Introduction
The International Agency for Research on Cancer
(IARC) has reported in recent years that colorectal cancer (CRC) is
the 3rd most common cancer type based on the number of cancer cases
worldwide. According to the estimates of world cancer incidence
rates, the number of CRC cases was ~1,360,000 (9.7% in total) for
the two sexes (1). Chemotherapy is an
important treatment strategy for CRC, and one of the key
chemotherapeutic drugs for treating metastatic CRC is irinotecan
(CPT-11). CPT-11, which is the standard drug for treatment of CRC,
can be converted by carboxylesterase to the active metabolite
SN-38, which has even greater antitumor activity, inhibiting DNA
topoisomerase I through the formation of stable topoisomerase I-DNA
cleavable complexes (2–4). The resulting DNA damage can lead to cell
cycle arrest and/or cell death by apoptosis (5). However, acquired or constitutive
resistance to SN-38 does occur, allowing for tumor progression
(6).
One cause of tumor initiation, progression, and drug
resistance acquisition is aberrant DNA hypermethylation. In tumors,
DNA hypermethylation mediates epigenetic changes that silence gene
expression without altering nucleotide sequences.
5-Aza-2′-deoxycytidine (DAC) is a DNA methyltransferase (DNMT)
inhibitor, a DNA demethylating agent, and a cell cycle-arresting
agent (7–9). Preliminary studies have been conducted
on the combined use of DNMT inhibitors with existing antitumor
agents (10–12). Accordingly, a preliminary experiment
was conducted on a combination of a specific antitumor agents,
including CPT-11, SN-38 or 5-FU with one of several epigenetic
modifiers including DAC in two different human CRC cell lines;
HCT116 and HT29 (13). Marked
enhancement of the antitumor activities of CPT-11 or SN-38 with DAC
was observed in HCT116 cells, but not in HT29 cells. The
potentiation of CPT-11/SN-38 by DAC was associated with decreased
expression of the B-cell lymphoma-2 (Bcl-2) protein. The Bcl-2
family includes anti-apoptotic (e.g., Bcl-2 and Bcl-extra large)
and pro-apoptotic (e.g., Bcl-associated X and Bcl-2 homologous
antagonist killer) proteins, which serve a crucial role in
mitochondria-driven cell death (14).
In addition, previous reports have shown that Bcl-2 can inhibit the
apoptosis induced by chemotherapeutic agents (15–17).
Wilms' tumor protein (WT1) was originally identified
as a tumor suppressor gene mapping to the chromosome 11p13 locus
(18). The WT1 gene product is a
transcription factor with a proline-glutamine rich domain at the
N-terminus and a zinc finger domain at the C-terminus. The WT1 gene
yields four alternative splice variants: WT1-A [17 amino acids
(AA)−/3 AA lysine-threonine-serine (KTS)−;
WT1-B [17 AA+/KTS−]; WT1-C [17
AA−/KTS+]; and WT1-D [17
AA+/KTS+] (19). Previous studies have indicated that
the WT1 protein upregulates or downregulates Bcl-2, depending on
the cell-type and/or isoform of WT1 (20,21). The
WT1 gene was also overexpressed in multiple types of solid tumor
and primary human leukemia (12,22–26), and
high expression levels of WT1 mRNA were associated with poor
prognosis in leukemia and breast cancer (22,27).
Mayo et al (21) revealed that the stable expression of
the WT1-B isoform resulted in elevated endogenous Bcl-2 protein in
rhabdoid cells. However, roles for WT1 and Bcl-2 in the
DAC-mediated potentiation of CPT-11/SN-38 antitumor activity have
not been elucidated in human CRC cells.
The present study aimed to clarify the association
between this potentiation of antitumor activity and the WT1-Bcl-2
pathway by RNA interference-mediated knockdown of WT1 using the
human CRC cell lines, HCT116 and HT29.
Materials and methods
Cell lines and culture conditions
Human colon carcinoma HCT116 (No. CCL-247) and human
colon adenocarcinoma HT29 (No. HTB-38) cell lines were obtained
from DS Pharma Biomedical Co., Ltd. (Osaka, Japan). These cell
lines were cultured in Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Hyclone; GE
Healthcare Life Sciences, Logan, UT, USA) and 1%
antibiotic-antimycotic (Gibco; Thermo Fisher Scientific, Inc.) at
37°C in a 5% CO2 incubator.
Regents
CPT-11 was purchased from Toronto Research
Chemicals, Inc. (Toronto, ON, Canada) and SN-38 was purchased from
Tocris Bioscience (Bristol, UK). CPT-11 and SN-38 were dissolved in
dimethyl sulfoxide and stored at −30°C. DAC was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany), dissolved in
Milli-Q water (Direct-Q UV; Merck KGaA), and stored at −30°C.
Drug exposure
For each experiment, HCT116 and HT29 cells were
exposed for 10 days to either vehicle alone (control), CPT-11
alone, SN-38 alone, DAC alone, CPT-11 plus DAC, or SN-38 plus DAC.
The drug concentrations used for the colony-forming assay were
62.5, 125, 250 or 500 nM CPT-11; 0.35, 0.5, 0.7 or 1.0 nM SN-38;
and 31.25 nM DAC in HCT116 cells. For HT29 cells, the drug
concentrations were 0.5 or 1.0 µM CPT-11; 1.0 or 2.5 nM SN-38; and
75 or 100 nM DAC. In western blot analysis and RNA interference
assays utilizing HCT116 cells, the concentrations used were 500 nM
CPT-11, 1.0 nM SN-38, and 31.25 nM DAC; the concentrations used for
HT29 cells were 500 nM CPT-11, 1.0 nM SN-38, and 75 nM DAC.
Colony-forming assay
HCT116 and HT29 cells were plated at a density of
20,000 and 5,000 cells per 60-mm dish, respectively. Following
incubation with each drug for 10 days, the colonies were stained
with 0.04% crystal violet overnight at room temperature and scored.
The scored colonies contained more than 50 cells for HCT116 and 30
cells for HT29.
