Introduction
Hepatocellular carcinoma (HCC) is the most frequent
subtype of primary liver cancer (PLC), which is the third leading
cause of cancer-associated mortality worldwide and results from
poor prognosis (1–3). A variety of methods are applied in the
treatment of HCC, including surgery, local treatment and liver
transplantation. However, <20% of patients are eligible for
effective therapies (4). Furthermore,
the 5-year survival rate worldwide of HCC is still <5%, as a
result of its high rate of recurrence and metastasis (5). HCC tumorigenesis is a complex multistep
process that is closely associated with multiple signaling pathways
and genes, and these factors can affect cell survival,
proliferation, invasion and metastasis (6,7). The role
of hepatitis B virus (HBV) or hepatitis C virus infection has been
extensively investigated and considered as one of the reasons in
the pathogenesis of HCC (8,9). Of note, increasing evidences demonstrate
that additional non-viral signaling pathways are involved in the
progression of the disease (10).
Thus, to explore the effects of these signaling pathways on cell
growth and invasion is conducive to identify the potential
mechanisms of carcinogenesis and effective therapeutic targets.
Signal transducer and activator of transcription 3
(STAT3) is one of the members of the STAT signaling protein family,
which consists of seven members (STAT1, STAT2, STAT3, STAT4,
STAT5a, STAT5b and STAT6) (11).
STAT3, being known as a transcription factor, is involved in
important physiology, including proliferative, anti-apoptotic,
metastatic and angiogenic effects (12). STAT3 has been most closely associated
with tumorigenesis (13,14). Previous studies demonstrated that
constitutive STAT3 activation was frequently detected in numerous
human cancers in vitro and in vivo (15–17).
Furthermore, STAT3 participated in the physiological and
pathological processes of HCC, including tumor cell survival,
proliferation, angiogenesis and metastasis (13). It has been previously demonstrated
that the inhibition of STAT3 activation (phosphorylation of STAT3)
reduces the expression of cyclooxygenase-2 (COX-2) in HCC cells
(18). Additionally, it has been
reported that STAT3 serves a pivotal role in malignancies
associated with inflammation due to the activation of genes that
promote cell proliferation, survival and invasion (19,20). The
activation of the STAT3 signaling pathway triggered by HBV
oncoproteins is associated with the carcinogenesis and progression
of HCC (21). In HCC, STAT3 is
constitutively activated, which promotes human cervical cancer
progression and poor prognosis (18,19).
Notably, STAT3 is also involved in the overexpression of COX-2 in
HCC (17).
Hydrogen sulfide (H2S) has been
classified as a novel gasotransmitter together with nitric oxide
(NO) and carbon monoxide (CO) (22).
In the liver, H2S can be catalyzed by both cystathionine
b-synthase (CBS) and cystathionine g-lyase (CSE) (23). Accumulating studies have demonstrated
that H2S is involved in the pathophysiological
progression of tumors (24–27). However, the potential mechanism of
H2S in cancer is unclear and controversial. Accumulating
evidences have demonstrated that H2S promotes cancer
progression, including proliferation, migration and invasion
(28–34). H2S can protect cancer cells
from chemopreventive agent β-phenylethyl isothiocyanate-induced
apoptosis (30) and promote
proliferation (30), which may be
mediated by the increase in Akt and extracellular signal-regulated
kinase (ERK) phosphorylation, and the decrease in
p21Waf1/Cip1 expression and NO production. A recent
study by our group revealed that exogenous H2S promotes
C6 glioma cell growth through the activation of the p38
MAPK/ERK1/2-COX-2 signaling pathway (32). Furthermore, in PLC/PRF/5 cells,
exogenous H2S exerts proliferation, anti-apoptosis,
angiogenesis and migration effects via amplifying the activation of
the nuclear factor (NF)-κB signaling pathway (24). Those results indicate that
H2S promotes cancer cell growth. Notably, H2S
post-conditioning effectively protects isolated
ischemia/reperfusion rat hearts via activation of the Janus kinase
2 (JAK2)/STAT3 signaling pathway (33). However, whether the STAT3-COX-2
signaling pathway contributes to the growth effect of exogenous
H2S on HCC cells remains unclear.
The present study was therefore designed to
determine the effect of H2S on the activation of the
STAT3-COX-2 signaling pathway in HCC (cell line, PLC/PRF/5) cells
and to investigate whether exogenous H2S could induce
proliferation and anti-apoptosis via amplification of the
STAT3-COX-2 signaling pathway in PLC/PRF/5 cells.
Materials and methods
Materials
NaHS, Hoechst 33258, AG490 and NS-398 were purchased
from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Cell Counting
Kit-8 (CCK-8) was supplied by Dojindo Molecular Technologies, Inc.
(Kumamoto, Japan). All antibodies were supplied by Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Cell culture
Human hepatoma PLC/PRF/5 cells were supplied by Sun
Yat-sen University Experimental Animal Center (Guangzhou, China).
The PLC/PRF/5 cells were grown in RPMI 1640 medium (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) under an atmosphere of 5% CO2 and at 37°C with 95%
air. The PLC/PRF/5 cells were collected following the indicated
treatments and their total RNA and protein contents were extracted
for further analyses.
Western blot analysis
Prior to western blot analysis, the following was
performed: Exposure of PLC/PRF/5 cells for the indicated times (3,
6, 9, 12 and 24 h) to 500 µmol/l NaHS; co-treatment of PLC/PRF/5
cells with 500 µmol/l NaHS and 30 µmol/l AG490 for 24 h; exposure
of PLC/PRF/5 cells for the indicated times (3, 6, 9, 12 and 24 h)
to 500 µmol/l NaHS; PLC/PRF/5 cells were co-treated with 500 µmol/l
NaHS and 20 µmol/l NS-398 for 24 h; exposure of PLC/PRF/5 cells for
the indicated times (1, 3, 6, 9, 12 and 24 h) to 500 µmol/l NaHS;
co-treatment of PLC/PRF/5 cells with 500 µmol/l NaHS and 30 µmol/l
AG490 for 24 h; and PLC/PRF/5 cells were co-treated with 500 µmol/l
NaHS and 20 µmol/l NS-398 for 24 h. Following the aforementioned
treatments, the cells were harvested and lysed with a cell lysis
solution (Beyotime Institute of Biotechnology, Shanghai, China) at
4°C for 30 min. Total proteins were quantified using a Pierce BCA
Protein Assay kit (cat. no. P0010S; Beyotime Institute of
Biotechnology). Loading buffer (Sigma-Aldrich; Merck KGaA) was
added to the cytosolic extracts, and upon boiling for 6 min,
equivalent volumes of supernatant from each sample were
fractionated by 10% SDS-PAGE, followed by transfer of the proteins
onto polyvinylidene difluoride membranes. The membranes were
blocked with 5% fat-free milk for 60 min in fresh blocking buffer
[0.1% Tween-20 in TBS (TBST)] at room temperature, and next
incubated with anti-phosphorylated (p)-STAT3 antibody (cat. no.
9145; 1:1,000 dilution), anti-STAT3 antibody (SAB1406487; 1:1,000
dilution), anti-COX-2 antibody (cat. no. 4842; 1:1,000 dilution) or
anti-cleaved caspase-3 antibody (cat. no. 9661; 1:1,000 dilution)
in freshly prepared TBS-T with 3% free-fat milk overnight with
gentle agitation at 4°C. The membranes were washed for 5 min with
TBS-T three times and then incubated with a horseradish
peroxidase-conjugated goat anti-rabbit secondary antibody
(SAB3701044; Sigma-Aldrich; Merck KGaA) at 1:2,500 dilution in
TBS-T with 3% fat-free milk for 1.5 h at room temperature. Then,
the membranes were washed three times with TBS-T for 5 min each.
