Introduction
The B-cell lymphoma/leukemia-2 (Bcl-2) family
proteins are central regulators of cell death having both anti- and
pro-apoptotic biochemical action. In humans, six anti-apoptotic
members of this family have been identified, Bcl-2,
Bcl-xL, Bcl-B, Bcl-W, Bfl-1, and Mcl-1, further divided
into three groups on the basis of their Bcl-2 homology, in which
the BH3 domain explicate the main role in the anti-apoptotic
signaling (1).
Bcl-2 and the anti-apoptotic proteins Mcl-1 and
Bcl-xL were found to be co-expressed at relatively high
levels in a substantial proportion of heterogeneous breast tumors,
including clinically aggressive basal-like cancers (2–5). The
anti-apoptotic Bcl-2 proteins neutralize the cell-killing function
of the pro-apoptotic family members engaging their BH3 domains.
Therefore, small-molecule designed to target BH3 domain such as the
mimetic ABT-737, −263 and −199 showed therapeutic potential for
treating cancer (6–8). Although, ABT-263 evidenced promising
results for solid tumor such as small lung cancer cells and other
non-hematological malignancies, the clinical trial was early
stopped due to its severe side effect (7). Finally, only ABT-199 was recently
approved (April 2016) by US-FDA as Venetoclax for the
pharmacological treatment of Chronic Lymphocytic Leukemia (8). Currently, the activity of Bcl-2 family
inhibitors is the subject of interest in an intense area of
research concerning therapeutic agents against cancer, as
demonstrated by the recent patent literature (9). Cancer, a complex genetic disease
resulting from mutation of oncogenes or tumour suppressor genes,
can be developed due to alteration of signalling pathways; it has
been well known to have numerous links to programmed cell death
(PCD). Despite the remarkable progress in the classification of the
different cell death modes according to the morphological
presentation, signalling pathways and type of stimuli, cell death
in vivo often comprises a complex interplay between
apoptosis, necrosis/necroptosis, a novel form of
caspase-independent PCD, and autophagy (10). Apoptosis, necrosis/necroptosis and
autophagy can all occur independent of, or simultaneously with,
each other. In some situations, a specific stimulus evokes only one
of the processes but in other situations, a combined cell death
phenotype is observed in response to the same stimulus (10). In this view, it was found that
Bcl-xL/Bcl-2 plays an important role in autophagic
process (11), thanks to its binding
with Beclin 1, an autophagy-related protein (12,13). The
interaction between Beclin 1 and Bcl-xL/Bcl-2 can be
inhibited by the small-molecule ABT-737 at low doses, which
stimulates autophagy without inducing apoptosis (14). Furthermore, it was discovered that a
novel Bcl-xL inhibitor, Z36, induces autophagic cell
death, but not apoptosis, in in vitro cancer model, blocking
the interaction between Bcl-xL and Beclin-1 (15). The autophagic process promotes also
necrosis in apoptosis-deficient cells and although necrosis has
been considered a passive form of cell death, there is now the
evidence that, instead, it represents a different type of PCD,
orchestrated by autophagy, demonstrable only when apoptosis is
inhibited (16–18).
JY-1-106 is a mimetic of the BH3 α-helical ‘death
domain’ of the pro-apoptotic Bcl-2 proteins (19). In many cancer types, excess
Bcl-xL and Mcl-1 bind to the BH3 domain inactivating the
function of pro-apoptotic Bcl-2 proteins which promotes cell
survival (20,21). JY-1-106, based on a trisarylamide
framework, inhibits Bcl-xL and Mcl-1 by binding the
hydrophobic groove on the surfaces of those proteins which in turn
sequesters the anti-apoptotic proteins through binding their
hydrophobic groove that would normally bind BH3 domains. In this
manner, JY-1-106 promotes apoptosis by disrupting the interaction
of Bcl-xL and Mcl-1 with the pro-apoptotic protein Bcl-2
homologous antagonist/killer-1 (Bak-1) in multiple cancer cell
lines, sensitizes tumor cells to conventional chemotherapeutic
agents and inhibits tumor growth in a xeno-graft model of lung
cancer (19).
