Introduction
Primary liver cancer, which consists mainly of
hepatocellular carcinoma (HCC), is the third-leading cause of
cancer mortality worldwide, following lung and stomach cancer,
owing to its poor prognosis and frequent relapse and metastasis
(1,2).
Although much is already known about the major pathogenic factors
behind HCC, including chronic hepatitis B and hepatitis C infection
(3,4),
the definitive mechanisms behind HCC have not been elucidated. Only
30–40% of patients with HCC are eligible for potentially radical
therapies (5), meaning the overall
survival rate of patients with HCC remains low. Therefore, the
identification of novel therapeutic targets to improve and develop
treatment strategies for HCC.
The phosphoinositide 3-kinase (PI3K)/RAC
seine/threonine-protein kinase (AKT)/mechanistic target of
rapamycin (mTOR) signaling pathway is a promising therapeutic
target owing to its frequent dysregulation in HCC and the critical
functions it has in regulating cell survival, proliferation,
apoptosis, migration and angiogenesis through phosphorylation of
distinct protein substrates (6,7). AKT is a
key molecule in the PI3K/AKT/mTOR signaling pathway, which has been
shown to serve notable functions in the regulation of cell
viability and to be closely associated with a variety of disorders
caused by dysfunctional cellular proliferation (8,9). Three
isoforms of AKT (AKT1, AKT2, and AKT3), which share >80%
sequence homology, have been identified in mammals (10,11). It
has been reported that overexpression of AKT is associated with
decreased disease-free survival rates and development of primary
carcinomas of the prostate, breast and ovary (12,13).
However, the action of individual AKT isoforms in different
molecular subtypes of HCC has not been extensively evaluated. Lee
et al (14) revealed that AKT1
serves a critical function in angiogenesis; AKT1 also has a crucial
effect on cell survival (14–17). However, the precise molecular
mechanisms by which AKT1 promotes cell proliferation and regulates
apoptosis (18,19) remain largely unclear.
High expression of activated AKT can be detected in
HCC, and AKT may promote cell proliferation and regulation of cells
apoptosis in HCC (20,21). The present study confirmed a potential
function for AKT1 in promoting proliferation and inhibiting
apoptosis of HCC. Subsequent mechanism investigations revealed that
AKT1 served a notable function in cell proliferation and
anti-apoptosis by directly regulating the expression of phosphatase
and tensin homolog (PTEN) and Notch1. The present study revealed
that the specific inhibition of AKT1 may be therapeutically
viable.
Materials and methods
Cell culture and plasmid
transfection
The human HL-7702 and SMMC-7721 cell lines were
purchased from the Shanghai Institutes for Biological Sciences
(Chinese Academy of Science, Shanghai, China). HL-7702 and
SMMC-7721 cells were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum containing penicillin (100 U/ml)/streptomycin
(100 mg/ml) (Gibco; Thermo Fisher Scientific, Inc.) and incubated
at 37°C in a humidified atmosphere containing 5% CO2.
The pEGFP-N1-AKT1 plasmid was synthesized by Bioworld Technology,
Inc. (St. Louis. Park, MN, USA). AKT1-RNAi plasmid was synthesized
by Shanghai Genechem Co., Ltd. (Shangahi, China). A blank plasmid,
an expression plasmid coding for AKT1-enhanced cyan fluorescent
protein (pEGFP-N1-AKT1) and a plasmid containing short hairpin RNA
(sh)-AKT (AKT1-RNAi plasmid) were transfected into cells using
Effectene transfection reagent (Qiagen, Inc., Valencia, CA, USA),
according to the manufacturer's protocol. SMMC-7721 cells were
seeded into a 6-well plate (2×105 cells/well).
Transfection was performed when the cell confluence reached 40–50%
and cells were collected 48 h following transfection for subsequent
experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay
Total RNA was extracted from HCC cells with TRIzol
(Thermo Fisher Scientific, Inc.). RNA was reverse-transcribed into
cDNA using a PrimeScript RT reagent kit (Takara Bio, Inc.). cDNA
samples were subjected to qPCR using the SYBR Premix Ex Taq kit
(Takara Bio, Inc.). The thermocycling conditions were as follows:
40 cycles of pre-denaturation at 95°C for 30 sec, annealing at 95°C
for 5 sec and final extension at 60°C for 30 sec. Relative gene
expression data were calculated using the 2−ΔΔCq method
(22). All reactions were performed
in triplicate and all experiments were performed three times. GAPDH
was used as a reference gene. The primers are presented in Table I.
 | Table I.Reverse transcription-quantitative
polymerase chain reaction primers. |
Table I.
Reverse transcription-quantitative
polymerase chain reaction primers.
| Gene | Sequence |
|---|
| AKT1 |
|
| Forward |
5′-CACAAACGAGGGGAGTACATC-3′ |
| Reverse |
5′-GCCATCATTCTTGAGGAGGAAGT-3′ |
| PTEN |
|
| Forward |
5′-AGGGACGAACTGGTGTAATGA-3′ |
| Reverse |
5′-CTGGTCCTTACTTCCCCATAGAA-3′ |
| Notch1 |
|
| Forward |
5′-ACTGTGTAGGACCTGGTGGAC-3′ |
| Reverse |
5′-TTGTAGGTGTTGGGGAGGTC-3′ |
| GAPDH |
|
| Forward |
5′-TCATGGGTGTGAACCATGAGAA-3′ |
| Reverse |
5′-GGCATGGACTGTGGTCATGAG-3′ |
Western blot analysis
SMMC-7721 cells were transfected with the
pEGFP-N1-AKT1, AKT1-RNAi and blank plasmids for 48 h. SMMC-7721
cells were lysed using radioimmunoprecipitation assay buffer
(Beyotime Institute of Biotechnology, Haimen, China). The protein
concentration was determined using a bicinchoninic acid assay kit
(Beyotime Institute of Biotechnology). A total of 20 µg protein was
separated by SDS-PAGE (10% gel) and transferred onto polyvinylidene
fluoride membranes. Following blocking with 5% skimmed milk for 2 h
at room temperature, membranes were incubated with primary
antibodies at 4°C overnight. Primary antibodies included: Anti AKT1
(rabbit monoclonal; dilution, 1:1,000; cat no. 2938), PTEN (mouse
monoclonal; dilution, 1:1,000; cat no. 9556), Notch1 (rabbit
monoclonal; dilution, 1:1,000; cat no. 3608), cyclin D1 (rabbit
monoclonal; dilution, 1:1,000; cat no. 2922), Bcl2 (rabbit
monoclonal; dilution, 1:1,000; cat no. 3498), GAPDH (rabbit
monoclonal; dilution, 1:1,000; cat no. 5174) (all primary
antibodies from Cell Signaling Technology, Inc., Danvers, MA, USA).