Analysis of drug combination
effects
Isobologram analysis was performed using CompuSyn
software version 1.0 (ComboSyn, Inc., Paramus, NJ, USA), which
enabled the calculation of a combination index (CI) according to
the Chou-Talalay CI-Isoblogram theory (28). To assess the combination effects of
CPT-11 or SN-38 with DAC, colony-forming assay data were converted
to a fraction of growth inhibition by each drug alone or by the
drug combinations as compared with control cells. There are two
methods of CompuSyn software analyses: constant ratio and
non-constant ratio analyses. A constant ratio requires the ratio of
CPT-11 and DAC concentrations in combination experiments to be
constant e.g., 500 nM CPT-11 and 31.25 nM DAC or 250 nM CPT-11 and
15.625 nM DAC etc., where CPT-11 concentration is consistently
16-fold higher than that of DAC, and constant throughout a series
of combination experiments. However, in the experimental conditions
of the present study, drug concentrations were either 125, 250, or
500 nM CPT-11, with 3.9 nM DAC; 125, 250, or 500 nM CPT-11, with
7.8 nM DAC; 125, 250, or 500 nM CPT-11, with 15.625 nM DAC; 125,
250, or 500 nM CPT-11, with 31.25 nM DAC. Furthermore, for SN-38
and DAC in the present study, the drug concentrations were as
follows: Drug concentrations were either 0.35, 0.5, 0.7 or 1.0 nM
SN-38, plus 3.9 nM DAC; 0.35, 0.5, 0.7 or 1.0 nM SN-38, plus 7.8 nM
DAC; 0.35, 0.5, 0.7 or 1.0 nM SN-38, plus 15.625 nM DAC; 0.35, 0.5,
0.7 or 1.0 nM SN-38, plus 31.25 nM DAC. Therefore, drug
concentration ratios for the present study were non-constant.
CompuSyn automatically created a normalized isobologram by a
non-constant ratio analysis (29).
The combination index, CI, is theoretically calculated by CompuSyn
software according to Equation 1.
CI=(D)1(Dx)1+(D)2(Dx)2=(D)1(Dm)1[fa/(1-fa)]1/m1+(D)2(Dm)2[fa/(1-fa)]1/m2
(Equation 1), where (Dx)1, and
(Dx)2 are characteristic parameters for either CPT-11 or
SN-38, and DAC, respectively, and fa, a fraction of growth
inhibition (i.e., 1-colony formation rate). By the median-effect
plot (28,29), log (Dm)1, and
1/m1 were computationally calculated using various
concentrations [i.e., (D)1] of DAC in single drug
treatments. Similarly, log (Dm)2, and 1/m2
were computationally calculated using various concentrations [i.e.,
(D)2] of CPT-11 or SN-38. Furthermore, in combination
experiments, CI values were computationally calculated using drug
concentrations that were used for combination experiments i.e.,
concentration of DAC, (D)1, and concentration of CPT-11
or SN-38 (D)2, and experimentally observed fa.
(Dm)1, m1, (Dm)2, and
m2, are constants obtained from single drug treatment
experiments. Normalized isobolograms, whose X-axis was
(D)1/(Dx)1, and Y-axis was
(D)2/(Dx)2 were created.
Western blotting
HCT116 and HT29 cells were cultured for 6 days
following initiation of the drug treatment and solubilized in
radioimmunoprecipitation assay buffer (50 mM Tris-HCl, 150 mM NaCl,
0.5% deoxycholate, 1% NP-40 and 0.1% SDS). The protein samples were
separated by 10% SDS-PAGE (equal amounts of total protein, 20 µg
per lane) and transferred onto PVDF membranes. Protein
concentrations of the samples were quantified by Bradford assay
using Bio-Rad Protein Assay Dye Reagent Concentrate (catalog no.
#5000006, Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
membranes were blocked with Blocking One (Nacalai, Kyoto, Japan) at
room temperature for 90 min and rinsed with TBS-T (Tris-buffered
saline with 0.1% Tween-20). The membranes were then incubated
overnight at 4°C with primary antibodies for Bcl-2 (1:500 dilution,
kindly provided by Professor Chihaya Maesawa of Iwate Medical
University, Morioka, Japan), WT1 (1:200 dilution; cat. no. M3561,
Clone 6F-H2; Dako; Agilent Technologies, Inc., Santa Clara, CA,
USA), or β-actin (1:1,000 dilution; cat. no. A1978, Sigma-Aldrich;
Merck KGaA) in Can Get Signal Solution 1 (Toyobo Life Science,
Osaka, Japan). The blots were incubated for 90 min with horseradish
peroxidase-conjugated mouse-IgG secondary antibody (1:1,000
dilution; cat. no. 616529; Invitrogen; Thermo Fisher Scientific,
Inc.) in Can Get Signal Solution 2 (Toyobo Life Science) at room
temperature for 2 h. The blots were then washed with TBS-T and
visualized with a ChemiDox™ XRS+ with Image Lab™ software version
4.0 (Bio-Rad Laboratories, Inc.) following incubation with a
chemiluminescent reaction using Clarify™ Western ECL substrate
(Bio-Rad Laboratories, Inc.). Following detection, the protein band
intensity was quantified using ImageJ software version 1.48
(National Institutes of Health, Bethesda, MD, USA).
RNA interference
Small interfering RNA (siRNA) was used to
downregulate WT1 gene expression in HCT116 cells by transfection of
RNA oligonucleotides with Lipofectamine™ RNAiMAX (Invitrogen;
Thermo Fisher Scientific, Inc.). siRNA sequences were constructed
to target human WT1 mRNA (Sequences 5′-3′, forward:
CCAAAGGAGACAUACAGGUGUGAAA; and reverse: UUUCACACCUGUAUGUCUCCUUUGG:
catalog no. HSS111390; Invitrogen; Thermo Fisher Scientific, Inc.),
and control siRNAs were designed by scrambling those nucleotide
sequences. Control siRNAs were not homologous to any other gene
(MISSION® siRNA Universal Negative Control #1,
SIC001-10; Sigma-Aldrich; Merck KGaA). The cells were plated in 3
ml of DMEM (100,000 cells per ml) on a 60-mm dish with or without a
100 nM (final concentration) siRNA mixture in the presence of 10 µl
Lipofectamine. At 12 and 24 h following the addition of
WT1-targetting siRNA to the cells, the WT1 and Bcl-2 protein
expression levels were analyzed by western blotting. Following
detection, the protein band intensity was quantified using ImageJ
software version 1.48.
Statistical analysis
Data on each colony formation rate of drug-treated
HCT116 cells were expressed as the mean ± standard deviation. Data
were compared between CPT-11 or SN-38 alone and in combination with
DAC using Student's t-test. In the same way, data on each colony
formation rate of drug-treated HT29 cells were expressed as the
mean and SD, and compared between each anticancer drug alone and
combination with DAC using one-way analysis of variance (ANOVA)
followed by Tukey's test.