Retinoids are molecules derivative by metabolism of
vitamin A. One of its natural isoforms, the all-trans
Retinoic acid (RA), is able to evocate autophagy and apoptosis
depending on the doses administered (22). RA differentiation properties
stimulates synthetic chemical approaches for several other
compounds, which explicates their biochemical effects through two
types of nuclear receptors, retinoic acid receptors (RARs) and
retinoid X receptors (RXRs), consisting of α, β, and γ isoforms
(23). Targeting this latter class of
receptor, RXRs, with combined treatment with PPARγ agonist, we were
able to induce cell death in breast cancer models with different
estrogen receptors (ERs) profile and p53 expression and
functioning. Therefore, combination therapies could represent a
strategy for lowering single drug dose (24,25).
Pre-clinical observations indicate that, beside triple negative
profile, breast cancer cell display also resistance to retinoids
due to the high expression of RARγ (26,27).
Herein we tested the effect of JY-1-106 Bcl-xL/Mcl-1
inhibitor in combination with a RARγ selective retinoid SR11253,
highlighting massive autophagy and necrosis but not apoptosis in
triple-negative breast cancer (TNBC) cells MDA-MB-231, the most
aggressive breast cancer subtype used as a basal-like tumor cell
model.
Materials and methods
Chemicals
JY-1-106 was synthesized by Dr. S. Fletcher's lab
(University of Maryland, MD, USA), all-trans-retinoic acid
(RA), Am580,
4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)
carboxamido] benzoic acid and SR11253 (or MM11253),
6-[2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1,3-dithiolan-2-yl]
2-naphthalenecarboxylic acid were purchased by Sigma-Aldrich; Merck
KGaA (Darmstadt, Germany) and Tocris Bioscience (Bristol, UK).
Cell culture and MTT proliferation
assay
The human epithelial breast carcinoma MDA-MB-231
cells were purchased from the American Type Culture Collection
(ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) (Corning Incorporated, Corning, NY,
USA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and maintained in
a humidified 5% CO2 incubator at 37°C as recommended by
ATCC. Cell viability was determined by measuring the reduction of
3-(4,5-dimethylthiasol-2-yl)-2,4,-diphenyltetrazolium bromide (MTT)
by mitochondrial succinate dehydrogenase. Briefly, cells were
incubated with various concentrations of JY-1-106 (2–20 µM) alone
or with DMSO (as a vehicle) at different time point in 96-well
plates. Cells were treated with SR11253 200 nM, Am580 200 nM and RA
1 µM individually or together with JY-1-106 (16 µM) for 96 h. The
optical density (OD) was calculated as the difference between the
absorbance at the reference wavelength (620 nm) and the absorbance
at the test wavelength (570 nm). Percent viability was calculated
as (OD of drug treated sample/OD of control) ×100.
DAPI and MDC staining
Changing in morphology for nuclei swelling or
autophagic vacuoles formation were assessed by
4,6-diamidino-2-phenylindole (DAPI) or monodansylcadave rine (MDC)
staining. MDA-MB-231 were grown on covers lip in 24-well plates and
treated with 16 µM JY-1-106 and 200 nM SR11253 alone or in
combination at different time points. Cells were fixed with 4%
paraformaldehyde (Thermo Fisher Scientific, Inc.) and imaged using
by a fluorescent microscope. At least five visual fields were
analyzed under fluorescence microscope for each sample.
Lactate dehydrogenase (LDH) assay
LDH activity from the cytosol was quantified
spectrophotometrically as an index of plasma membrane injury and
cytotoxicity. Cell-free culture supernatants of MDA-MB-231 cells
were collected after 48, 72, or 96 h incubation with 16 µM JY-1-106
and 200 nM SR11253 individually or in combination and analyzed by
Cytotoxicity Detection KitPLUS (LDH) (Roche Applied
Science, Penzberg, Germany) according to the manufacturer's
instructions. The absorbance was measured with a microtiter plate
reader at a test wavelength of 492 nm, and a reference wavelength
of 690 nm.
Quantitative real-time RT-PCR
Expression levels of target genes were determined by
real-time RT-PCR. Cells were grown in 10 cm dishes to 70–80%
confluence, and exposed to 16 µM JY-1-106 and 200 nM SR11253
individually or together for 96 h incubation. Total RNA was
isolated and purified by spin protocol using the RNeasy Mini kit
(Qiagen China Co., Ltd., Shanghai, China) and QIAshredder (Qiagen
China Co., Ltd.) according to the manufacturer's instructions. Two
micrograms of total RNA were reverse-transcribed using components
of a High Capacity cDNA Reverse Transcription kit (Thermo Fisher
Scientific, Inc.). Following reverse transcription, quantitative
PCR amplification was performed on an StepOnePlus™
System (Thermo Fisher Scientific, Inc.) using TaqMan Universal PCR
Master Mix (Thermo Fisher Scientific, Inc.). For each target gene,
RARα, RARγ, p53 and β-actin (as endogenous control), a validated
predesigned TaqMan® Gene Expression Assays (Thermo
Fisher Scientific, Inc.) was used.