The immunoreactive signals were visualized using enhanced
chemiluminescence detection. In order to quantify the protein
expression, the X-ray films were scanned and analyzed with ImageJ
1.47i software (National Institutes of Health, Bethesda, MD, USA).
The experiment was conducted three times.
Measurement of cell viability
Cells were seeded in 96-well plates at a density of
1×104 cells/ml and incubated at 37°C. Prior to the CCK-8
assay to assess cell viability, the following was performed:
PLC/PRF/5 cells were treated with different concentrations (100,
200, 300, 400 and 500 µmol/l) of NaHS; treatment of PLC/PRF/5 cells
with 500 µmol/l NaHS for the indicated times (12, 24, 36 and 48 h);
co-treatment of PLC/PRF/5 cells with 500 µmol/l NaHS and different
doses of AG490 (1, 10, 20 and 30 µmol/l) for 24 h; and PLC/PRF/5
cells were co-treated with 500 µmol/l NaHS and NS-398 (0.01–0.5
µmol/l) for 24 h. Following the above treatments, 10 µl CCK-8
solution at 1/10 dilution was added to each well, and then the
plate was incubated for 1.5 h at 37°C. Absorbance at 450 nm was
determined using a microplate reader (Molecular Devices, Sunnyvale,
CA, USA). The means of the optical density (OD) values of three
wells in Fig. 2A-D were used to
calculate the percentage of cell viability according to the
following formula: Cell viability (%)=(OD treatment group/OD
control group) ×100%. The experiment was carried out three
times.
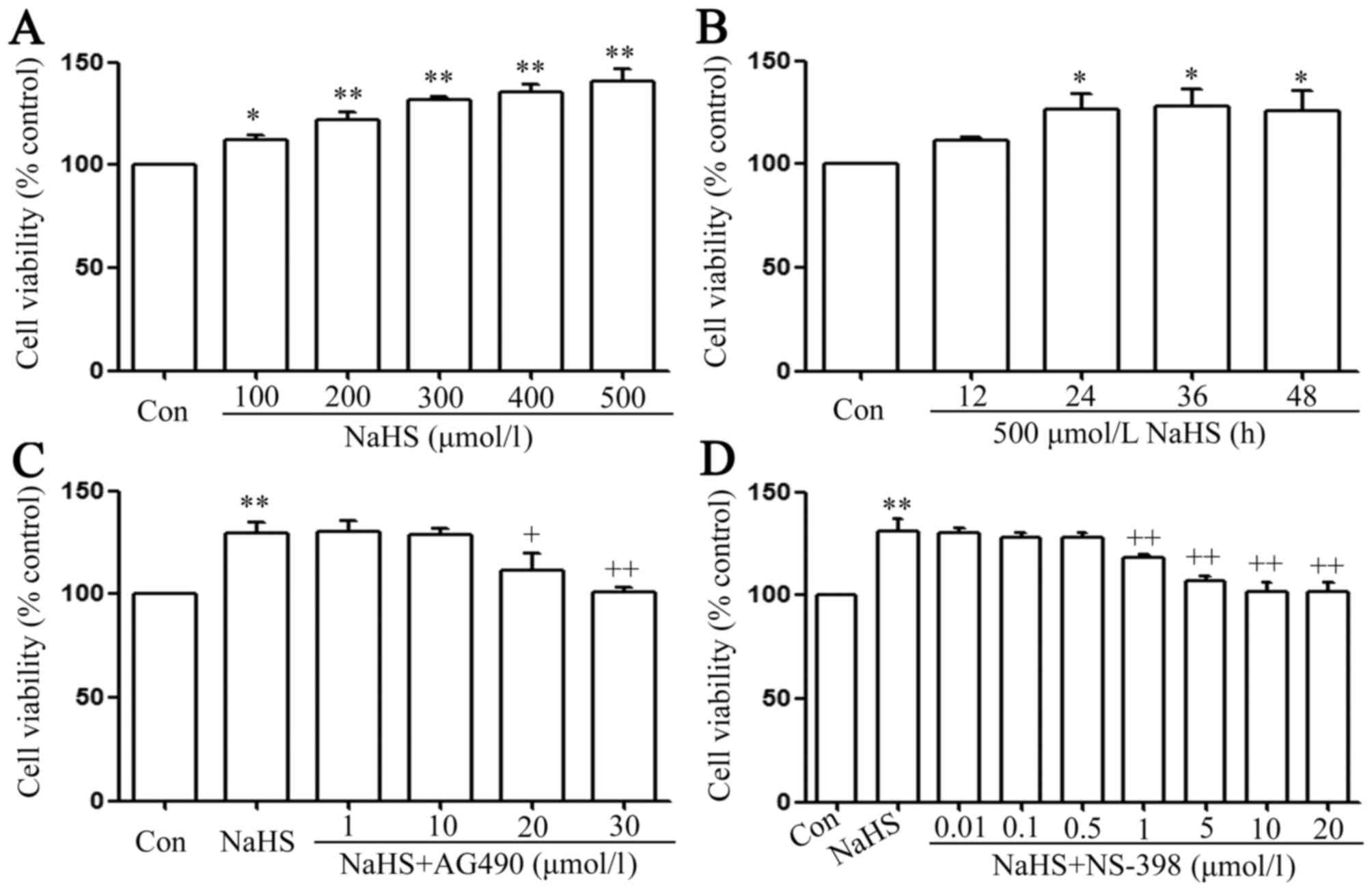 | Figure 2.The signal transducer and activator
of transcription 3-cyclooxygenase-2 signaling pathway serves a
function in NaHS-induced increase in cell viability in PLC/PRF/5
cells. (A) PLC/PRF/5 cells were treated with different
concentration (100, 200, 300, 400 and 500 µmol/l) of NaHS. (B)
Treatment of PLC/PRF/5 cells with 500 µmol/l NaHS for the indicated
times (12, 24, 36 and 48 h). (C) co-treatment of PLC/PRF/5 cells
with 500 µmol/l NaHS and different doses of AG490 (1, 10, 20 and 30
µmol/l) for 24 h. (D) PLC/PRF/5 cells were co-treated with 500
µmol/l NaHS and NS-398 (0.01–0.5 µmol/l) for 24 h. Cell viability
was detected by Cell Counting Kit-8 assay. Data are presented as
the mean ± standard error of the mean (n=5). *P<0.05,
**P<0.01 vs. the control group. +P<0.05,
++P<0.01 vs. with the NaHS-treated group. Con,
control. |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-PCR was carried out in 200-µl sterile tubes
(Eppendorf, Hamburg, Germany). Approximately 2 µg total RNA, 1 µl
oligo (dT) (Sigma-Aldrich; Merck KGaA), 1 µl dNTP, and
diethylpyrocarbonate were placed into the PCR System (cat. no.
204174; Qiagen, Inc., Valencia, CA, USA) for reaction at 65°C for 5
min. Once the reaction had ended, the tubes were stored on ice.