The membrane was washed three times with Tris-buffered saline with
0.1% Tween-20 (TBST) for 15 min three times, then incubated with
anti-rabbit (1:10,000; cat no. 7074; Cell Signaling Technology,
Inc.) or anti-mouse (1:10,000; cat no. 7076; Cell Signaling
Technology, Inc.) secondary antibodies for 2 h at room temperature.
The protein bands were visualized using enhanced chemiluminescence
(ECL Plus kit; Thermo Fisher Scientific, Inc.). The protein
expression was detected using Image-Pro Plus software (version 6.0;
Media Cybernetics, Inc., Rockville, MD, USA).
Colony forming assay
For the colony forming assay, 2,000 cells in the
blank plasmid, pEGFP-N1-AKT1 plasmid and AKT1-RNAi plasmid
transfection groups were added to a 6-well plate and incubated in a
humid incubator at 37°C with 5% CO2. After 10 days in
culture, cells were fixed with 100% methanol for 30 min at room
temperature and stained with 0.2% crystal violet for 15 min at room
temperature. Colonies (>50 cells) were then counted using light
microscopy (magnification, ×40).
MTT cell proliferation assay
For the MTT assay, SMMC-7721 cells were seeded into
a 96-well plate (6,000 cells/well), incubated for 24 h, and
separately transfected with blank plasmid, pEGFP-N1-AKT1 plasmid or
AKT1-RNAi plasmid for 48 h. Once the medium was replaced with 100
µl culture liquid containing 10% fetal bovine serum, 20 µl MTT
solution was added and plates were incubated for another 4 h,
followed by the addition of 150 µl dimethyl sulfoxide. The
absorbance was measured at 490 nm using a microplate reader to
determine the number of viable cells in each well.
Flow cytometric analysis
SMMC-7721 cells were seeded into 6-well plates for
~12 h until confluence reached 50%. Next, cells were transfected
with blank, pEGFP-N1-AKT1 or AKT1-RNAi plasmids, as aforementioned.
After 48 h, cells were washed with ice-cold PBS and fixed with 70%
ice-cold ethanol at 4°C overnight. Subsequently, propidium iodide
(PI; Beyotime Institute of Biotechnology) was added to transfected