Data on the protein expression level of Bcl-2 and
WT1 in the various drug-treated cells were compared between
control, CPT-11 or SN-38 alone, and combination with DAC treatments
by one-way ANOVA followed by Dunnett's test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Enhancement of antitumor activity of
CPT-11 and SN-38 by DAC in HCT116 cells
The antitumor effects of CPT-11 and its active
metabolite SN-38 were investigated with the potential enhancer DAC
in the HCT116 and HT29 human CRC cell lines by assessing colony
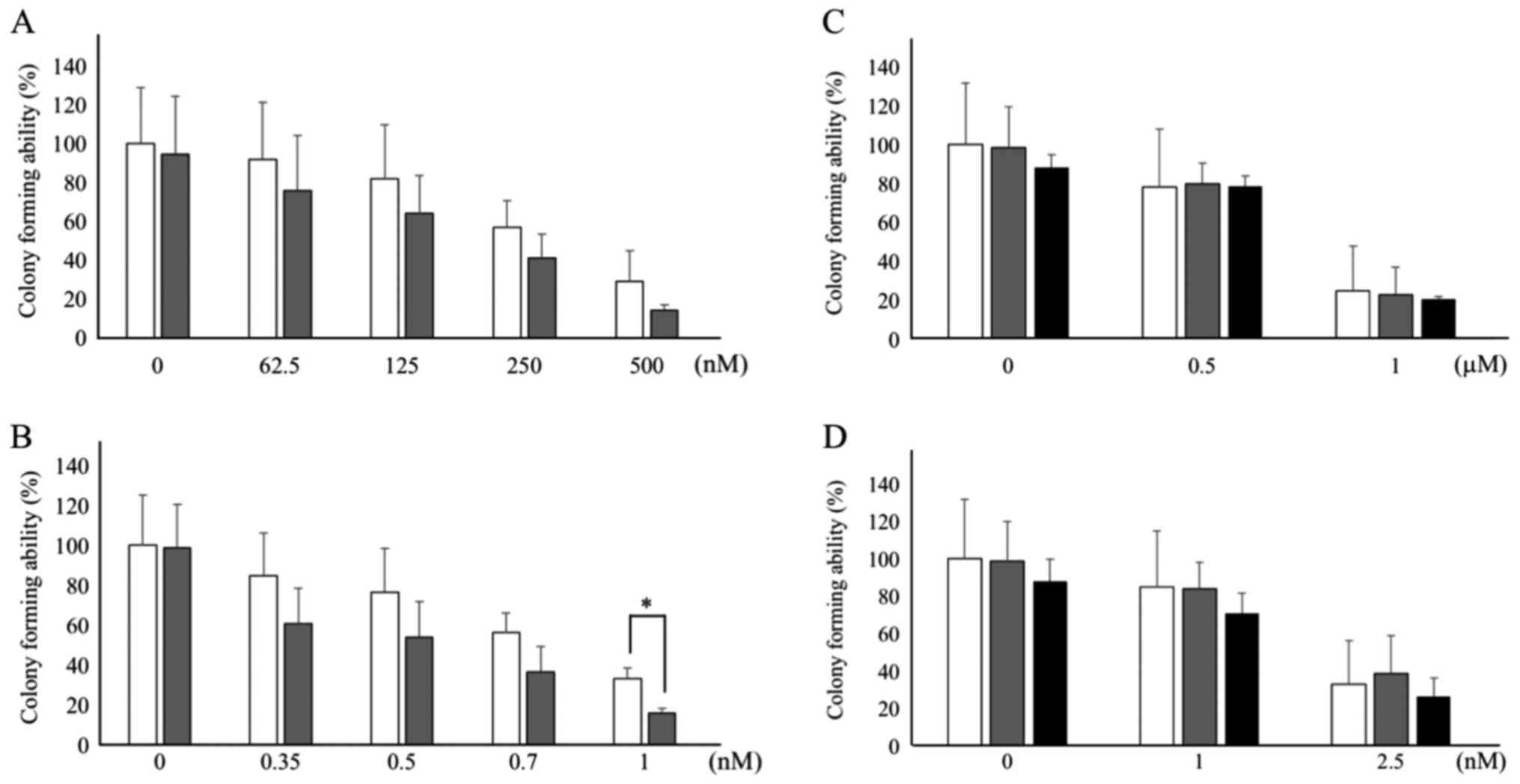
formation. As depicted in Fig. 1A and
B, concentration-dependent antitumor activity of CPT-11
(62.5–500 nM) and SN-38 (0.35–1.0 nM) was observed. DAC (31.25 nM)
exhibited a tendency to potentiate the antitumor activity of CPT-11
(not statistically significant; Fig.
1A) and SN-38 (with statistical significance (P<0.05) at 1.0
nM, Fig. 1B) in HCT116 cells. CPT-11
suppressed colony formation to 91.6% at 62.5 nM, to 81.6% at 125
nM, to 56.7% at 250 nM, and to 28.7% at 500 nM in the absence of
CPT-11. On the other hand, the presence of DAC and CPT-11
suppressed colony formation to 75.5% at 62.5 nM, to 63.7% 125 nM,
to 40.8% 250 nM and to 14.1% at 500 nM. SN-38 suppressed colony
formation to 84.8% at 0.35 nM, to 76.6% at 0.5 nM, to 56.0% at 0.7
nM, and to 33.1% and 1.0 nM without DAC. The presence of DAC plus
SN-38 suppressed colony formation to 60.9% at 0.35 nM, to 53.7% at
0.5 nM, to 36.4% at 0.7 nM, and to 15.6% at 1.0 nM. The antitumor
effect elicited by the combination of 1.0 nM SN-38 and 31.25 nM DAC
was stronger than that at 1.0 nM SN-38 alone with statistical
significance (P<0.05). Treatment with DAC alone only slightly
inhibited colony formation in HCT116 cells, with no statistical
significance observed (Fig. 1A and
B). By contrast, HT29 cells were ~2-fold less sensitive to
CPT-11 (0.5–1.0 µM) and 2.5-fold less sensitive to SN-38 (1.0–2.5
nM) than HCT116 cells (Fig. 1C and
D). In the absence of DAC, CPT-11 suppressed colony formation
to 78.0% at 0.5 µM and to 24.6% at 1 mM, whereas SN-38 suppressed
colony formation to 85.0% at 1.0 nM and to 33.1% at 2.5 nM. In
combination with DAC, no enhancement of antitumor activity was
observed for either CPT-11 or SN-38 (Fig.
1C and D).
Synergistic antitumor activity induced
by SN-38 and DAC in HCT116 cells
Fig. 1 demonstrates
that the antitumor activity of CPT-11 and SN-38 was enhanced by DAC
in HCT116 cells. These data indicate that CPT-11 or SN-38 combined
with DAC might synergize to inhibit cell survival. To examine
synergism, the enhancement of CPT-11- and SN-38-mediated antitumor
activity was analyzed in the presence of various concentrations of
DAC by creating isobolograms with CompuSyn software. Using this
isobologram type, the combined effects of the two drugs were
summarized as follows: CI values <1.0 (points in the lower
left), =1.0 (points on the hypotenuse) and >1.0 (points in the
upper right) respectively indicated synergistic, additive and
antagonistic effects (28,29). The combination of CPT-11 and DAC was
mostly additive when evaluated using isobolograms (Fig. 2A). The normalized isobologram for the
combination of SN-38 and DAC demonstrates synergism of the
antitumor activity (Fig. 2B). The
ranges of concentrations exhibiting synergism were 0.35–1.0 nM for
SN-38 and 3.9–7.8 nM for DAC.
