
reason for this is that the cell lines used were different. It has
been reported that HER2 protein overexpression and gene
amplification are more heterogeneous in gastric cancer than in
breast cancer (5). In gastric and
gastroesophageal cancer, HER2 overexpression varies and prognostic
relevance is inconsistent (5), which
may explain, to a certain extent, the inconsistency in the
mechanisms of autophagy in different Tzb-refractory cells.
Additionally, Tzb resistance time and the method used to achieve
Tzb resistance differ to those used previously.
The Akt/mTOR signaling pathway is an important
negative regulator of autophagic flux (23–25). In
the present study, the phosphorylation of Akt and mTOR in
Tzb-refractory cells and parental NCI 87 cells was examined. The
data demonstrated that Tzb may activate the Akt/mTOR pathway to
inhibit autophagic flux.
Everolimus has been approved by the US Food and Drug
Administration for the treatment of renal and breast cancer
(26). The present study demonstrated
that everolimus induces the cell death of parental gastric cancer
cells and Tzb-refractory cells, suggesting that the mTOR inhibitor
inhibits Tzb-refractory cell growth. Fuereder et al
(27) reported that another mTOR
inhibitor, BEZ235, inhibited the growth of NCI-N87, but not that of
MK45 or MKN28 (HER2-negative gastric cancer) xenografts.
Furthermore, Zhu et al (28)
reported that BEZ235 exerts extensive antitumor activity in
HER2-positive gastric cancer (NCI-N87 and SNU216) in vitro
and in vivo. These results implied that mTOR may serve an
important function in HER2-positive gastric cancer and that the
resistance of HER2 to Tzb may, at least partially, be due to mTOR
activation.
In conclusion, the present study suggests that
autophagy may be involved in the resistance of human gastric cancer
cells to Tzb. Furthermore, it was also revealed that the Akt/mTOR
pathway is involved in the Tzb resistance of gastric cancer cell
lines and that the autophagy inducer, everolimus, is able to
inhibit cell growth in these cell lines. These results suggest that
Tzb-refractory cells prevent cell apoptosis via autophagic flux
inhibition, and that everolimus may overcome resistance to Tzb in
HER2-postive gastric cancer. Although these effects are not fully
understood and the details of such mechanisms require further
investigation, autophagy inhibited by Tzb appears to provide cells
with the capacity to evolve and develop Tzb resistance, thereby
preventing gastric cancer cell death.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81503102), the
Natural Science Foundation of Ningbo (grant no. 2015A610230), the
Oncology Key Special Subject of Ningbo (grant no. 2016-B05) and the
Medical Science and Technology of Clinical Research Projects in
Zhejiang Province (grant no. 2015kyB345).
References
|
1
|
Wang Q, Zhang X, Shen E, Gao J, Cao F,
Wang X, Li Y, Tian T, Wang J, Chen Z, et al: The anti-HER3 antibody
in combination with trastuzumab exerts synergistic antitumor
activity in HER2-positive gastric cancer. Cancer Lett. 380:20–30.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gravalos C and Jimeno A: HER2 in gastric
cancer: A new prognostic factor and a novel therapeutic target. Ann
Oncol. 19:1523–1529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arienti C, Zanoni M, Pignatta S, Del Rio
A, Carloni S, Tebaldi M, Tedaldi G and Tesei A: Preclinical
evidence of multiple mechanisms underlying trastuzumab resistance
in gastric cancer. Oncotarget. 7:18424–18439. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: Correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abrahao-Machado LF and Scapulatempo-Neto
C: HER2 testing in gastric cancer: An update. World J
Gastroenterol. 22:4619–4625. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bang YJ, Van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): A phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhi X and Zhong Q: Autophagy in cancer.
F1000prime Rep. 7:182015. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao Z, Han F, Yang S, Wu J and Zhan W:
Oxamate-mediated inhibition of lactate dehydrogenase induces
protective autophagy in gastric cancer cells: Involvement of the
Akt-mTOR signaling pathway. Cancer Lett. 358:17–26. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pan WR, Chen YL, Hsu HC and Chen WJ:
Antimicrobial peptide GW-H1-induced apoptosis of human gastric
cancer AGS cell line is enhanced by suppression of autophagy. Mol
Cell Biochem. 400:77–86. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu M, Lao Y, Xu N, Wang X, Tan H, Fu W,
Lin Z and Xu H: Guttiferone K induces autophagy and sensitizes
cancer cells to nutrient stress-induced cell death. Phytomedicine.