Then, 4 µl 5X First-Strand Buffer (cat. no. 19051; Qiagen, Inc.)
and 2 µl DL-dithiothreitol were successively added for reaction at
37°C for 2 min. Subsequently 1 µl reverse transcriptase (M-MLV RT;
cat. no. 1701; Promega Corporation, Madison, WI, USA) was added to
every tube for reaction at 37°C for 50 min and then at 70°C for 15
min. The primers used for RT-qPCR were as follows: STAT3 forward
5′-ACCTCCAGGACGACTTTGAT-3′ and reverse 5′-TGTCTTCTGCACGTACTCCA-3′;
COX-2 forward 5′-CTGTATCCCGCCCTGCTGGTG-3′ and reverse
5′-ACTTGCGTTGATGGTGGCTGTCTT −3′; and GAPDH forward
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse 5′-TGGTGAAGACGCCAGTGGA-3′.
RT-qPCR was performed with the Applied Biosystems 7500 Fast
Real-Time PCR System (Life technologies, Carlsbad, CA, USA).
Quantitative gene amplifications were performed using the following
thermocycling conditions: Initial denaturation for 5 min at 95°C,
40 cycles of denaturation at 95°C for 5 sec and annealing and
extension at 60°C for 20 sec. After normalizing to the GAPDH gene,
expression levels for each target gene were calculated using the
comparative threshold cycle (2−ΔΔCq) method (35).
Hoechst 33258 nuclear staining for
evaluation of apoptosis
Apoptotic cell death was evaluated by Hoechst 33258
staining followed by photofluorography. First, PLC/PRF/5 cells were
plated onto 35-mm dishes at a density of 1×106
cells/well. Subsequently, the following was performed: PLC/PRF/5
cells were treated with RPMI 1640 medium for 24 h; treatment of
PLC/PRF/5 cells with 500 µmol/l NaHS for 24 h; co-treatment of
PLC/PRF/5 cells with 500 µmol/l NaHS and 30 µmol/l AG490 for 24 h;
PLC/PRF/5 cells were co-treated with 500 µmol/l NaHS and 20 µmol/l
NS-398 for 24 h; treatment of PLC/PRF/5 cells with 30 µmol/l AG490
for 24 h; treatment of PLC/PRF/5 cells with 20 µmol/l NS-398 for 24
h; treatment of PLC/PRF/5 cells with 500 µmol/l NaHS for the
indicated times (1, 3, 6, 9, 12 and 24 h); co-treatment of
PLC/PRF/5 cells with 500 µmol/l NaHS and 30 µmol/l AG490 or 20
µmol/l NS-398 for 24 h. Following the aforementioned treatments,
the cells were fixed with 4% paraformaldehyde in 0.1 mol/l PBS (pH
7.4) for 10 min at 4°C. The slides were then washed three times
with PBS. Upon staining with 5 mg/ml Hoechst 33258 for 15 min, the
cells were washed three times with PBS. Finally, the cells were
visualized under a fluorescence microscope (Bx50-FLA; Olympus
Corporation, Tokyo, Japan). Viable PLC/PRF/5 cells displayed a
uniform blue fluorescence throughout the nucleus and a normal
nuclear size. By contrast, apoptotic PLC/PRF/5 cells exhibited
condensed, distorted or fractured nuclei. The experiment was
repeated three times.
Transwell migration assay
PLC/PRF/5 cells were seeded in 96-well plates at a
density of 1×104 cells/ml, incubated at 37°C and added
to the upper chamber of a Transwell membrane (Transwell Permeable
Support with a 5.0-µm polycarbonate membrane, 6.5-mm insert and
24-well plate; Costar; Corning Life Sciences, Tewksbury, MA, USA).
Next, 500 µl of 10% FBS-RPMI-1640 (Sigma-Aldrich; Merck KGaA) was
added to each bottom chamber. After 24 h of incubation at 37°C, the
cells that had migrated to the lower chamber were counted.
Triplicate experiments were performed with each group, and the
means and standard error of the mean were calculated under a fully
automated inverted microscope.
ELISA for detection of VEGF in the
culture supernatants
PLC/PRF/5 cells were cultured in 96-well plates.
PLC/PRF/5 cells were co-conditioned with 500 µmol/l NaHS and 30
µmol/l AG490 or 20 µmol/l NS-398 for 24 h. Following these
treatments, the level of VEGF in the culture media was evaluated
using a Human VEGF ELISA kit (cat. no. RAB0507; Sigma-Aldrich;
Merck KGaA) according to the manufacturer's protocol. The
experiment was performed ≥5 times.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Differences between groups were analyzed by one-way
analysis of variance using SPSS 13.0 software (SPSS, Inc., Chicago,
IL, USA), followed by a least significant difference post hoc
comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
NaHS activates the STAT3-COX-2
signaling pathway in PLC/PRF/5 cells
As shown in Fig. 1A and
C, exposure of PLC/PRF/5 cells for the indicated times (3, 6,
9, 12 and 24 h) to 500 µmol/l NaHS markedly enhanced the expression