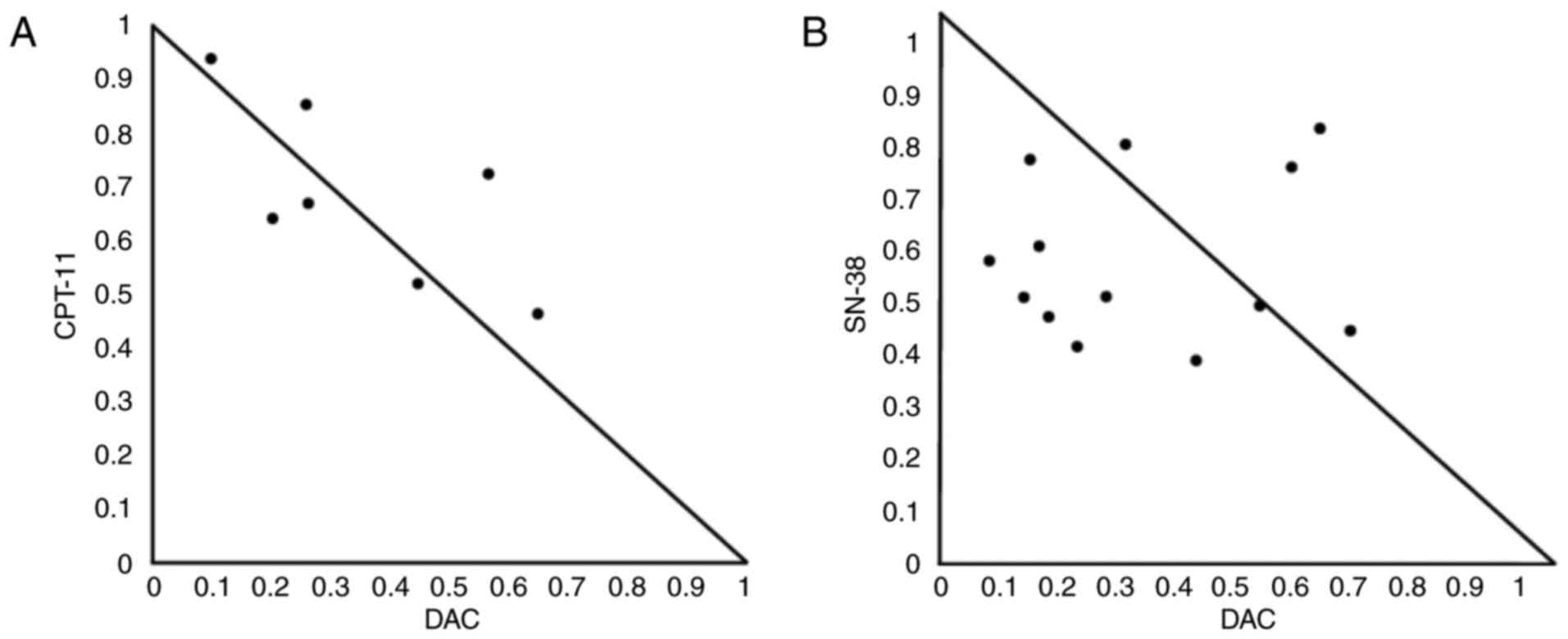 | Figure 2.Normalized isobologram for
CPT-11/SN-38 and DAC in HCT116 cells. CI for various combinations
of DAC (3.9–31.25 nM) and either CPT-11 (125, 250 and 500 nM) or
SN-38 (0.35, 0.5, 0.7 and 1.0 nM). Cells were treated with various
concentrations of (A) CPT-11 and DAC, or (B) SN-38 and DAC. The
combination effects can be summarized as follows: CI<1, dots
located lower left; CI=1, dots on the hypotenuse; and CI>1, dots
located upper right; these results indicate synergistic, additive,
and antagonistic effects, respectively. DAC,
5-aza-2′-deoxycytidine; CPT-11, irinotecan; SN-38,
7-ethyl-10-hydroxycamptothecin; CI, combination index. |
Protein expression levels of Bcl-2 in
HCT116 cells and HT29 cells treated with CPT-11, SN-38, and DAC,
alone and in combination
Several lines of evidence have indicated that
intracellular Bcl-2 protein levels are associated with the
resistance of cancer cells to CPT-11 and SN-38 (30,31). These
previous studies indicated that Bcl-2 expression is associated with
cancer cell sensitivities to anticancer drugs. Therefore, the
expression of Bcl-2 protein in HCT116 and HT29 cells was examined.