Relative gene expression levels were normalized to
the basal, untreated sample chosen as calibrator. Final results are
expressed as folds of difference in gene expression relative to
β-actin mRNA and calibrator, calculated following the ΔCq method,
as follows:
Relative expression
(folds)=2−(ΔCqsample−ΔCqcalibrator)
where ΔCt values of the sample and calibrator were
determined by subtracting the average Ct value of the β-actin mRNA
reference gene from the average Ct value of the analyzed gene.
Statistical analysis
Results were expressed as mean ± SD from four
independent experiments. Statistical differences were determined by
one-way analysis of variance (ANOVA) followed by Dunnet's method.
P<0.05 (*), P<0.01 (**) and P<0.001 (***) were considered
to indicate a statistically significant difference.
Results
The Bcl-xL/Mcl-1 inhibitor
JY-1-106 affects cell viability in MDA-MB-231 cells
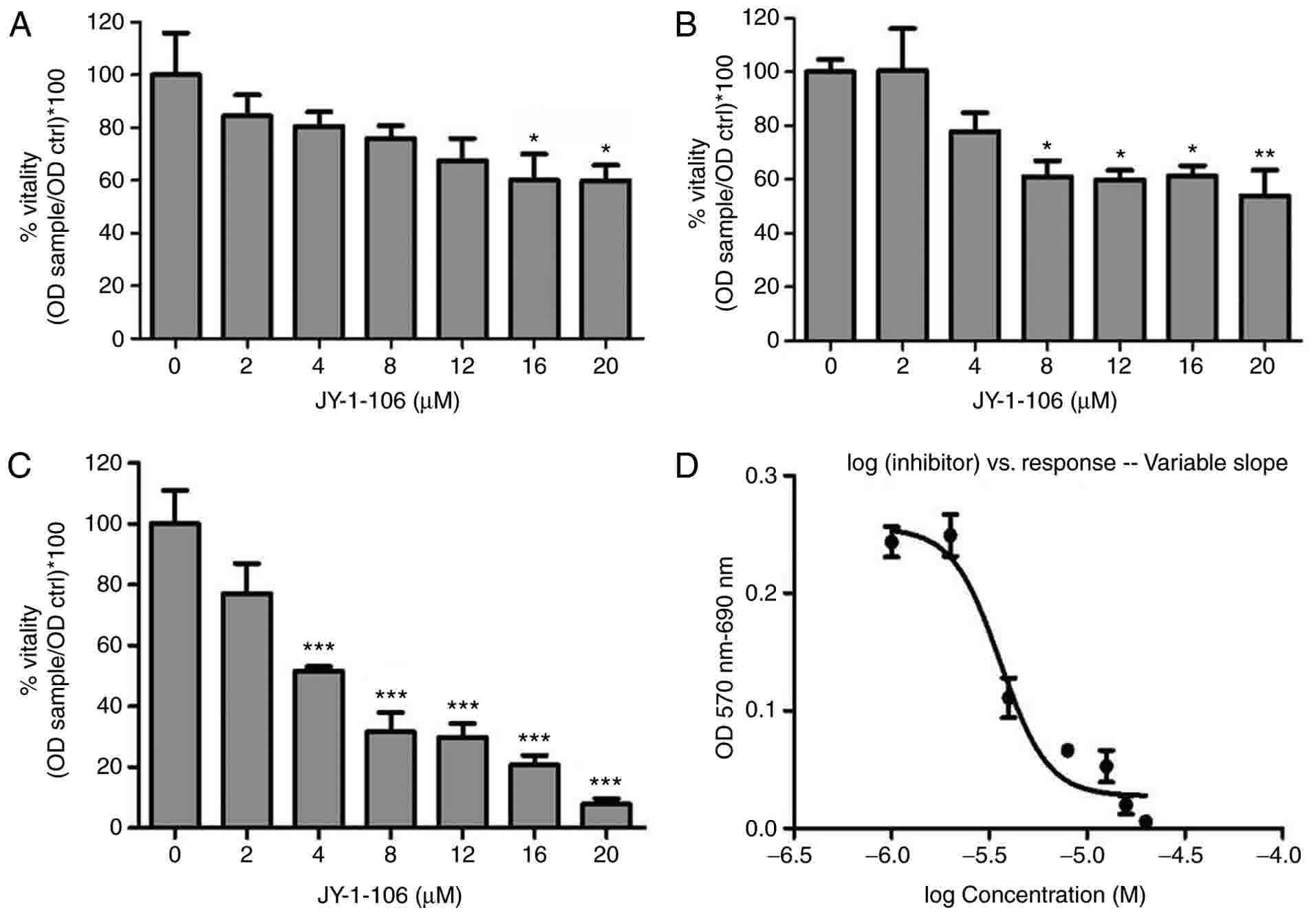
Cytotoxic effect of JY-1-106 Bcl-xL/Mcl-1
inhibitor on MDA-MB-231 cells was tested in a range from 2 to 20 µM
for 24 (A), 48 (B), or 72 h (C). Cell viability was quantified by
MTT assay. As shown in Fig. 1, a dose
and time-dependent reduction of vitality. Cell survival declined
drastically from 48 to 72 h as highlighted in Fig. 1B and C. EC50 of 3.6 µM was
calculated at 72 h as shown in Fig.
1D.