cells for 30 min at 4°C in the dark. Finally, an Aria II flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA) was used to
detect the distribution of cells in the cell cycle. The proportion
of cells in different phrases was analyzed using FlowJo software
(version 7.6; FlowJo LLC, Ashland, OR, USA).
Analysis of apoptosis
At 48 h post-transfection, SMMC-7721 cells were
suspended in binding buffer and were hatched with an
Annexin-V-fluorescein isothiocyanate (FITC)/PI apoptosis detection
kit (Beyotime Institute of Biotechnology) for 30 min at room
temperature in the dark. Flow cytometry was performed using a flow
cytometer. The results were analyzed using FlowJo software (version
7.6; FlowJo LLC, Ashland, OR, USA).
Statistical analysis
Student's t-test or one-way ANOVA followed by least
significant difference or Dunnett's test. Statistical analyses were
conducted using GraphPad Prism 6 (GraphPad Software, Inc., La
Jolla, CA, USA) and PASW Statistics 18 (SPSS, Inc., Chicago, IL,
USA). Results are presented as the mean ± standard error of the
mean. P<0.05 was considered to indicate a statistically
significant difference. All experiments were performed in
triplicate.
Results
Expression of AKT1 is upregulated in
HCC
Western blot analysis and RT-qPCR were used to
examine the expression levels of AKT1 in HCC SMMC-7721 cell line,
as compared with the expression level in normal liver cell line
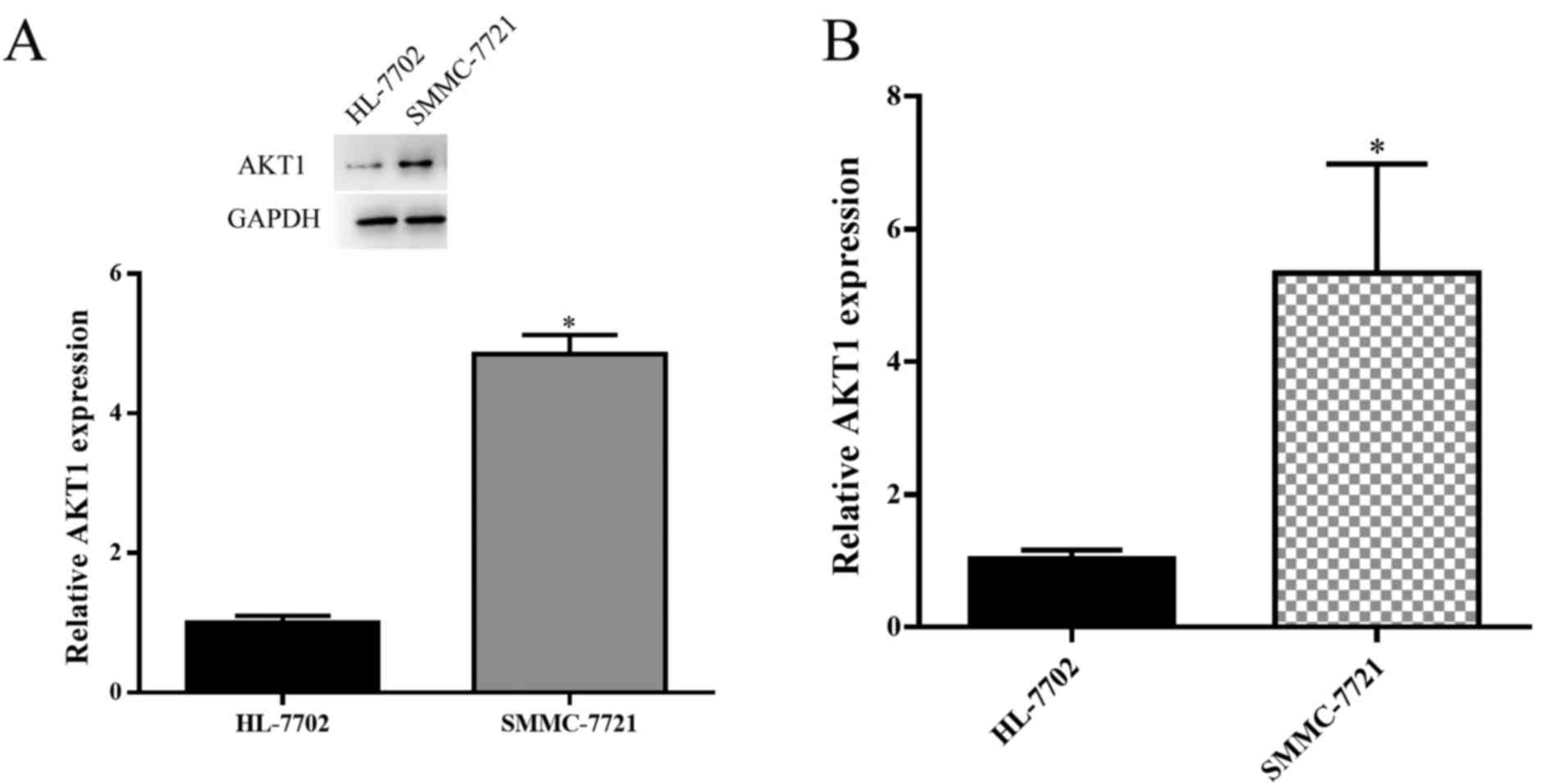
HL-7702. The results demonstrated that AKT1 was significantly
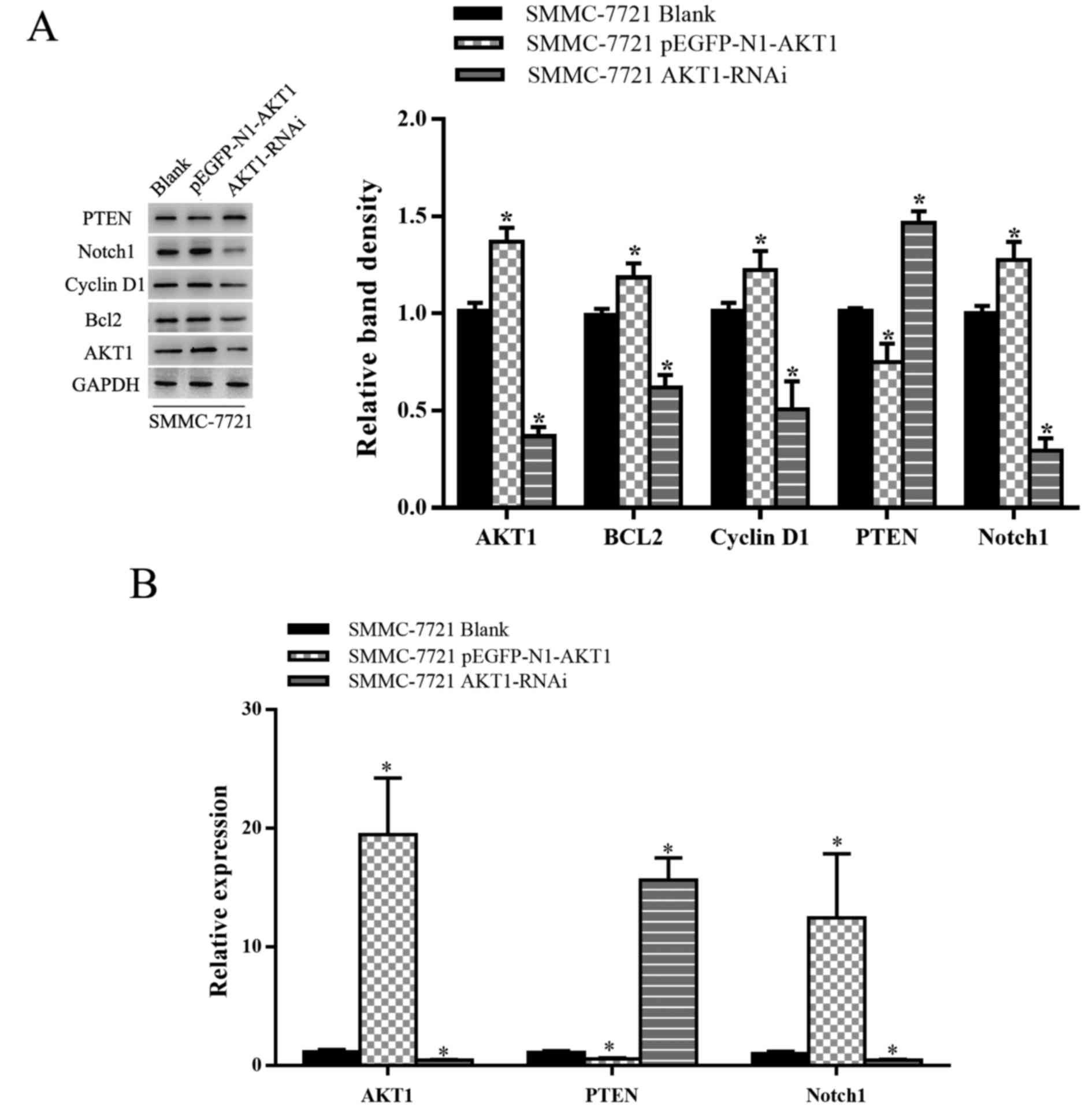
upregulated in SMMC-7721 cells (P<0.05; Fig. 1A and B). As AKT1 expression was
elevated in HCC cells, AKT1 appears to have a positive effect on
HCC progression. The transfection efficiency of various plasmids
[blank plasmid, AKT1-upregulation plasmid (pEGFP-N1-AKT1 plasmid)
and AKT1-downregulation plasmid (AKT1-RNAi plasmid)] was then
assessed. Transfection with the AKT1-upregulation plasmid promoted
the expression of AKT1 and AKT-1-downregulation plasmid inhibited
the expression of AKT1 (P<0.05; Fig.