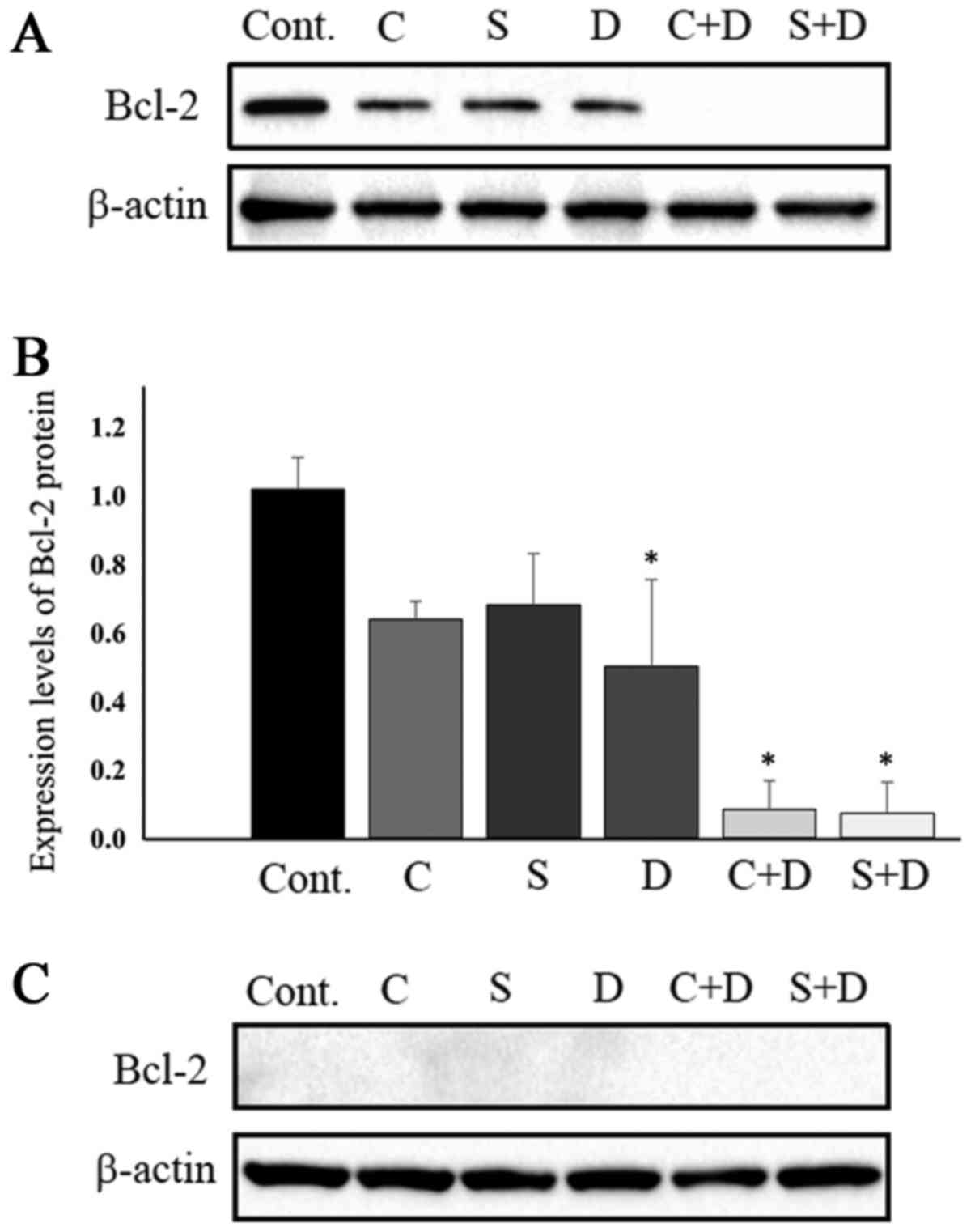
Fig. 3A and B depict Bcl-2 protein
expression in HCT116 cells. Bcl-2 protein levels were marginally
downregulated in cells exposed to 0.5 µM CPT-11, 1.0 nM SN-38 and
31.25 nM DAC. Combination treatment of 31.25 nM DAC with either 0.5
µM CPT-11 or 1.0 nM SN-38 resulted in Bcl-2 protein levels that
were almost under the limit of detection. Protein band intensity
was determined using by ImageJ software following normalization to
β-actin. Bcl-2 protein expression levels were suppressed to 62.7%
with CPT-11 alone, 66.7% with SN-38 alone, 50.0% with DAC alone,
7.8% with CPT-11 plus DAC, and 6.9% with SN-38 plus DAC, when
compared with control samples (Fig.
3B). The combination of DAC and either CPT-11 or SN-38 most
strongly inhibited colony formation by HCT116 cells, an observation
consistent with previous work demonstrating that cancer cell
resistance to CPT-11 was associated with Bcl-2 overexpression in
the human lung cancer cell SBC-3/Bcl-2 subline and the human
leukemia cell multidrug resistant HL-60-Vinc subline (30,31).
Changes in Bcl-2 protein expression in HT29 cells were also
examined following exposure to the drugs, where no DAC-mediated
potentiation of CPT-11/SN-38 antitumor activity was observed. Bcl-2
protein levels in the HT29 cells were under the limit of detection,
at least 10 times lower than those in the HCT116 cells (Fig. 3C). Under these conditions, possible
changes in Bcl-2 protein expression for cells treated with CPT-11,
SN-38, DAC, CPT-11 with DAC, and SN-38 with DAC were not
detectable.
WT1 protein expression levels in two
CRC cell lines treated with CPT-11, SN-38 and DAC, alone and in
combination
WT1 was reported to reverse antitumor drug-induced
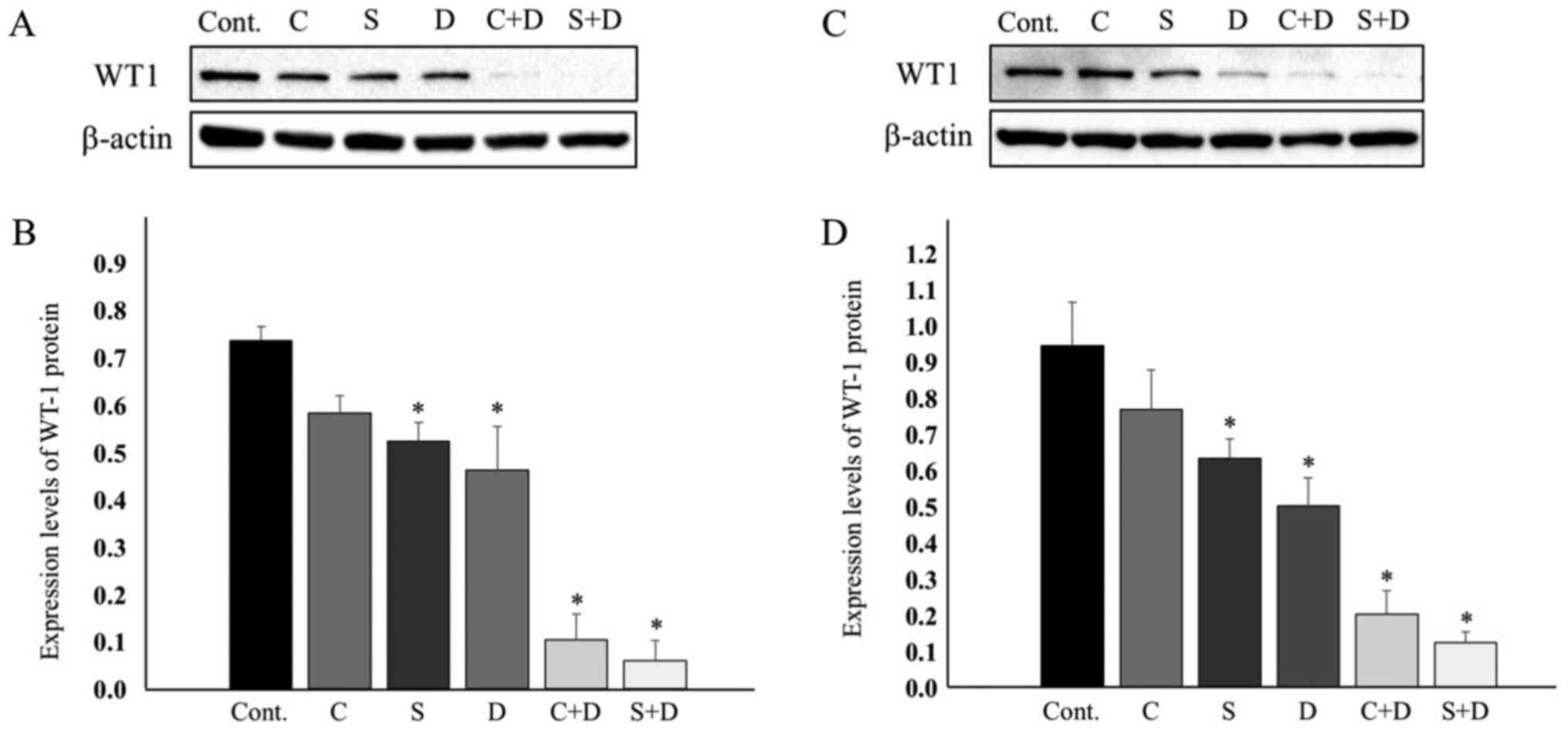
apoptosis by transcriptionally upregulating Bcl-2 (21). The expression of the WT1 protein was
examined in HCT116 and HT29 cells. Fig.