RARγ antagonist in combination with
Bcl-xL/Mcl-1 inhibitor JY-1-106 reduces cell viability
inducing autophagy and necrosis in MDA-MB-231 cells
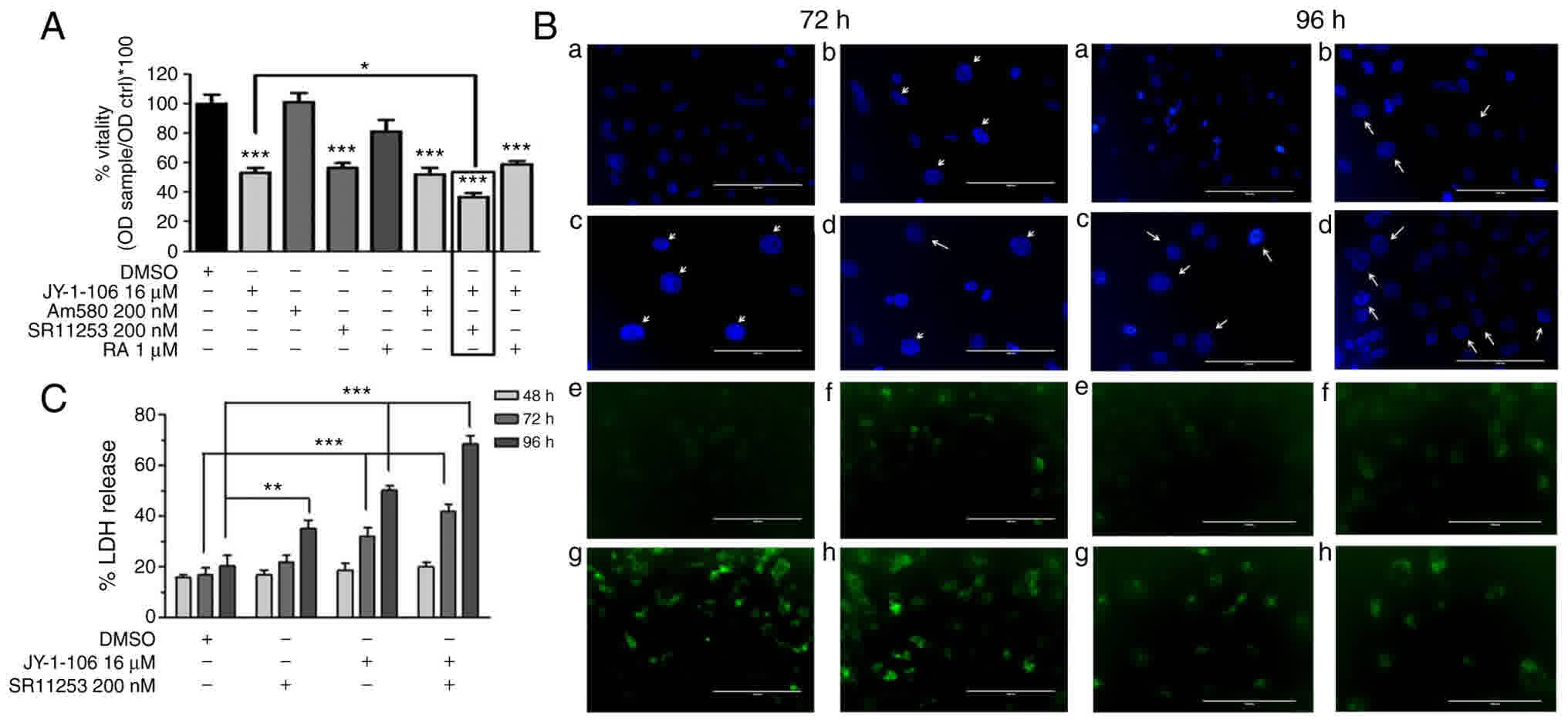
First, we tested the effects of RA 1 µM, Am580 (a
RARα agonist) 200 nM and SR11253 (a RARγ antagonist) 200 nM alone
and then together with JY-1-106 (16 µM) on MDA-MB-231 for 96 h as
shown in Fig. 2A. RA or Am580 alone
did not alter the MDA-MB-231cell growth while SR11253 decreased
cell vitality by 42%. Combined doses of JY-1-106 at 16 µM with
SR11253 200 nM show a greater effect on reducing cells viability.
From this, MDA-MB-231 were treated with JY-1-106 at 16 µM together
with SR11253 at 200 nM for 72 and 96 h of incubation. DAPI staining
analysis did not show any chromatin condensation displaying,
instead gross abnormalities in nuclear morphology (increased size
and irregular shape) as it shown in Fig.
2B (a-d, blue staining). To better understand the mechanisms
involved in TNBC cell death, we stained MDA-MB-231 cells with MDC,
visualizing autophagosome (autophagic vacuoles) development as
shown in Fig. 2B (e-h, green
staining) with massive formation at 72 and 96 h incubation. Lactate
dehydrogenase (LDH) activity in the culture media was measured as
an index of plasma membrane damage, as well as a necrotic marker.
Damaged cells after 72 and 96 h with JY-1-106 alone or in