2A and B).
Upregulation of AKT1 promotes HCC cell
proliferation, whereas downregulation of AKT1 inhibits
proliferation
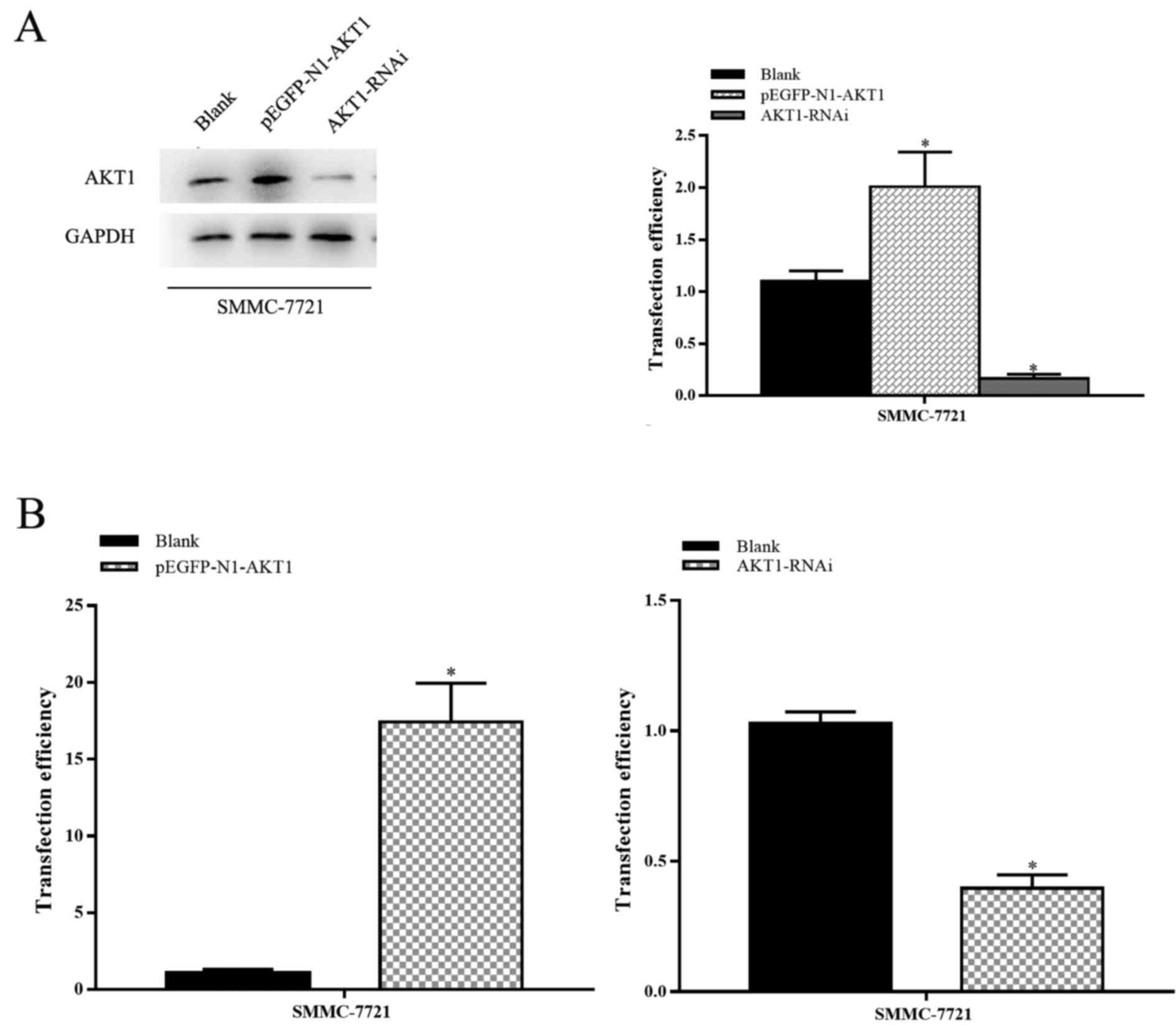
To identify the function of AKT1 in HCC progression,
SMMC-7721 cells were transfected with pEGFP-N1-AKT1 or AKT1-RNAi
plasmids. The expression of AKT1 protein in pEGFP-N1-AKT1
plasmid-transfected cells was upregulated by 150–300%, whereas in
AKT1-RNAi plasmid-transfected cells it was downregulated by
400–1,000%, compared with cells transfected with an empty vector
(P<0.05; Fig. 2A and B). The
transfection efficiency was confirmed by RT-qPCR and western blot
analysis.