level of p-STAT3, reaching a maximal peak at 9 h, while the
expression level of STAT3 was not altered. Furthermore, exposure of
the cells to 500 µmol/l NaHS for 24 h markedly increased STAT3 mRNA
expression (Fig. 1E). This indicates
that the STAT3 signaling pathway is activated in NaHS-induced
PLC/PRF/5 cell growth.
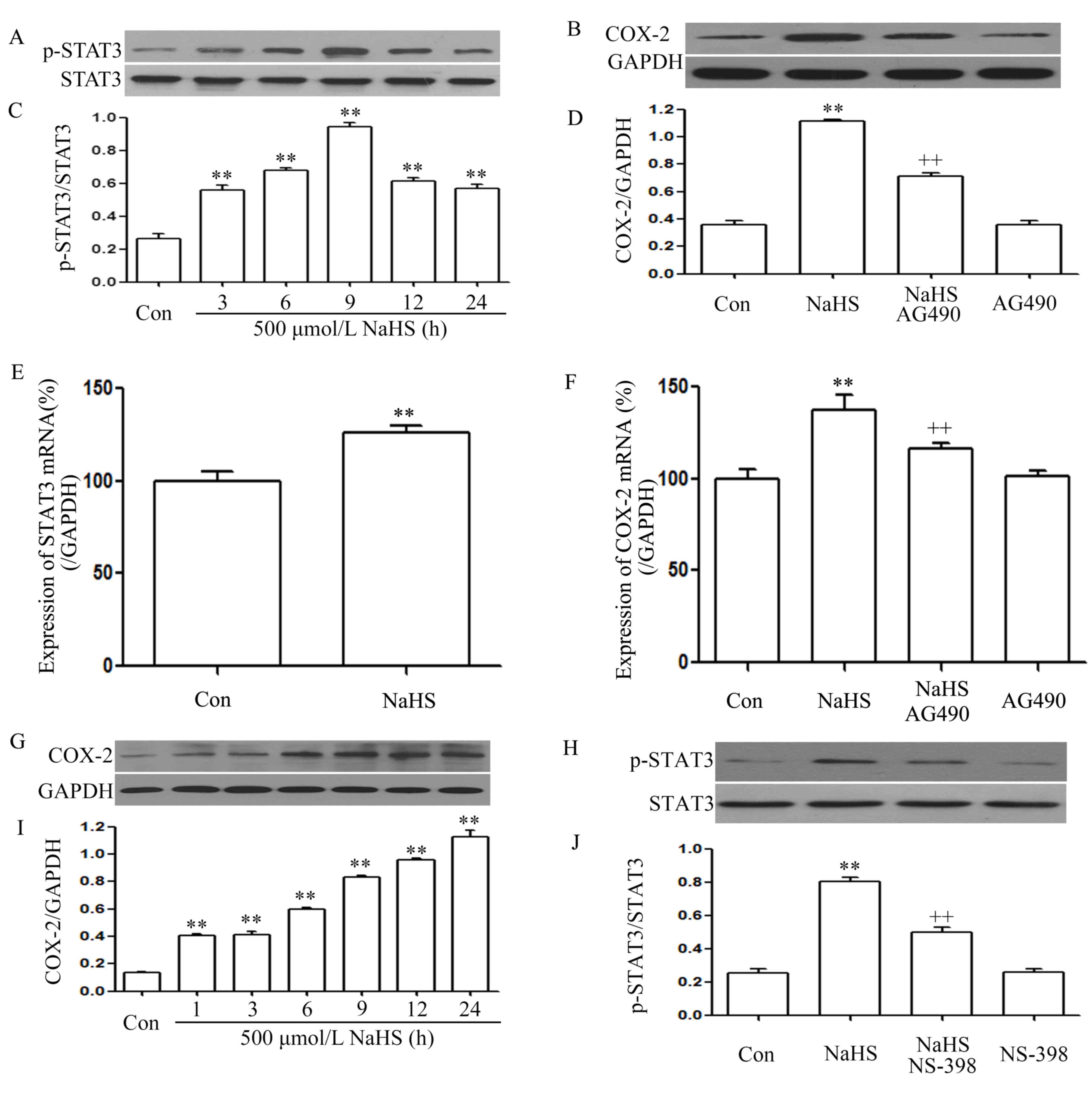 | Figure 1.NaHS activates the STAT3-COX-2
signaling pathway in PLC/PRF/5 cells. (A) Exposure of PLC/PRF/5
cells for the indicated times (3, 6, 9, 12 and 24 h) to 500 µmol/l
NaHS; (B) co-treatment of PLC/PRF/5 cells with 500 µmol/l NaHS and
30 µmol/l AG490 for 24 h; (C) the rate of p-STAT3/STAT3; (D) the
rate of COX-2/GAPDH; (E) the expression of STAT3 mRNA; (F) the
expression of COX-2 mRNA; (G) exposure of PLC/PRF/5 cells for the
indicated times (3, 6, 9, 12 and 24 h) to 500 µmol/l NaHS; (H)
PLC/PRF/5 cells were co-treated with 500 µmol/l NaHS and 20 µmol/l
NS-398 for 24h; (I) the rate of COX-2/GAPDH; (J) the rate of
p-STAT3/STAT3. The expression levels of p-STAT3 (A, C, H and J) and
COX-2 (B, D, G and I) were semiquantified by western blot assay.
(C, D, I and J) Densitometric analysis of the p-STAT3 expression
levels shown in panels A, B, G and H, respectively. The mRNA
expression levels of (E) STAT3 and (F) COX-2 in PLC/PRF/5 cells
were examined by semiquantitative reverse transcription-polymerase
chain reaction. GAPDH mRNA was used as a loading control. Data are
presented as means ± standard error of the mean (n=3). **P<0.01
vs. the control group. ++P<0.01 vs. the group treated
with NaHS, a donor of H2S. Con, control; STAT3, signal
transducer and activator of transcription 3; COX-2,
cyclooxygenase-2; p-, phosphorylated. |
As shown in Fig. 1G and
I, exposure of PLC/PRF/5 cells for the indicated times (3, 6,
9, 12 and 24 h) to 500 µmol/l NaHS markedly enhanced the expression
level of COX-2, reaching a maximal peak at 24 h. Exposure of the
cells to 500 µmol/l NaHS for 24 h markedly increased COX-2 mRNA
expression (Fig. 1F). This indicates
that the COX-2 signaling pathway was also activated in the
NaHS-induced PLC/PRF/5 cell growth.
Notably, co-treatment of PLC/PRF/5 cells with 500
µmol/l NaHS and 30 µmol/l AG490 for 24 h considerably suppressed
the NaHS-induced increase in the expression levels of COX-2
(Fig. 1B and D) and COX-2 mRNA
(Fig. 1F). Alone, treatment of cells
with 30 µmol/l AG490 for 24 h did not alter the basal expression
level of COX-2 mRNA. This indicates that COX-2 was located
downstream of STAT3 in the signaling pathway. Notably, co-treatment
of PLC/PRF/5 cells with 500 µmol/l NaHS and 20 µmol/l NS-398 for 24
h suppressed the expression of p-STAT3 (Fig. 1H and J). Therefore, it can be deduced
that there was interaction between the STAT3 and COX-2 signaling
pathways.
The STAT3-COX-2 signaling pathway
participates in the NaHS-induced increase in cell viability in
PLC/PRF/5 cells
As shown in Fig. 2A,
doses of NaHS from 100 to 500 µmol/l markedly promoted cell
proliferation, leading to an increase in cell viability and
reaching a maximal peaking at 500 µmol/l. Treatment of PLC/PRF/5
cells with 500 µmol/l NaHS for the indicated times (12, 24, 36 and
48 h) markedly promoted cell proliferation, reaching the maximal
proliferative effect at 24 h. Based on the above results, PLC/PRF/5
cells were treated with 500 µmol/l NaHS for 24 h in all subsequent
experiments. As shown in Fig. 2C, the
increased cell viability was suppressed by co-treatment with 500
µmol/l NaHS and different doses of AG490 (a specific inhibitor of
the STAT3 signaling pathway) (36)
for 24 h. Doses of AG490 from 1 to 10 µmol/l did not change cell
viability. On the contrary, doses of AG490 from 20 to 30 µmol/l
significantly suppressed cell proliferation, leading to a decrease
in cell viability. According to the above results, PLC/PRF/5 cells
were co-treated with 500 µmol/l NaHS and 30 µmol/l AG490 for 24 h
in all subsequent experiments. Doses of NS-398 from 0.01 to 0.5
µmol/l did not change cell viability, while doses of NS-398 from 1
to 20 µmol/l significantly suppressed cell proliferation, leading
to a decrease in cell viability, which reached a minimum at 10 and
20 µmol/l. According to the above results, PLC/PRF/5 cells were
co-treated with 500 µmol/l NaHS and 20 µmol/l NS-398 for 24 h in
all subsequent experiments.