combination with SR11253 increased LDH activity compared to control
(Fig. 2C).
Gene expression analysis of RARα, RARγ
and p53 under JY-1-106 and SR11253 on MDA-MB-231
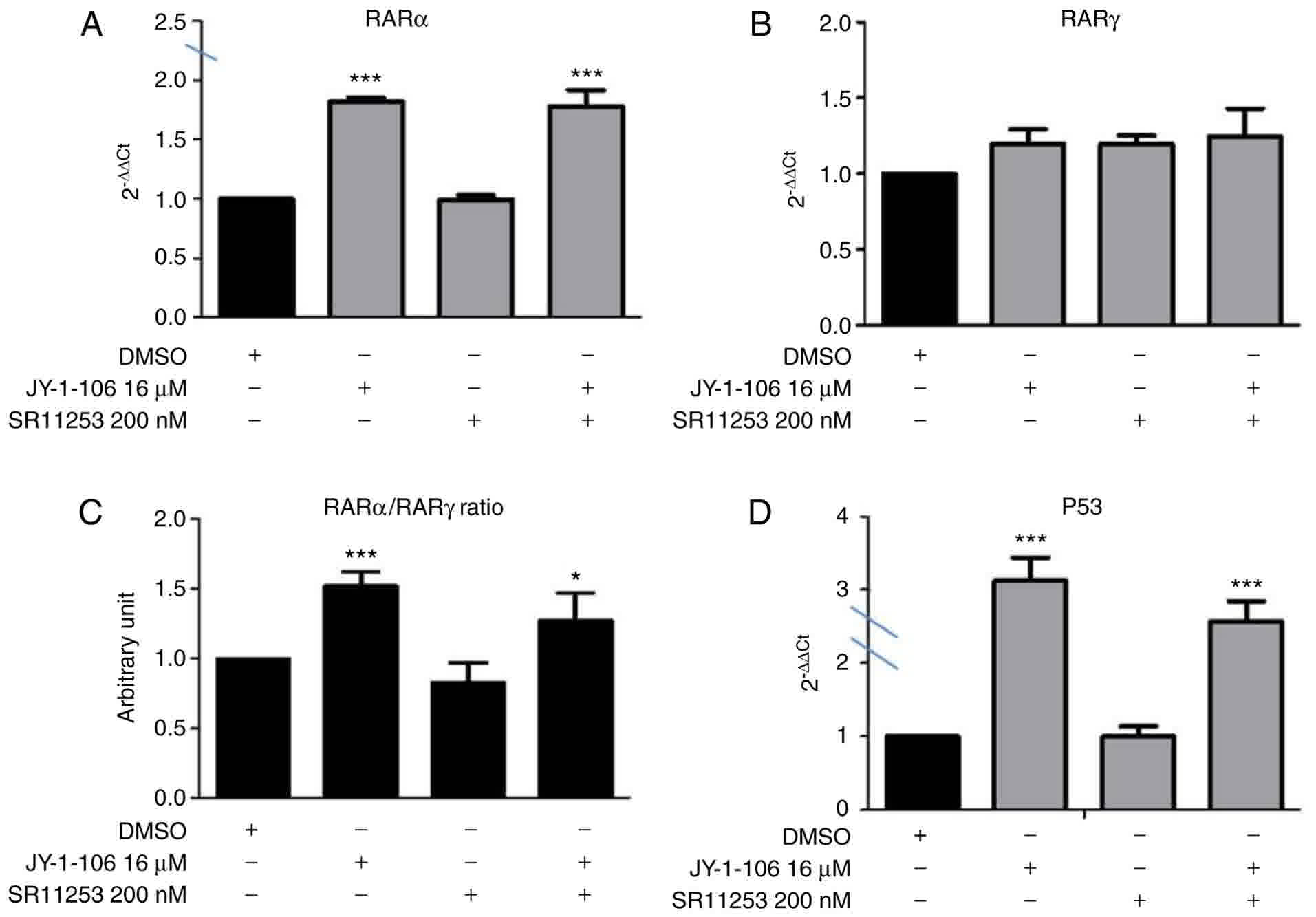
Gene expression analysis with combined treatment
JY-1-106 and SR11253 was conducted in the TNBC cells MDA-MB-231. As
shown in Fig. 3A RARα, but not RARγ
(Fig. 3B), was enhanced already by
JY-1-106 at 16 µM individually. The combination of it with SR11253
at 200 nM did not enhanced this expression (Fig. 3A and B). The RARα/RARγ ratio
calculated was therefore in favor of RARα, (Fig. 3C) suggesting an increased
susceptibility to the cell death. Therefore, P-53 was strongly
up-regulated as well using JY-1-106 (16 µM) alone or together with
SR11253 (200 nM) at the same time point (Fig. 3D).
Discussion
The heterogeneity of breast cancer classified them
in base of a specific biochemical markers profile based on estrogen
receptors (ER), progesterone receptors (PR) as well as human
epidermal growth factor receptor 2 (HER2) expression to address
pharmacological intervention. The TNBC did not express any of these
receptors (ER-/PR-/HER2-) therefore, is the most aggressive breast
cancer type (28).
Clinical/Pharmacological protocols for TNBC are limited to surgery,
radiation, and systemic chemotherapy due to the lack of more