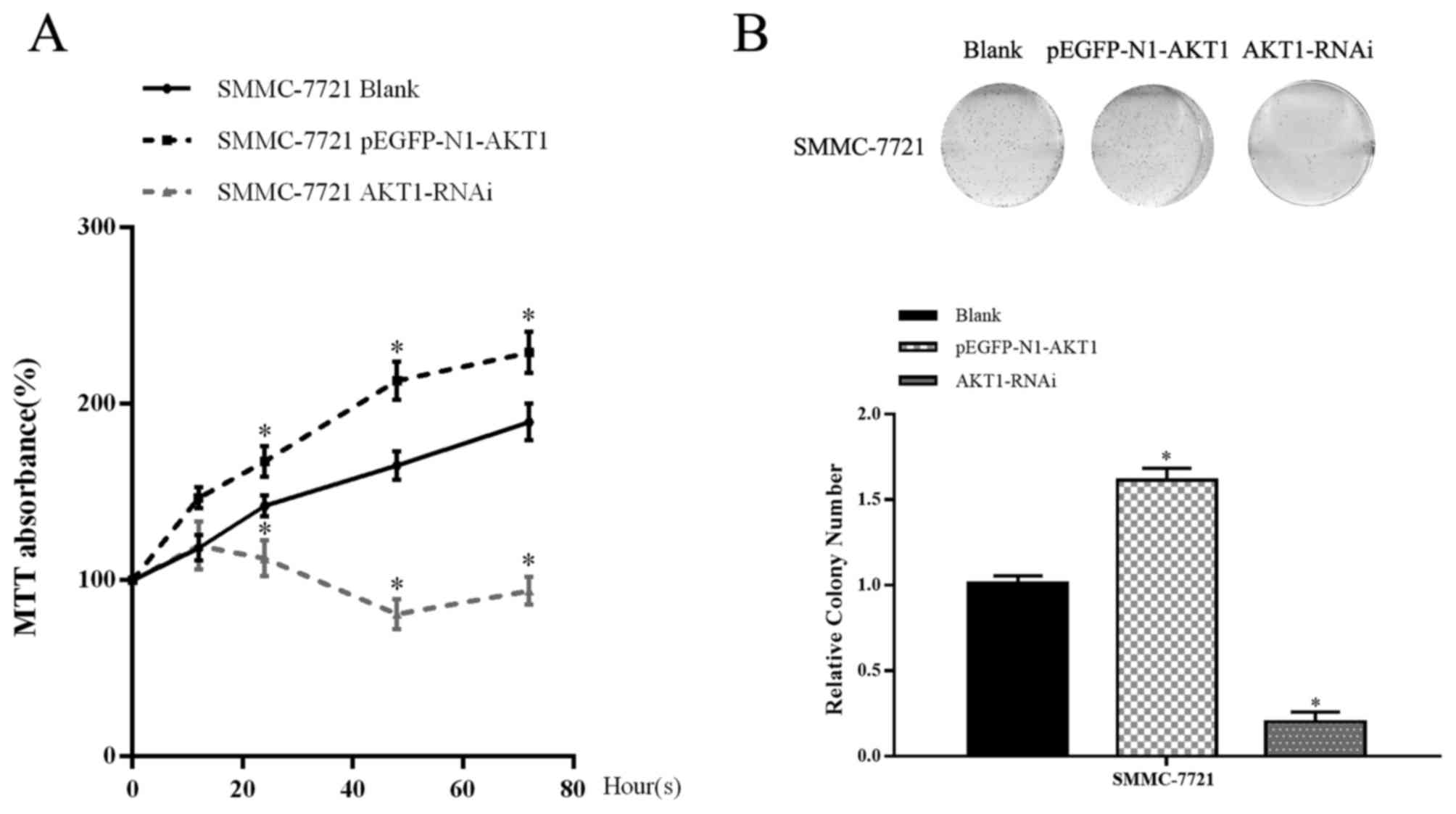
MTT and colony formation assays were performed to
assess the function of AKT1 on HCC cell proliferation. These assays
revealed that the overexpression of AKT1 resulted in a significant
increase in viability compared with control group in SMMC-7721
cells. In line with this, downregulation of AKT1 suppressed cell
proliferation (P<0.05; Fig. 3A and
B). These results demonstrated that AKT1 effectively promoted
the proliferation of HCC cells, whereas knockdown of AKT1 inhibited
proliferation. These results indicate that the upregulation of AKT1
expression promoted cell proliferation, whereas its downregulation
restrained this proliferation.
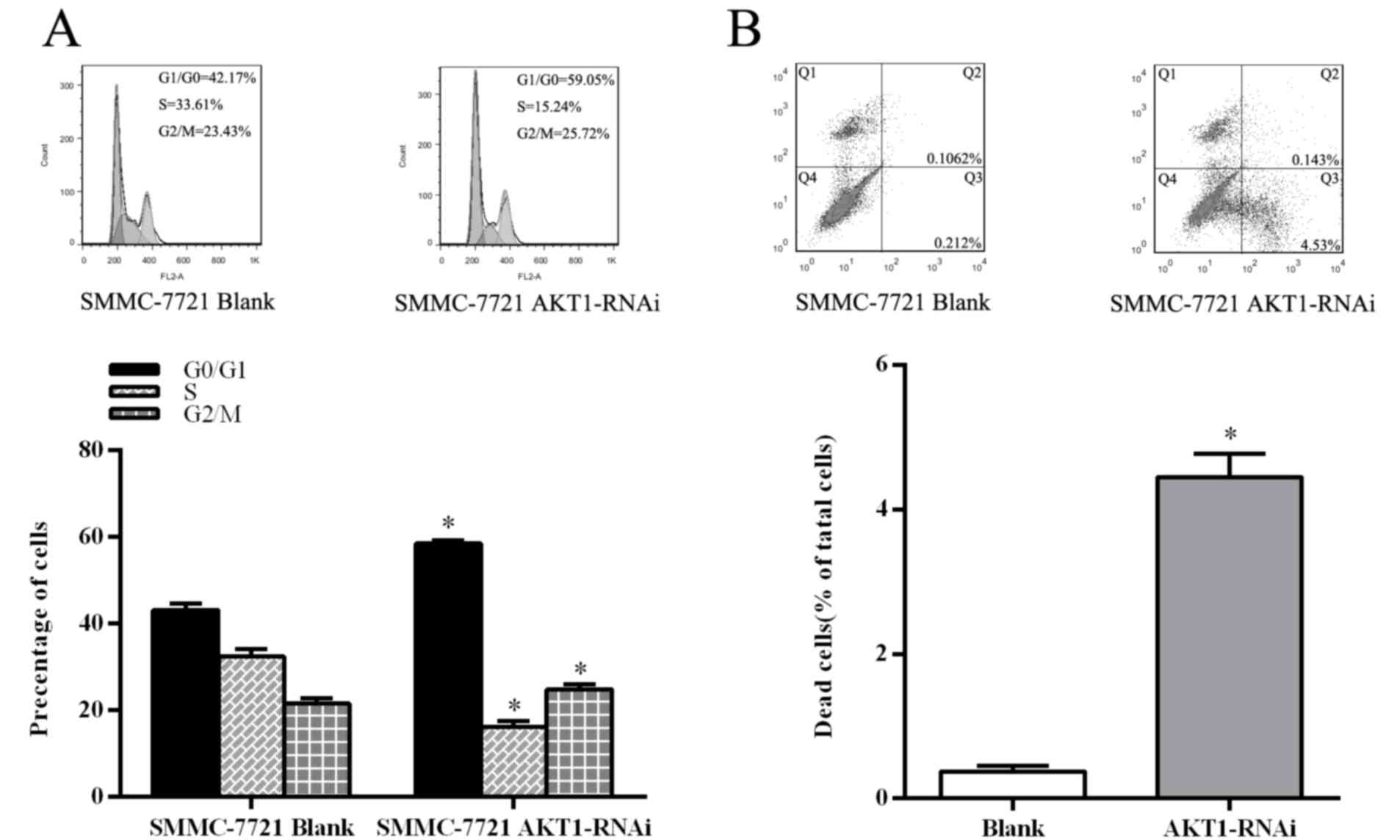
Deactivation of AKT1 suppresses cell
cycle and induces apoptosis in HCC cells
Suppression of AKT1 in SMMC-7721 by transfection
with the AKT1-RNAi plasmid confirmed the function of AKT1 in HCC
cells. Flow cytometric analysis revealed a marked increase in the
percentage of cells at G1/G0 phase and a
decrease in the percentage of cells in S phase in the AKT1-RNAi
plasmid transfected cells, compared with those transfected with the
blank vector (P<0.05; Fig. 4A).