The STAT3-COX-2 signaling pathway
participates in NaHS-induced anti-apoptosis in PLC/PRF/5 cells
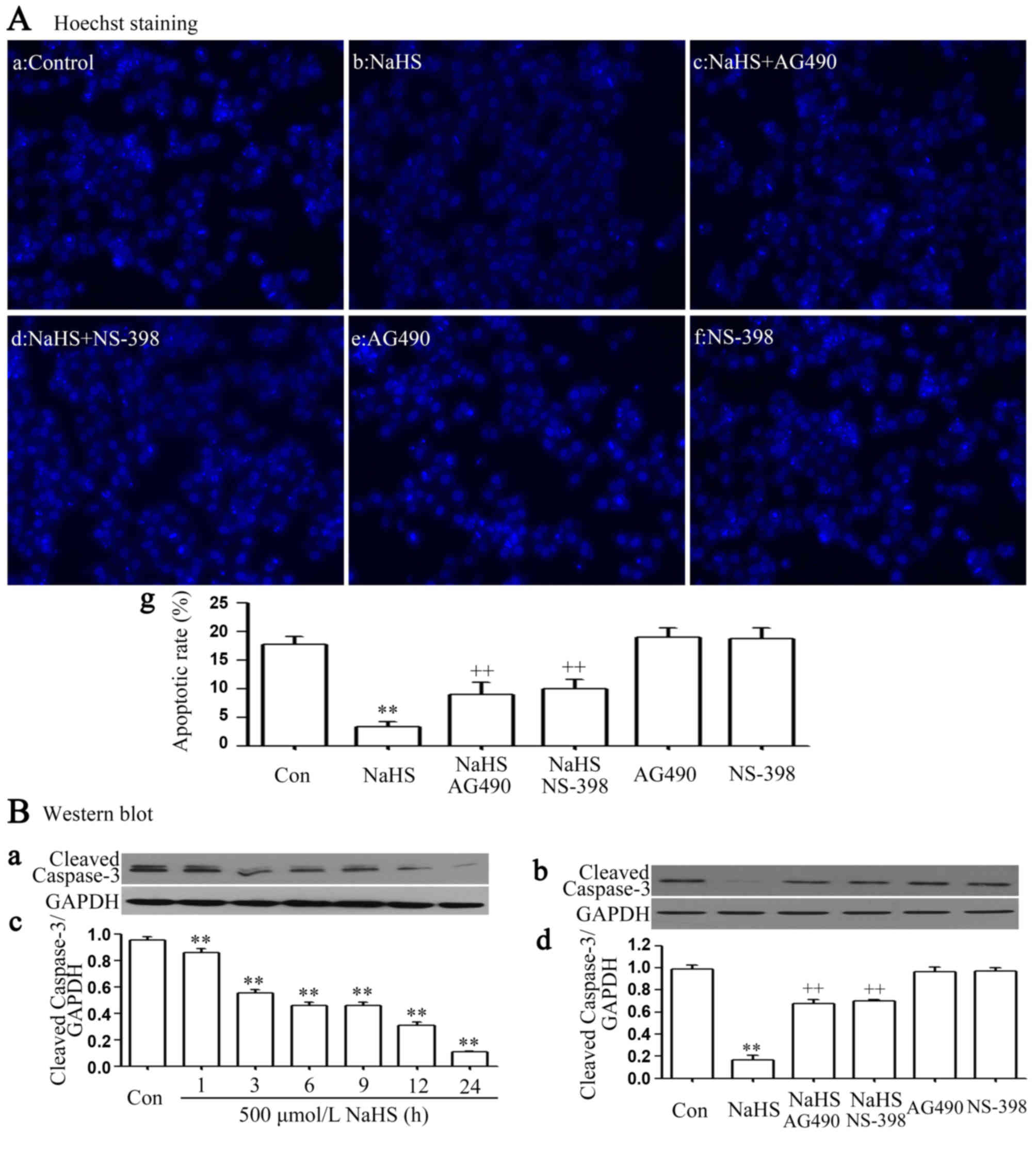
It was demonstrated that exposure of cells to 500
µmol/l NaHS for 24 h markedly enhanced cell proliferation, as
evidenced by a decrease in the number of apoptotic cells (Fig. 3Ab and Ag). In addition, the above
anti-apoptosis was nearly completely inhibited by co-treating
PLC/PRF/5 cells with 500 µmol/l NaHS and 30 µmol/l AG490 or 20
µmol/l NS-398 for 24 h. Furthermore, exposure of cells to 500
µmol/l NaHS for 24 h markedly decreased cleaved caspase-3
expression (Fig. 3Ba and Bc), and the
NaHS-induced decrease in the expression level of cleaved caspase-3
was inhibited by co-treating PLC/PRF/5 cells with 500 µmol/l NaHS
and 30 µmol/l AG490 or 20 µmol/l NS-398 for 24 h (Fig. 3Bb and d).
The STAT3-COX-2 signaling pathway
participates in NaHS-induced migration in PLC/PRF/5 cells
As shown in Fig. 4,
NaHS significantly enhanced the expression levels of matrix
metalloproteinase-2 (MMP-2) (Fig. 4A and
C) and promoted migration (Fig.
4G) in PLC/PRF/5 cells. These NaHS-induced effects were
inhibited by co-treating PLC/PRF/5 cells with 500 µmol/l NaHS and
30 µmol/l AG490 or 20 µmol/l NS-398 for 24 h (Fig. 3Bb and d).
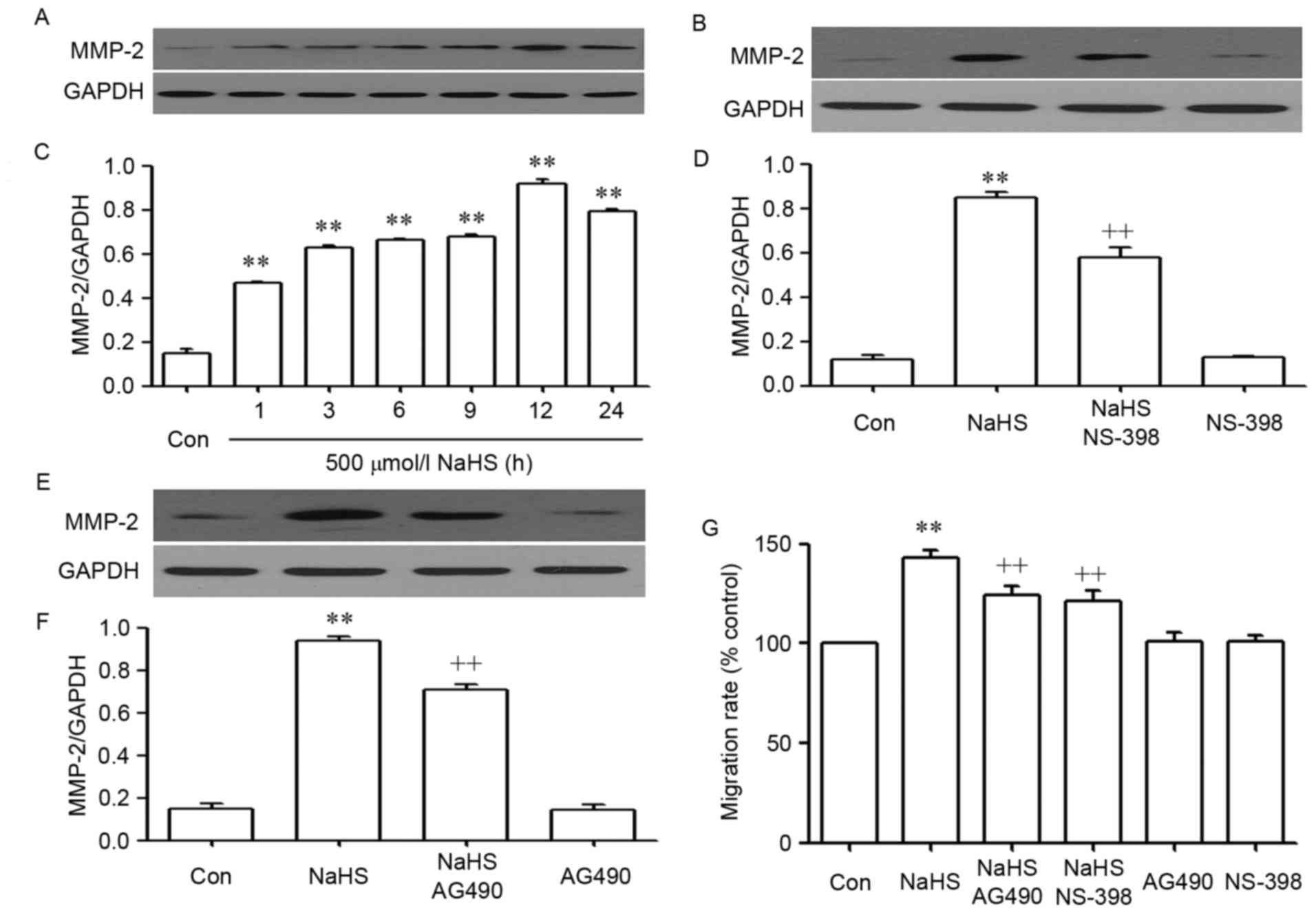 | Figure 4.The STAT3-COX2 signaling pathway
participates in NaHS-induced migration in PLC/PRF/5 cells. (A)
Exposure of PLC/PRF/5 cells for the indicated times (1, 3, 6, 9, 12
and 24 h) to 500 µmol/l NaHS; (B) the rate of MMP-2/GAPDH; (C)
co-treatment of PLC/PRF/5 cells with 500 µmol/l NaHS and 30 µmol/l
AG490 for 24 h; (D) the rate of MMP-2/GAPDH; (E) PLC/PRF/5 cells
were co-treated with 500 µmol/l NaHS and 20 µmol/l NS-398 for 24h;
(F) the rate of MMP-2/GAPDH; (G) PLC/PRF/5 cells were co-treated
with 500 µmol/l NaHS and 30 µmol/l AG490, or 20 µmol/l NS-398 for
24 h. (A-F) The expression levels of MMP-2 were semiquantified by
western blot assay. (C, D and F) Densitometric analysis of the
p-STAT3 expression levels shown in panels A-C, respectively. (G)
Cell migration was evaluated by Transwell migration assay, and the
migration rate was calculated under a fully automated inverted
microscope. Data are presented as the mean ± standard error of the
mean (n=3). **P<0.01 vs. the control group.