4A indicates that the WT1 protein was marginally downregulated
in the HCT116 cells exposed to 0.5 µM CPT-11, 1.0 nM SN-38, and
31.25 nM DAC. Most evidently, the combination of 31.25 nM DAC and
either 0.5 µM CPT-11 or 1.0 nM SN-38 resulted in WT1 protein levels
that were as low as the limit of detection. The WT1 protein levels
were decreased, as estimated by ImageJ software following
normalization to β-actin. WT1 expression was suppressed to 79.2% at
0.5 µM CPT-11, 71.1% at 1.0 nM SN-38, 62.9% at 31.25 nM DAC, 14.0%
with CPT-11 plus DAC, and 8.0% with SN-38 plus DAC (Fig. 4A and B).
These downregulation profiles of WT1 expression were
quite similar to those of the Bcl-2 protein (Fig. 3A and B). Low-level expression of Bcl-2
in HT29 cells led us to examine whether expression of WT1, a Bcl-2
regulator, was similarly low in HT29 cells. Notably, WT1 protein
expression was observed at a level close to that observed in HCT116
cells. The expression level of WT1 decreased to 81.4% upon
treatment with 0.5 µM CPT-11, to 67.2% upon treatment with 1.0 nM
SN-38, 53.2% upon treatment with 75 nM DAC, 21.4% upon treatment
with 0.5 µM CPT-11 and 75 nM DAC, and 13.1% upon treatment with 1.0
nM SN-38 and 75 nM DAC (Fig. 4C and
D).
Knockdown of WT1 suppressed Bcl-2
protein expression in HCT116 cells
Since the association between WT1 and Bcl-2 in CRC
cells remains unclear, a WT1-targeted siRNA was utilized to assess
WT1 function in HCT116 cells. Knockdown of WT1 was confirmed at the
protein level from 12 and 24 h following treatment with 100 nM
siRNA, with 88.2% suppression of WT1 at 12 h as compared with
non-transfected control and 88.8% suppression as compared with
control (scrambled) siRNA-transfected cells. At 24 h, the extent of
WT1 suppression was as low as 66.1% as compared to the
non-transfected control, and as low as 71.2% as compared with the
control siRNA-transfected cells (Fig.
5). WT1 expression was markedly reduced within 12 h of the
application of the WT1 siRNA and was rapidly restored by 24 h.
Anti-apoptotic Bcl-2 protein levels at 12 and 24 h were also
suppressed following the application of WT1 siRNA. Bcl-2 proteins
were suppressed to 44.6% when compared with the non-transfected
control and 55.8% when compared with the control siRNA-transfected
cells at 12 h. At 24 h, the extent of Bcl-2 suppression was 62.3%
when compared with the non-transfected control and 66.9% compared
with the control siRNA-transfected group (Fig. 5). These results clearly indicated that
WT1 functions as a positive regulator of Bcl-2 in human CRC HCT116
cells.
Discussion
The present study revealed that two different human
colon cancer cell lines, HCT116 and HT29, exhibited different
profiles for the DAC-mediated potentiation of CPT-11/SN-38
antitumor activity, as measured by colony-formation assays. A
statistically significant antitumor potentiation of CPT-11/SN-38 by
DAC was demonstrated in HCT116 cells (Fig. 1A and B); however, no appreciable
effect was observed by the same combination in HT29 cells (Fig. 1C and D). HCT116 cells continuously
exposed to various concentrations of CPT-11/SN-38 and 31.25 nM DAC
for 10 days had a plating efficiency lower than that of the
respective control cells, which were treated with CPT-11/SN-38
alone (Fig. 1A and B). In these
experiments, it is noteworthy that 31.25 nM DAC exhibited
essentially no cytotoxicity in HCT116 cells. This DAC concentration
was at least one order of magnitude lower than clinically achieved
plasma concentrations (~360-660 nM) in a phase I clinical trial
study (1 h infusion with a dose of 45 mg/m2) when used
in combination with carboplatin in solid tumors, performed in the
United Kingdom (32) and that
achieved in a phase I/II study of DAC (1 h infusion with a dose of
15–20 mg/m2) in patients with myelodysplastic syndrome,
performed in Japan (33). The
potentiation of CPT-11/SN-38 antitumor activity by DAC was also
examined to identify possible synergism, according to the
Chou-Talalay method utilizing the CI-Isoblogram theory (28). The results of this analysis indicated
that while a combination of CPT-11 and DAC was additive, a
combination of SN-38 and DAC yielded synergistic effects in HCT116
cells.
Differences in the expression of proteins with
pro-apoptotic or anti-apoptotic functions were examined in HCT116
and HT29 cells. Notably, the level of Bcl-2 protein, an
apoptosis-suppressing factor, was markedly different in the two
cell lines (Fig. 3A and C). Bcl-2
protein expression was marginally downregulated by 37.3, 33.3, and
50.0% by the single-drug administrations of CPT-11, SN-38, and DAC,
respectively. By contrast, Bcl-2 proteins were heavily
downregulated in the drug combination groups with a 92.2% reduction
induced by treatment with CPT-11 and DAC and a 93.1% reduction by
treatment with SN-38 and DAC in HCT116 cells following a 6-day
exposure (Fig. 3A). By contrast,
Bcl-2 protein levels in HT29 cells were barely detectable (Fig. 3C).
WT1 was reported to function as a Bcl-2
transcriptional regulatory factor (20,21) and is
overexpressed in several solid tumors (12,23–26).