Furthermore, inhibition of AKT1 led to a decrease in B-cell
lymphoma-2 (Bcl-2) and cyclin D1 expression (P<0.05; Fig. 5A). Subsequently, apoptosis analysis
using flow cytometry an increase in the percentage of annexin
V-FITC-positive HCC cells in those transfected with AKT1-RNAi
compared with the control cells (P<0.05; Fig. 4B). These results demonstrated that
inhibition of AKT1 suppresses the cell cycle in HCC cells and
promotes apoptosis.
AKT1 promotes HCC cell survival and
proliferation by targeting PTEN in vivo
PTEN is a dual lipid/protein phosphatase, acting as
a tumor suppressor by negatively regulating the AKT signaling
pathway; it is inactivated in a number of cancer types (23,24). PTEN
is crucial for inhibiting cell proliferation downstream of Notch
(25–27). The upregulation of Notch1 in SMMC-7721
cells increased the expression of Notch1 compared with that in
control cells. The RT-qPCR and western blot assays were
additionally performed to examine the effects of AKT1 on the
endogenous expression of PTEN and Notch1. As shown in Fig. 5, AKT1 downregulation was associated
with a significant increase and decrease in PTEN and Notch1 mRNA
and protein levels, respectively, in SMMC-7721 cells, with AKT1
overexpression inducing the opposite result. It is possible that
Notch1 and PI3K-AKT signaling are closely linked to the control of
cell growth and proliferation via PTEN and differential regulation
of AKT1 signaling promotes cell growth, in part through Notch1.
Discussion
HCC is one of the most common malignant tumors of
digestive tract carcinoma, the incidence of which has been
increasing recently (28–31). The survival rate and prognosis of
patients with HCC are poor, partly owing to local recurrence,
metastasis and multi-drug resistance of tumor cells (32,33).
Furthermore, HCC is refractory to treatment as it is usually
diagnosed at advanced stages (34).
Accordingly, it is necessary to seek novel therapeutic targets for
the treatment of HCC. The present study investigated the link
between AKT1 and HCC and assessed the effect of differential
regulation of AKT1 on the proliferation and apoptosis of cells and
the specific regulatory mechanisms involved. Previous studies have
indicated that AKT is closely associated with cell survival,
proliferation, apoptosis, migration and angiogenesis in HCC
(6,17). AKT was frequently dysregulated in
hepatoma cell lines and human HCC tissues (35). AKT isoforms are highly expressed in
the majority of cancer types, including those of the lung, breast
and colon, and HCC; however, the isoform-specific functions in the
occurrence and development of HCC remain unclear. As a member of
the AKT family, AKT1 expression has been proved to be dysregulated
in cancer cells; this dysregulation of AKT1 is associated with
cancer cell survival, proliferation and metabolism (15,16).
Previous evidence demonstrated that AKT1 participates in the
initiation, progression and metastasis of malignant tumors
(36–38). Thus, AKT1 has emerged as a novel
potential target of anticancer drugs.
The present study analyzed the functional relevance
of AKT1 action on the HCC SMMC-7721 cell line, on proliferation,
apoptosis and cell cycle. First, the AKT1 expression in different
HCC subtypes was screened and found that AKT1 was high expression
in SMMC-7721 HCC cell line. Compared with the HCC, the expression
of AKT1 was higher in SMMC-7721 cells. This result is in line with
that of a previous study showing that AKT was high expression in
oncological patients (38,39). To identify the precise impact of AKT1
on the occurrence and development of HCC, colony formation, MTT,
and flow cytometry assays were performed. The results of these
analyses demonstrated that AKT1 had significant impact on cellular
survival and proliferation. Silencing AKT1 significantly stimulated
apoptosis and suppressed the cell cycle, whereas increasing AKT1
expression promoted HCC cells proliferation.
To investigate the mechanism of action of AKT1 in
HCC, we increased and decreased AKT1 expression levels in HCC cell
lines using the pEGFP-N1-AKT1 and AKT1-RNAi plasmids, respectively.
Compared with the control, the level of AKT1 expression was
significantly enhanced in pEGFP-N1-AKT1 plasmid-transfected cells
and significantly decreased in cells transfected with the AKT1-RNAi
plasmid. When the AKT1-RNAi plasmid was transfected into the HCC
cell line, PTEN expression was significantly increased and Notch1
was evidently decreased, indicating that that PTEN and Notch1 may
be the target of AKT1 in HCC cells. Recent studies have shown that
Notch1 inhibits HCC through the upregulation of PTEN and the
subsequent inactivation of focal adhesion kinase; PTEN is a
phosphatase that can negatively regulate Notch (25,40). The
expression of AKT1, which is negatively associated with PTEN
expression, was positively associated with Notch1 expression in
AKT1-RNAi plasmid-transfected cells. Additionally, the
pEGFP-N1-AKT1 plasmid-transfected cells were in the opposite
direction. In addition, the present study revealed that the
downregulation of AKT1 expression in the HCC SMMC-7721 cell line by
transfection with the AKT1-RNAi plasmid suppressed cell survival
and proliferation, and promoted apoptosis.