22:902–910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qian HR and Yang Y: Functional role of
autophagy in gastric cancer. Oncotarget. 7:17641–17651. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cufi S, Vazquez-Martin A,
Oliveras-Ferraros C, Corominas-Faja B, Urruticoechea A,
Martin-Castillo B and Menendez JA: Autophagy-related gene 12
(ATG12) is a novel determinant of primary resistance to
HER2-targeted therapies: Utility of transcriptome analysis of the
autophagy interactome to guide breast cancer treatment. Oncotarget.
3:1600–1614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rodriguez CE, Reidel SI, Bal de Kier Joffé
ED, Jasnis MA and Fiszman GL: Autophagy protects from
trastuzumab-induced cytotoxicity in HER2 overexpressing breast
tumor spheroids. PLoS One. 10:e01379202015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cufi S, Vazquez-Martin A,
Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, López-Bonet E,
Martin-Castillo B, Joven J and Menendez JA: The anti-malarial
chloroquine overcomes primary resistance and restores sensitivity
to trastuzumab in HER2-positive breast cancer. Sci Rep. 3:24692013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vazquez-Martin A, Oliveras-Ferraros C and
Menendez JA: Autophagy facilitates the development of breast cancer
resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS
One. 4:e62512009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bartlett BJ, Isakson P, Lewerenz J,
Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert
DR, Simonsen A and Finley KD: p62, Ref(2)P and ubiquitinated
proteins are conserved markers of neuronal aging, aggregate
formation and progressive autophagic defects. Autophagy. 7:572–583.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pamarthy S, Jaiswal MK, Kulshreshtha A,
Katara GK, Gilman-Sachs A and Beaman KD: The vacuolar ATPase
a2-subunit regulates Notch signaling in triple-negative breast
cancer cells. Oncotarget. 6:34206–34220. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Manic G, Obrist F, Kroemer G, Vitale I and
Galluzzi L: Chloroquine and hydroxychloroquine for cancer therapy.
Mol Cell Oncol. 1:e299112014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lui A, New J, Ogony J, Thomas S and
Lewis-Wambi J: Everolimus downregulates estrogen receptor and
induces autophagy in aromatase inhibitor-resistant breast cancer
cells. BMC Cancer. 16:4872016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin CI, Whang EE, Donner DB, Du J, Lorch
J, He F, Jiang X, Price BD, Moore FD Jr and Ruan DT: Autophagy
induction with RAD001 enhances chemosensitivity and
radiosensitivity through Met inhibition in papillary thyroid
cancer. Mol Cancer Res. 8:1217–1226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Said A, Bock S, Lajqi T, Muller G and
Weindl G: Chloroquine promotes IL-17 production by CD4+
T cells via p38-dependent IL-23 release by monocyte-derived
Langerhans-like cells. J Immunol. 193:6135–6143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Spears LD, Tran AV, Qin CY, Hobbs SB,
Burns CA, Royer NK, Zhang Z, Ralston L and Fisher JS: Chloroquine
increases phosphorylation of AMPK and Akt in myotubes. Heliyon.
2:e000832016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O'Reilly KE, Rojo F, She QB, Solit D,
Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al:
mTOR inhibition induces upstream receptor tyrosine kinase signaling
and activates Akt. Cancer Res. 66:1500–1508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang XL, Lin FJ, Guo YJ, Shao ZM and Ou
ZL: Gemcitabine resistance in breast cancer cells regulated by
PI3K/AKT-mediated cellular proliferation exerts negative feedback
via the MEK/MAPK and mTOR pathways. OncoTargets Ther. 7:1033–1042.
2014.
|
|
25
|
Zhang L, Wang H, Xu J, Zhu J and Ding K:
Inhibition of cathepsin S induces autophagy and apoptosis in human
glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K
and JNK signaling pathways. Toxicol Lett. 228:248–259. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saran U, Foti M and Dufour JF: Cellular
and molecular effects of the mTOR inhibitor everolimus. Clin Sci
(Lond). 129:895–914. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fuereder T, Wanek T, Pflegerl P,
Jaeger-Lansky A, Hoeflmayer D, Strommer S, Kuntner C, Wrba F,
Werzowa J, Hejna M, et al: Gastric cancer growth control by BEZ235
in vivo does not correlate with PI3K/mTOR target inhibition but
with [18F]FLT uptake. Clin Cancer Res. 17:5322–5332. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu Y, Tian T, Zou J, Wang Q, Li Z, Li Y,
Liu X, Dong B, Li N, Gao J and Shen L: Dual PI3K/mTOR inhibitor
BEZ235 exerts extensive antitumor activity in HER2-positive gastric
cancer. BMC Cancer. 15:8942015. View Article : Google Scholar : PubMed/NCBI
|