++P<0.01 vs. the NaHS-treated group. MMP-2, matrix
metalloproteinase-2; Con, control. |
The STAT3-COX-2 signaling pathway
participates in the NaHS-induced production of VEGF in PLC/PRF/5
cells
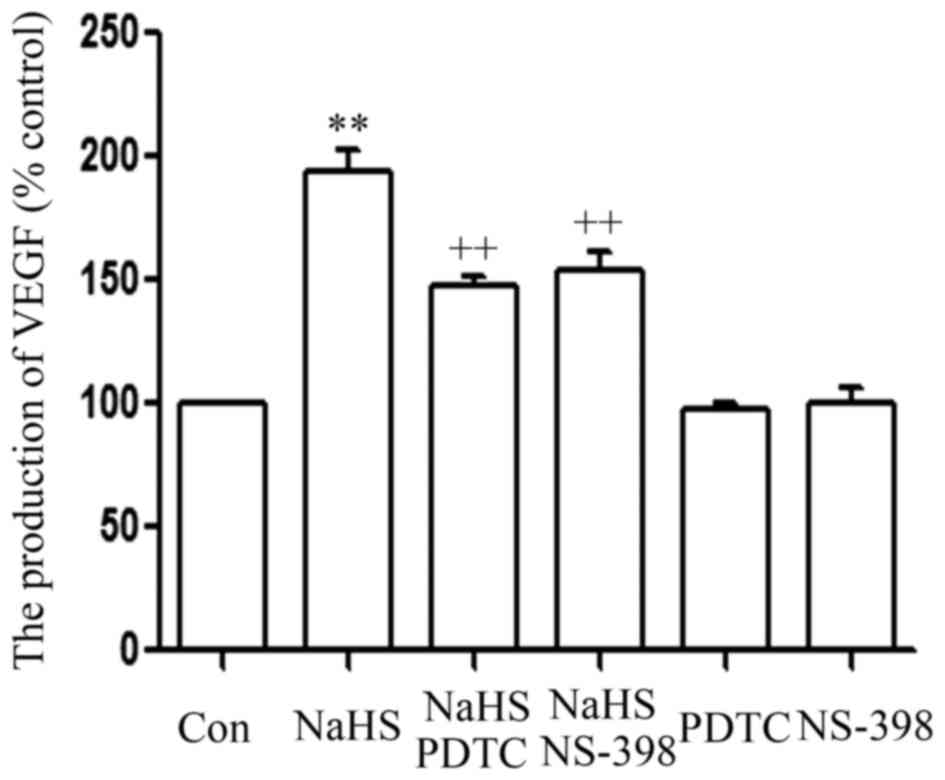
As shown in Fig. 5,
the level of VEGF was markedly increased in NaHS-treated PLC/PRF/5
cells, compared with that in the control group (P<0.01).
However, this increase in the level of VEGF was significantly
suppressed by co-treatment of cells with 500 µmol/l NaHS and 30
µmol/l AG490 or 20 µmol/l NS-398 for 24 h.
Discussion
Previous studies have demonstrated that STAT3
(13,16) and COX-2 (24) are associated with the progression of
tumors. Inhibition of the STAT3 (34)
or COX-2 (37) signaling pathways can
contribute to the inhibitory effect of tumor growth. The present
study extends these previous findings and provides new evidence
that the STAT3-COX-2 signaling pathway is associated with the
growth of PLC/PRF/5 cells, as evidenced by the increased expression
levels of STAT3 and COX-2. Importantly, the present study has
demonstrated for the first time that exogenous H2S
promotes PLC/PRF/5 cell proliferation and anti-apoptosis by
activating the STAT3-COX-2 signaling pathway.
H2S has been classified as a novel
gasotransmitter together with NO and CO (22). Its broad range of physiological
functions, including cardioprotective (38), angiogeneic (39), antioxidant (40), and pro- and anti-inflammatory
activities (38), are attracting
widespread attention at present. In the liver, H2S can
exert hepatoprotective effects via miR-34a-mediated modulation of
the nuclear factor erythroid 2-related factor 2 signaling pathway
(41). Recently, our group
demonstrated that exogenous H2S promoted PLC/PRF/5 cell
proliferation, anti-apoptosis, angiogenesis and migration by
amplifying the activation of the NF-κB signaling pathway (24). To further investigate the effect of
exogenous H2S on PLC/PRF/5 cells, PLC/PRF/5 cells were
treated with 500 µmol/l NaHS for 24 h. The results revealed that
exogenous H2S increased PLC/PRF/5 cell growth,
angiogenesis and migration, as evidenced by an increase in cell
viability, migration, expression of MMP-2 and production of VEGF,
and a decrease in apoptotic rate and expression of caspase-3 (one
of the apoptotic factors) (42),
which is consistent with the findings of our previous study
(23).
Furthermore, it was observed that the expression
levels of p-STAT3, STAT3 mRNA, COX-2 protein and COX-2 mRNA were
upregulated in NaHS-treated PLC/PRF/5 cells. This indicates that
exogenous H2S can activate the STAT3 and COX-2 signaling
pathways in PLC/PRF/5 cells. Previous studies have shown that the
STAT3 (13–17) and COX-2 (32,43–45)
signaling pathways are associated with tumorigenesis. The present
study hypothesized that the STAT3 and COX-2 signaling pathways may
be involved in the effects of exogenous H2S on the
growth of PLC/PRF/5 cells. In order to corroborate our hypothesis,
PLC/PRF/5 cells were co-treated with NaHS and AG490 (an inhibitor
of STAT3) (36) or NS-398. Our data
revealed that co-treatment of PLC/PRF/5 cells with NaHS and AG490
or NS-398 markedly alleviated the NaHS-induced cell growth effects,
including proliferation, angiogenesis, migration and
anti-apoptosis. These results suggest that NaHS-induced PLC/PRF/5
cell growth is at least in part associated with the activated STAT3
and COX-2 signaling pathways.