Previous studies revealed that the Notch1 and the
PI3K/AKT signaling pathways interact in more complex signaling
networks, centered on one of the principal downstream targets of
Notch1 (41,42). PTEN protein is a tumor suppressor gene
that is mutated in a large number of cancer types at high frequency
(43–46). Notch1 negatively regulates PTEN at the
transcriptional level (47). The
Notch signaling pathway, a highly conserved pathway composed of
Notch1-4 receptors (48), is critical
for a wide variety of cells and tissues through its regulation of
growth, differentiation and apoptosis. Notch1 is a receptor that
tends to be highly expressed in human HCC (40). In the present study, RT-qPCR and
western blot analysis revealed that Notch1 acted as a tumor
promoter by modulating the PI3K/AKT pathway in HCC, and that PTEN
is a possible intermediary of this signaling. The downregulation of
AKT1 by the AKT1-RNAi plasmid in SMMC-7721 cells increased PTEN
expression and decreased the expression of Notch1. These results
contributed to cell apoptosis, increasing the expression of Bcl-2
and reducing the expression of cyclin D1, inducing G1
cell cycle arrest. AKT1 overexpression elicited an opposite effect
to AKT1 knockdown. PTEN expression appears to serve a notable
function in restraining the proliferation of HCC in AKT1.
In conclusion, the results of the present study
indicated that silencing AKT1 is highly effective in upregulating
expression of the tumor-suppressing gene PTEN and reducing
expression of Notch1 expression, dampening tumor growth and
inducing apoptosis. Targeting AKT1 suppresses HCC growth and
provides a basis for designing novel therapeutic strategies for
HCC.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferenci P, Fried M, Labrecque D, Bruix J,
Sherman M, Omata M, Heathcote J, Piratsivuth T, Kew M, Otegbayo JA,
et al: Hepatocellular carcinoma (HCC): A global perspective. J Clin
Gastroenterol. 44:239–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li J, Wang K, Chen X, Meng H, Song M, Wang
Y, Xu X and Bai Y: Transcriptional activation of microRNA-34a by
NF-kappa B in human esophageal cancer cells. BMC Mol Biol.
13:42012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gupta P, Cairns MJ and Saksena NK:
Regulation of gene expression by microRNA in HCV infection and
HCV-mediated hepatocellular carcinoma. Virol J. 11:642014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Enguita-Germán M and Fortes P: Targeting
the insulin-like growth factor pathway in hepatocellular carcinoma.
World J Hepatol. 6:716–737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhaskar PT and Hay N: The two TORCs and
Akt. Dev Cell. 12:487–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toker A and Yoeli-Lerner M: Akt signaling
and cancer: Surviving but not moving on. Cancer Res. 66:3963–3966.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vara Fresno JA, Casado E, de Castro J,
Cejas P, Belda-Iniesta C and González-Barón M: PI3K/Akt signalling
pathway and cancer. Cancer Treat Rev. 30:193–204. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brodbeck D, Hill MM and Hemmings BA: Two
splice variants of protein kinase B gamma have different regulatory
capacity depending on the presence or absence of the regulatory
phosphorylation site serine 472 in the carboxyl-terminal
hydrophobic domain. J Biol Chem. 276:29550–29558. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brodbeck D, Cron P and Hemmings BA: A
human protein kinase Bgamma with regulatory phosphorylation sites
in the activation loop and in the C-terminal hydrophobic domain. J
Biol Chem. 274:9133–9136. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grottke A, Ewald F, Lange T, Nörz D,
Herzberger C, Bach J, Grabinski N, Gräser L, Höppner F, Nashan B,
et al: Downregulation of AKT3 increases migration and metastasis in
triple negative breast cancer cells by upregulating S100A4. PLoS
One. 11:e01463702016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sun M, Wang G, Paciga JE, Feldman RI, Yuan
ZQ, Ma XL, Shelley SA, Jove R, Tsichlis PN, Nicosia SV and Cheng
JQ: AKT1/PKBalpha kinase is frequently elevated in human cancers
and its constitutive activation is required for oncogenic
transformation in NIH3T3 cells. Am J Pathol. 159:431–437. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee MY, Luciano AK, Ackah E,
Rodriguez-Vita J, Bancroft TA, Eichmann A, Simons M, Kyriakides TR,
Morales-Ruiz M and Sessa WC: Endothelial Akt1 mediates angiogenesis
by phosphorylating multiple angiogenic substrates. Proc Natl Acad
Sci U S A. 111:12865–12870. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol
K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al: Growth
retardation and increased apoptosis in mice with homozygous
disruption of the Akt1 gene. Genes Dev. 15:2203–2208. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cho H, Thorvaldsen JL, Chu Q, Feng F and
Birnbaum MJ: Akt1/PKBalpha is required for normal growth but
dispensable for maintenance of glucose homeostasis in mice. J Biol
Chem. 276:38349–38352. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu N, Lao Y, Zhang Y and Gillespie DA:
Akt: A double-edged sword in cell proliferation and genome
stability. J Oncol. 2012:9517242012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu Z, Xu Z, Disante G, Wright J, Wang M,
Li Y, Zhao Q, Ren T, Ju X, Gutman E, et al: miR-17/20 sensitization
of breast cancer cells to chemotherapy-induced apoptosis requires
Akt1. Oncotarget. 5:1083–1090. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Green BD, Jabbour AM, Sandow JJ, Riffkin
CD, Masouras D, Daunt CP, Salmanidis M, Brumatti G, Hemmings BA,
Guthridge MA, et al: Akt1 is the principal Akt isoform regulating
apoptosis in limiting cytokine concentrations. Cell Death Differ.