Preliminary studies reported that WT1 exists in four isoforms, each
of which can regulate the Bcl-2 gene in a positive or a negative
way, depending on the cancer cell type (20,21). The
heterologous expression of WT1 in HeLa cells led to the repression
of Bcl-2 promoters, demonstrating negative regulation of Bcl-2 by
WT1 in HeLa cells (20). Rhabdoid
cell lines stably expressing the WT1-B isoform [17
AA+/KTS−] resulted in increased expression of
Bcl-2 proteins, indicating positive regulation of Bcl-2 expression
by WT1 in a Rhabdoid cell model (21). Tatsumi et al (34), using a WT1-downregulating short
hairpin RNA as a potent apoptosis-inducing agent, demonstrated that
WT1 isoforms with exon 5 [17 AA+/KTS+ and 17
AA+/KTS−] were anti-apoptotic proteins in
WT1-expressing cell-lines (including fibrosarcoma HT-1080, lung
cancer LU99B, gastric cancer AZ-521, and glioblastoma A172 cells),
but not WT1-non-expressing cell lines (including gastric cancer
MKN28, cervical cancer HelaAG and lung cancer PC14 cells).
To understand the role of WT1 on Bcl-2 expression in
a human CRC cell line, WT1 protein expression levels in HCT116
cells and associated changes in Bcl-2 protein levels were analyzed
using a WT1-targeted siRNA. Downregulation of WT1 protein
expression was observed at 12 h after the application of the WT1
siRNA, although protein expression was restored after 24 h. Bcl-2
protein expression was also examined at 12 and 24 h. While WT1
protein was forcibly downregulated to 11–12% by WT1-targeting siRNA
at 12 h after the addition of the WT1 siRNA, Bcl-2 protein
expression was downregulated to 45–56%. At 24 h after the WT1 siRNA
addition, expressions of WT1 and Bcl-2 were restored to 66–71 and
62–67%, respectively. These results led to the conclusion that WT1
positively regulated Bcl-2 expression in the HCT116 human CRC cell
line.
In experiments using HCT116 cells, an additive
antitumor effect was observed when a combination of CPT-11 and DAC
was used, whereas synergism was observed when a combination of
SN-38 and DAC was used. By contrast, no such potentiation of
antitumor activity was observed in the human CRC HT29 cell line. In
HCT116 cells, it was demonstrated that WT1 acts as a positive Bcl-2
regulator. Expression of WT1 and Bcl-2 were markedly downregulated
in cells exposed to a combination of CPT-11 or SN-38 with DAC, when
compared with those in cells exposed to CPT-11, SN-38 or DAC alone.
Given the low level of cytotoxicity observed in HCT116 cells with
31.25 nM DAC alone, it was notable that downregulation of WT1 and
Bcl-2 was dependent on the presence of this non-cytotoxic
concentration of DAC. Detailed molecular mechanisms for the
DAC-mediated potentiation of CPT-11 or SN-38 cytotoxicity and the
resultant downregulation of WT1 and Bcl-2 in HCT116 cells have not
been elucidated. However, the HT29 cells, in which the expression
level of Bcl-2 was much lower than that in HCT116 cells, failed to
exhibit potentiation of the antitumor activity. We hypothesize that
even though WT1 is expressed in HT29 cells, the low level of Bcl-2
expression might be insufficient for the potentiation of this
activity, indicating a requirement for participation of the
WT1-Bcl-2 pathway in this process.
In conclusion, the known DNA methyltransferase
inhibitor DAC sensitized the human CRC HCT116 cell line to CPT-11
and SN-38, likely through the downregulation of the WT1-Bcl-2
pathway. The extent of the DAC-dependent sensitization may be
associated with Bcl-2 expression levels in CRC cells, which is
dependent on the characteristics of the individual carcinoma
cells.
Acknowledgements
The authors would like to thank Dr Shinji Yasuhira
and Professor Chihaya Maesawa (Department of Tumor Biology,
Institute of Biomedical Science, Iwate Medical University, Morioka,
Japan) for providing anti-Bcl-2 antibodies for western blot
analysis.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CPT-11
|
irinotecan
|
|
CRC
|
colorectal cancer
|
|
DAC
|
5-aza-2′-deoxycytidine
|
|
DNMT
|
DNA methyltransferase
|
|
SN-38
|
7-ethyl-10-hydroxycamptothecin
|
References
|
1
|
Stewart BW and Wild CP: International
Agency for Research on Cancer. WHO: World Cancer Report 2014.
http://www.thehealthwell.info/node/725845January
18–2018
|
|
2
|
Kawato Y, Aonuma M, Hirota Y, Kuga H and
Sato K: Intracellular roles of SN-38, a metabolite of the
camptothecin derivative CPT-11, in the antitumor effect of CPT-11.
Cancer Res. 51:4187–4191. 1991.PubMed/NCBI
|
|
3
|
Tanizawa A, Fujimori A, Fujimori Y and
Pommier Y: Comparison of topoisomerase I inhibition, DNA damage,
and cytotoxicity of camptothecin derivatives presently in clinical
trials. J Natl Cancer Inst. 11:836–842. 1994. View Article : Google Scholar
|
|
4
|
Hsiang YH, Lihou MG and Liu LF: Arrest of
replication forks by drug-stabilized topoisomerase I -DNA cleavable
complexes as a mechanism of cell killing by camptotecin. Cancer
Res. 49:5077–5082. 1989.PubMed/NCBI
|
|
5
|
Te Poele RH and Joel SP:
Schedule-dependent cytotoxicity of SN-38 in p53 wild-type and
mutant colon adenocarcinoma cell lines. Br J Cancer. 81:1285–1293.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Allen WL and Johnston PG: Role of genomic
markers in colorectal cancer treatment. J Int Oncol. 20:4545–4552.
2005.
|
|
7
|
Paz MF, Fraga MF, Avila S, Guo M, Pollan
M, Herman JG and Esteller M: A systematic profile of DNA
methylation in human cancer cell lines. Cancer Res. 63:1114–1121.
2003.PubMed/NCBI
|
|
8
|
Gnyszka A, Jastrzebski Z and Flis S: DNA
methyltransferase inhibitors and their emerging role in epigenetic
therapy of cancer. Anticancer Res. 33:2989–2996. 2013.PubMed/NCBI
|
|
9
|
Tsai HC, Li H, Van Neste L, Cai Y, Robert
C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al:
Transient low doses of DNA-demethylating agents exert durable
antitumor effects on hematological and epithelial tumor cells.
Cancer Cell. 21:430–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ishiguro M, Iida S, Uetake H, Morita S,
Makino H, Kato K, Takagi Y, Enomoto M and Sugihara K: Effect of
combined therapy with low-dose 5-aza-2′-deoxycytidine and
irinotecan on colon cancer cell line HCT-15. Ann Surg Oncol.