A novel finding of the present study is the
interaction between STAT3 and COX-2 in PLC/PRF/5 cells. COX-2 is
ever-present as a downstream effector of the STAT3 signaling
pathway in various cancer cells (46–48). A
previous study has demonstrated that the inhibition of STAT3
activation reduces the expression of COX-2 in SMMC-7721 cells
(18). This indicates that COX-2 is a
downstream effector of the STAT3 signaling pathway in liver cancer.
Additionally, COX-2 is located upstream of the STAT3 signaling
pathway in various cancer cells. Liu et al (49) observed that COX-2/prostaglandin E2
regulated JAK2/STAT3 signaling in colorectal cancer cells (49). Furthermore, Xiong et al
(50) revealed that there is a
positive feedback loop between the STAT3 and COX-2 genes that may
contribute to Helicobacter pylori-associated human gastric
tumorigenesis (50). However, the
exact association between STAT3 and COX-2 in cell proliferation,
migration and apoptosis in PLC/PRF/5 cells is not completely
understood. The present study provided novel evidence that there is
a positive interaction between the STAT3 and COX-2 signaling
pathways, which may be an important mechanism responsible for cell
proliferation and anti-apoptosis in PLC/PRF/5 cells. This mechanism
is supported by the following results: i) Treatment of PLC/PRF/5
cells with NaHS and AG490 attenuated the expression level of COX-2;
ii) treatment of PLC/PRF/5 cells with NaHS and NS-398 attenuated
the expression level of p-STAT3; and iii) exposure of PLC/PRF/5
cells to AG490 or NS-398 induced growth inhibition and apoptosis,
as demonstrated by the decrease in cell viability, and the increase
in the number of apoptotic cells and cleaved caspased-3
expression.
To conclude, the present study provides novel
evidence that the activation of the STAT3-COX-2 signaling pathway
contributes to HCC carcinogenesis, including cell proliferation and
anti-apoptosis. Understanding the roles of such a signaling pathway
is important, as it may lead to the development of novel treatment
strategies designed to inhibit this signaling cascade in PLC/PRF/5
cells. In addition, the interaction between STAT3 and COX-2 in
PLC/PRF/5 cells may serve a crucial role in PLC/PRF/5
carcinogenesis, however understanding of any additional roles
remain unclear and must be investigated. Additionally, the present
study provides important new insight into the molecular mechanisms
underlying the promotion of cell proliferation and apoptosis by
H2S in PLC/PRF/5 cells. First, H2S increases
cell viability and reduces apoptosis. Second, NaHS-induced growth
inhibition and apoptosis appear to be linked to the inhibition of
the activation of the STAT3-COX-2 signaling pathway. In HCC, these
findings provide a novel insight into CBS- and CSE-derived
H2S as an endogenous tumor-promoting factor and
anticancer drug target.
References
|
1
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bruix J, Boix L, Sala M and Llovet JM:
Focus on hepatocellular carcinoma. Cancer Cell. 5:215–219. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lupberger J and Hildt E: Hepatitis B
virus-induced oncogenesis. World J Gastroenterol. 13:74–81. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Verslype C, Van Cutsem E, Dicato M, Arber
N, Berlin JD, Cunningham D, De Gramont A, Diaz-Rubio E, Ducreux M,
Gruenberger T, et al: The management of hepatocellular carcinoma.
current expert opinion and recommendations derived from the 10th
World Congress on Gastrointestinal Cancer, Barcelona, 2008. Ann
Oncol. 20 Suppl 7:Svii1–Svii6. 2009. View Article : Google Scholar
|
|
5
|
Wang H and Chen L: Tumor microenviroment
and hepatocellular carcinoma metastasis. J Gastroenterol Hepatol.
28 Suppl 1:S43–S48. 2013. View Article : Google Scholar
|
|
6
|
Iakova P, Timchenko L and Timchenko NA:
Intracellular signaling and hepatocellular carcinoma. Semin Cancer
Biol. 21:28–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Faivre S, Bouattour M and Raymond E: Novel
molecular therapies in hepatocellular carcinoma. Liver Int. 31
Suppl 1:S151–S160. 2011. View Article : Google Scholar
|
|
8
|
Chuang SC, La Vecchia C and Boffetta P:
Liver cancer: Descriptive epidemiology and risk factors other than
HBV and HCV infection. Cancer Lett. 286:9–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liang X, Bi S, Yang W, Wang L, Cui G, Cui
F, Zhang Y, Liu J, Gong X, Chen Y, et al: Epidemiological
serosurvey of hepatitis B in China-declining HBV prevalence due to
hepatitis B vaccination. Vaccine. 27:6550–6557. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zheng B, Zhu YJ, Wang HY and Chen L:
Gender disparity in hepatocellular carcinoma (HCC): Multiple
underlying mechanisms. Sci China Life Sc. 60:575–584. 2017.
View Article : Google Scholar
|
|
11
|
Levy DE and Darnell JE Jr: Stats:
Transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aggarwal BB, Kunnumakkara AB, Harikumar
KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B: Signal
transducer and activator of transcription-3, inflammation and
cancer: How intimate is the relationship? Ann N Y Acad Sci.
1171:59–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aggarwal BB, Sethi G, Ahn KS, Sandur SK,
Pandey MK, Kunnumakkara AB, Sung B and Ichikawa H: Targeting
signal-transducer-and-activator-of-transcription-3 for prevention
and therapy of cancer: modern target but ancient solution. Ann N Y
Acad Sci. 1091:151–169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao T, Ren H, Wang X, Liu P, Yan F, Jiang
W, Li Y, Li J, Gribben JG, Jia L and Hao J: Rituximab-induced HMGB1
release is associated with inhibition of STAT3 activity in human
diffuse large B-cell lymphoma. Oncotarget. 6:27816–27831. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mukthavaram R, Ouyang X, Saklecha R, Jiang
P, Nomura N, Pingle SC, Guo F and Makale M: Effect of the
JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres.
J Transl Med. 13:2692015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liao XH, Zheng L, He HP, Zheng DL, Wei ZQ,
Wang N, Dong J, Ma WJ and Zhang TC: STAT3 regulated ATR via
microRNA-383 to control DNA damage to affect apoptosis in A431
cells. Cell Signal. 27:2285–2295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He S, Lu G, Hou H, Zhao Z, Zhu Z, Lu X,
Chen J and Wang Z: Saikosaponin-d suppresses the expression of
cyclooxygenase-2 through the phospho-signal transducer and
activator of transcription 3/hypoxia-inducible factor-1α pathway in
hepatocellular carcinoma cells. Mol Med Rep. 10:2556–2562. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Loncle C, Bonjoch L, Folch-Puy E,
Lopez-Millan MB, Lac S, Molejon MI, Chuluyan E, Cordelier P, Dubus
P, Lomberk G, et al: IL-17 functions through the novel
REG3β-JAK2-STAT3 inflammatory pathway to promote the transition
from chronic pancreatitis to pancreatic cancer. Cancer Res.