20:1341–1349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ewald F, Nörz D, Grottke A, Bach J,
Herzberger C, Hofmann BT, Nashan B and Jücker M: Vertical targeting
of AKT and mTOR as well as dual targeting of AKT and MEK signaling
is synergistic in hepatocellular carcinoma. J Cancer. 6:1195–1205.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol
K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al: Growth
retardation and increased apoptosis in mice with homozygous
disruption of the akt1 gene. Genes Dev. 15:2203–2208. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee MS, Jeong MH, Lee HW, Han HJ, Ko A,
Hewitt SM, Kim JH, Chun KH, Chung JY, Lee C, et al: PI3K/AKT
activation induces PTEN ubiquitination and destabilization
accelerating tumourigenesis. Nat Commun. 6:77692015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He X, Saji M, Radhakrishnan D, Romigh T,
Ngeow J, Yu Q, Wang Y, Ringel MD and Eng C: PTEN lipid phosphatase
activity and proper subcellular localization are necessary and
sufficient for down-regulating AKT phosphorylation in the nucleus
in cowden syndrome. J Clin Endocrinol Metab. 97:E2179–E2187. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Serra H, Chivite I, Angulo-Urarte A, Soler
A, Sutherland JD, Arruabarrena-Aristorena A, Ragab A, Lim R,
Malumbres M, Fruttiger M, et al: PTEN mediates Notch-dependent
stalk cell arrest in angiogenesis. Nat Commun. 6:79352015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jo HS, Kang KH, Joe CO and Kim JW: Pten
coordinates retinal neurogenesis by regulating Notch signaling.
EMBO J. 31:817–828. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vo K, Amarasinghe B, Washington K,
Gonzalez A, Berlin J and Dang TP: Targeting notch pathway enhances
rapamycin antitumor activity in pancreas cancers through PTEN
phosphorylation. Mol Cancer. 10:1382011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bosch FX, Ribes J, Díaz M and Cléries R:
Primary liver cancer: Worldwide incidence and trends.
Gastroenterology. 127:S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luo G, Chao YL, Tang B, Li BS, Xiao YF,
Xie R, Wang SM, Wu YY, Dong H, Liu XD and Yang SM: miR-149
represses metastasis of hepatocellular carcinoma by targeting
actin-regulatory proteins PPM1F. Oncotarget. 6:37808–37823. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chow AK, Ng L, Lam CS, Wong SK, Wan TM,
Cheng NS, Yau TC, Poon RT and Pang RW: The enhanced metastatic
potential of hepatocellular carcinoma (HCC) cells with sorafenib
resistance. PLoS One. 8:e786752013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Llovet JM, Fuster J and Bruix J: The
Barcelona approach: Diagnosis, staging, and treatment of
hepatocellular carcinoma. Liver Transpl. 10:S115–120. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen JS, Wang Q, Fu XH, Huang XH, Chen XL,
Cao LQ, Chen LZ, Tan HX, Li W, Bi J and Zhang LJ: Involvement of
PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in
hepatocellular carcinoma: Association with MMP-9. Hepatol Res.
39:177–186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bellacosa A, Testa JR, Staal SP and
Tsichlis PN: A retroviral oncogene, akt, encoding a
serine-threonine kinase containing an SH2-like region. Science.
254:274–277. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bellacosa A, Franke TF, Gonzalez-Portal
ME, Datta K, Taguchi T, Gardner J, Cheng JQ, Testa JR and Tsichlis
PN: Structure, expression and chromosomal mapping of c-akt:
Relationship to v-akt and its implications. Oncogene. 8:745–754.
1993.PubMed/NCBI
|
|
38
|
Yang ZZ, Tschopp O, Hemmings-Mieszczak M,
Feng J, Brodbeck D, Perentes E and Hemmings BA: Protein kinase B
alpha/Akt1 regulates placental development and fetal growth. J Biol
Chem. 278:32124–32131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Skeen JE, Bhaskar PT, Chen CC, Chen WS,
Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H and Hay N: Akt
deficiency impairs normal cell proliferation and suppresses
oncogenesis in a p53-independent and mTORC1-dependent manner.
Cancer Cell. 10:269–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu YJ, Li HY, Qiu KJ, Li DC, Zhou JH, Hu
YH and Zhang FM: Downregulation of Notch1 inhibits the invasion of
human hepatocellular carcinoma HepG2 and MHCC97H cells through the
regulation of PTEN and FAK. Int J Mol Med. 34:1081–1086. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou W, Fu XQ, Zhang LL, Zhang J, Huang X,
Lu XH, Shen L, Liu BN, Liu J, Luo HS, et al: The
AKT1/NF-kappaB/Notch1/PTEN axis has an important role in
chemoresistance of gastric cancer cells. Cell Death Dis.
4:e8472013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hales EC, Orr SM, Gedman Larson A, Taub JW
and Matherly LH: Notch1 Receptor Regulates AKT protein activation
loop (Thr308) dephosphorylation through modulation of the PP2A
Phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell
acute lymphoblastic leukemia cells. J Biol Chem. 288:22836–22848.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang L, Wang WL, Zhang Y, Guo SP, Zhang J
and Li QL: Epigenetic and genetic alterations of PTEN in
hepatocellular carcinoma. Hepatol Res. 37:389–396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chow LM and Baker SJ: PTEN function in
normal and neoplastic growth. Cancer Lett. 241:184–196. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hu TH, Huang CC, Lin PR, Chang HW, Ger LP,
Lin YW, Changchien CS, Lee CM and Tai MH: Expression and prognostic
role of tumor suppressor gene PTEN/MMAC1/TEP1 in hepatocellular
carcinoma. Cancer. 97:1929–1940. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dong-Dong L, Xi-Ran Z and Xiang-Rong C:
Expression and significance of new tumor suppressor gene PTEN in
primary liver cancer. J Cell Mol Med. 7:67–71. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Palomero T, Sulis ML, Cortina M, Real PJ,
Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, et
al: Mutational loss of PTEN induces resistance to NOTCH1 inhibition
in T-cell leukemia. Nat Med. 13:1203–1210. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Miele L, Miao H and Nickoloff BJ: NOTCH
signaling as a novel cancer therapeutic target. Curr Cancer Drug
Targets. 6:313–323. 2006. View Article : Google Scholar : PubMed/NCBI
|