14:1752–1762. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ikehata M, Ogawa M, Yamada Y, Tanaka S,
Ueda K and Iwakawa S: Different effects of epigenetic modifiers on
the cytotoxicity induced by 5-fluorouracil, irinotecan or
oxaliplatin in colon cancer cells. Biol Pharm Bull. 37:67–73. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oji Y, Inohara H, Nakazawa M, Nakano Y,
Akahani S, Nakatsuka S, Koga S, Ikeba A, Abeno S, Honjo Y, et al:
Overexpression of the Wilms' tumor gene WT1 in head and neck
squamous cell carcinoma. Cancer Sci. 94:523–529. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okada K, Hakata S, Terashima J, Gamou T,
Habano W and Ozawa S: Combination of the histone deacetylase
inhibitor depsipeptide and 5-fluorouracil upregulates major
histocompatibility complex class II and p21 genes and activates
caspase-3/7 in human colon cancer HCT-116 cells. Oncol Rep.
36:1875–1885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsujimoto Y: Role of Bcl-2 family proteins
in apoptosis: Apoptosomes or mitochondria? Genes Cells. 3:697–707.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamesaki S, Kamesaki H, Jorgensen TJ,
Tanizawa A, Pommier Y and Cossman J: Bcl-2 protein inhibits
etoposide-induced apoptosis through its effects on events
subsequent to topoisomerase II-induced DNA strand breaks and their
repair. Cancer Res. 18:4251–4256. 1993.
|
|
16
|
Dole M, Nuñez G, Merchant AK, Maybaum J,
Rode CK, Bloch CA and Castle VP: Bcl-2 inhibits
chemotherapy-induced apoptosis in neuroblastoma. Cancer Res.
12:3253–3259. 1994.
|
|
17
|
Lou YF, Zou ZZ, Chen PJ, Huang GB, Li B,
Zheng DQ, Yu XR and Luo XY: Combination of Gefitinib and DNA
methylation inhibitor decitabine exerts synergistic anti-cancer
activity in colon cancer cells. PLoS One. 9:e977192014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Call KM, Glaser T, Ito CY, Buckler AJ,
Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, et al:
Isolation and characterization of a zinc finger polypeptide gene at
the human chromosome 11 Wilms' tumor locus. Cell. 60:509–520. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haber DA, Sohn RL, Buckler AJ, Pelletier
J, Call KM and Housman DE: Alternative splicing and genomic
structure of the Wilms tumor gene WT1. Proc Natl Acad Sci USA.
88:pp. 9618–9622. 1991; View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hewitt SM, Hamada S, McDonnell TJ,
Rauscher FJ III and Saunders GF: Regulation of the proto-oncogenes
bcl-2 and c-myc by the Wilms' tumor suppressor gene WT1. Cancer
Res. 55:5386–5389. 1995.PubMed/NCBI
|
|
21
|
Mayo MW, Wang CY, Drouin SS, Madrid LV,
Marshall AF, Reed JC, Weissman BE and Baldwin AS: WT1 modulates
apoptosis by transcriptionally upregulating the bcl-2
proto-oncogene. EMBO J. 18:3990–4003. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Inoue K, Ogawa H, Sonoda Y, Kimura T,
Sakabe H, Oka Y, Miyake S, Tamaki H, Oji Y, Yamagami T, et al:
Aberrant overexpression of the Wilms' tumor gene (WT1) in human
leukemia. Blood. 89:1405–1412. 1997.PubMed/NCBI
|
|
23
|
Oji Y, Miyoshi S, Maeda H, Hayashi S,
Tamaki H, Nakatsuka S, Yao M, Takahashi E, Nakano Y, Hirabayashi H,
et al: Overexpression of the Wilms' tumor gene WT1 in de novo lung
cancers. Int J Cancer. 100:297–303. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oji Y, Yamamoto H, Nomura M, Nakano Y,
Ikeba A, Nakatsuka S, Abeno S, Kiyotoh E, Jomgeow T, Sekimoto M, et
al: Overexpression of the Wilms' tumor gene WT1 in colorectal
adenocarcinoma. Cancer Sci. 94:712–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Loeb DM, Evron E, Patel CB, Sharma PM,
Niranjan B, Buluwela L, Weitzman SA, Korz D and Sukumar S: Wilms'
tumor suppressor gene (WT1) is expressed in primary breast tumors
despite tumor-specific promoter methylation. Cancer Res.
61:921–925. 2001.PubMed/NCBI
|
|
26
|
Oji Y, Nakamori S, Fujikawa M, Nakatsuka
S, Yokota A, Tatsumi N, Abeno S, Ikeba A, Takashima S, Tsujie M, et
al: Overexpression of the Wilms' tumor gene WT1 in pancreatic
ductal adenocarcinoma. Cancer Sci. 95:583–587. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyoshi Y, Ando A, Egawa C, Taguchi T,
Tamaki Y, Tamaki H, Sugiyama H and Noguchi S: High expression of
Wilms' tumor suppressor gene predicts poor prognosis in breast
cancer patients. Clin Cancer Res. 8:1167–1171. 2002.PubMed/NCBI
|
|
28
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chou TC: Drug combination studies and
their synergy quantification using Chou-Talalay method. Cancer Res.
70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ohmori T, Podack ER, Nishio K, Takahashi
M, Miyahara Y, Takeda Y, Kubota N, Funayama Y, Ogasawara H, Ohira
T, et al: Apoptosis of lung cancer cells caused by some anti-cancer
agents (MMC, CPT-11, ADM) is inhibited by BCL-2. Biochem Biophys
Res Commun. 192:30–36. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Palissot V, Belhoussine R, Carpentier Y,
Sebille S, Morjani H, Manfait M and Dufer J: Resistance to
apoptosis induced by topoisomerase I inhibitors in
multidrug-resistant HL60 leukemic cells. Biochem Biophys Res
Commun. 245:918–922. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Appleton K, Mackay HJ, Judson I, Plumb JA,
McCormick C, Strathdee G, Lee C, Barrett S, Reade S, Jadayel D, et
al: Phase I and pharmacodynamic trial of the DNA methyltransferase
inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol.
25:4603–4609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oki Y, Kondo Y, Yamamoto K, Ogura M, Kasai
M, Kobayashi Y, Watanabe T, Uike N, Ohyashiki K, Okamoto S, et al:
Phase I/II study of decitabine in patients with myelodysplastic
syndrome: A multi-center study in Japan. Cancer Sci. 103:1839–1847.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tatsumi N, Oji Y, Tsuji N, Tsuda A,
Higashio M, Aoyagi S, Fukuda I, Ito K, Nakamura J, Kitamura Y, et
al: Wilms' tumor gene WT1-shRNA as a potent apoptosis-inducing
agent for solid tumors. Int J Oncol. 32:701–711. 2008.PubMed/NCBI
|