75:4852–4862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rokavec M, Öner MG and Hermeking H:
lnflammation-induced epigenetic switches in cancer. Cell Mol Life
Sci. 73:23–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choudhari SR, Khan MA, Harris G, Picker D,
Jacob GS, Block T and Shailubhai K: Deactivation of Akt and STAT3
signaling promotes apoptosis, inhibits proliferation and enhances
the sensitivity of hepatocellular carcinoma cells to an anticancer
agent, Atiprimod. Mol Cancer Ther. 6:112–121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kilburn KH, Thrasher JD and Gray MR:
Low-level hydrogen sulfide and central nervous system dysfunction.
Toxicol Ind Health. 26:387–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kamoun P: Endogenous production of
hydrogen sulfide in mammals. Amino Acids. 26:243–254. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhen Y, Pan W, Hu F, Wu H, Feng J, Zhang Y
and Chen J: Exogenous hydrogen sulfide exerts
proliferation/anti-apoptosis/angiogenesis/migration effects via
amplifying the activation of NF-κB pathway in PLC/PRF/5 hepatoma
cells. Int J Oncol. 46:2194–2204. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu D, Si W, Wang M, Lv S, Ji A and Li Y:
Hydrogen sulfide in cancer: Friend or foe? Nitric Oxide. 50:38–45.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee ZW and Deng LW: Role of H2S donors in
cancer biology. Handb Exp Pharmacol. 230:243–265. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hellmich MR and Szabo C: Hydrogen sulfide
and cancer. Handb Exp Pharmacol. 230:233–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Szabo C, Coletta C, Chao C, Módis K,
Szczesny B, Papapetropoulos A and Hellmich MR: Tumor-derived
hydrogen sulfide, produced by cystathionine-β-synthase, stimulates
bioenergetics, cell proliferation and angiogenesis in colon cancer.
Proc Natl Acad Sci USA. 110:12474–12479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
DU SX, Xiao J, Guan F, Sun LM, Wu WS, Tang
H, DU JB, Tang CS and Jin HF: Predictive role of cerebrospinal
fluid hydrogen sulfide in central nervous system leukemia. Chin Med
J (Engl). 124:3450–3454. 2011.PubMed/NCBI
|
|
30
|
Rose P, Moore PK, Ming SH, Nam OC,
Armstrong JS and Whiteman M: Hydrogen sulfide protects colon cancer
cells from chemopreventative agent betaphenylethyl isothiocyanate
induced apoptosis. World J Gastroenterol. 11:3990–3997. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cai WJ, Wang MJ, Ju LH, Wang C and Zhu YC:
Hydrogen sulfide induces human colon cancer cell proliferation:
Role of Akt, ERK and p21. Cell Biol Int. 34:565–572. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhen Y, Zhang W, Liu C, He J, Lu Y, Guo R,
Feng J, Zhang Y and Chen J: Exogenous hydrogen sulfide promotes C6
glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2
pathways. Oncol Rep. 34:2413–2422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luan HF, Zhao ZB, Zhao QH, Zhu P, Xiu MY
and Ji Y: Hydrogen sulfide postconditioning protects isolated rat
hearts against ischemia and reperfusion injury mediated by the
JAK2/STAT3 survival pathway. Braz J Med Biol Res. 45:898–905. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu A, Huang JJ, Jin XJ, Li JP, Tang YJ,
Huang XF, Cui HJ, Xu WH and Sun GB: Curcumin suppresses
invasiveness and vasculogenic mimicry of squamous cell carcinoma of
the larynx through the inhibition of JAK-2/STAT-3 signaling
pathway. Am J Cancer Res. 5:278–288. 2014.PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu YY, Guo M, Yang LQ, Zhou F, Yu C, Wang
A, Pang TH, Wu HY, Zou XP, Zhang WJ, et al: Regulation of CD44v6
expression in gastric carcinoma by the IL-6/STAT3 signaling pathway
and its clinical significance. Oncotarget. 8:45848–45861.
2017.PubMed/NCBI
|
|
37
|
Zeng L, Zhen Y, Chen Y, Zou L, Zhang Y, Hu
F, Feng J, Shen J and Wei B: Naringin inhibits growth and induces
apoptosis by a mechanism dependent on reduced activation of
NF-κB/COX-2-caspase-1 pathway in HeLa cervical cancer cells. Int J
Oncol. 45:1929–1936. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu W, Chen J, Lin J, Liu D, Mo L, Pan W,
Feng J, Wu W and Zheng D: Exogenous H2S protects H9c2 cardiac cells
against high glucose-induced injury and inflammation by inhibiting
the activation of the NF-κB and IL-1β pathways. Int J Mol Med.
35:177–186. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Coletta C, Papapetropoulos A, Erdelyi K,
Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I,
Martin E and Szabo C: Hydrogen sulfide and nitric oxide are
mutually dependent in the regulation of angiogenesis and
endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA.
109:9161–9166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kimura H: Hydrogen sulfide: From brain to
gut. Antioxid Redox Signal. 12:1111–1123. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Huang X, Gao Y, Qin J and Lu S: The role
of miR-34a in the hepatoprotective effect of hydrogen sulfide on
ischemia/reperfusion injury in young and old rats. PLoS One.
9:e1133052014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
You Q, Wu Z, Wu B, Liu C, Huang R, Yang L,
Guo R, Wu K and Chen J: Naringin protects cardiomyocytes against
hyperglycemia-induced injuries in vitro and in vivo. J Endocrinol.
230:197–214. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tegeder I, Niederberger E, Israr E,
Gühring H, Brune K, Euchenhofer C, Grösch S and Geisslinger G:
Inhibition of NF-kappaB and AP-1 activation by R- and
S-flurbiprofen. FASEB J. 15:2–4. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Seo KW, Coh YR, Rebhun RB, Ahn JO, Han SM,
Lee HW and Youn HY: Antitumor effects of celecoxib in COX-2
expressing and non-expressing canine melanoma cell lines. Res Vet
Sci. 96:482–486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu X, Cai M, Ji F and Lou LM: The impact
of COX-2 on invasion of osteosarcoma cell and its mechanism of
regulation. Cancer Cell Int. 14:272014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gao J, Tian J, Lv Y, Shi F, Kong F, Shi H
and Zhao L: Leptin induces functional activation of
cyclooxygenase-2 through JAK2/STAT3, MAPK/ERK and PI3K/AKT pathways
in human endometrial cancer cells. Cancer Sci. 100:389–395. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xu W, Chen GS, Shao Y, Li XL, Xu HC, Zhang
H, Zhu GQ, Zhou YC, He XP and Sun WH: Gastrin acting on the
cholecystokinin2 receptor induces cyclooxygenase-2 expression
through JAK2/STAT3/PI3K/Akt pathway in human gastric cancer cells.
Cancer Lett. 332:11–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gong J, Xie J, Bedolla R, Rivas P,
Chakravarthy D, Freeman JW, Reddick R, Kopetz S, Peterson A, Wang
H, et al: Combined targeting of STAT3/NF-κB/COX-2/EP4 for effective
management of pancreatic cancer. Clin Cancer Res. 20:1259–1273.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu X, Ji Q, Ye N, Sui H, Zhou L, Zhu H,
Fan Z, Cai J and Li Q: Berberine inhibits invasion and metastasis
of colorectal cancer cells via COX-2/PGE2 mediated JAK2/STAT3
Signaling pathway. PLoS One. 10:e01234782015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xiong H, Du W, Sun TT, Lin YW, Wang JL,
Hong J and Fang JY: A positive feedback loop between STAT3 and
cyclooxygenase-2 gene may contribute to Helicobacter
pylori-associated human gastric tumorigenesis. Int J Cancer.
134:2030–2040. 2014. View Article : Google Scholar : PubMed/NCBI
|