specific therapeutic targets (28).
TNBC cells line represent an important tool for screening and
searching better treatment for this type of cancer (27). Recently, the suppression of Mcl-1
expression by microRNA-101 was able to inhibits cell progression in
TNBC (29). Significantly, almost the
70% of breast cancers overexpress anti-apoptotic Bcl-2 family
members evidencing how inhibition of specific targets belong to
this family could represent an attractive way for breast cancer
treatment (30). Therefore, Mcl-1
represent an interesting target for TNBC treatment. In this study,
we tested the JY-1-106 a Bcl-xL/Mcl-1 inhibitor (α-helix
mimetic for BH3 domain) in MDA-MB-231 TNBC cells line. Although
preliminary studies, our results show that this compound, in
combination with SR11253, has the capability to strongly reduce
MDA-MB-231 cell viability, stimulating autophagy and necrosis.
Autophagosome is evidenced by MDC staining (Fig. 2B, green staining) and, as consequence,
the inducing cell death by necrosis by DAPI staining and LDH
release (Fig. 2B, blue staining and
C). The existence of a ‘programmed necrosis’ seems to be
conditional only to apoptosis inhibition (18,31,32). It is
worth to note that a novel form of PCD, named necroptosis, was
recently discovered, although the molecular mechanisms of this
process need to be further elucidated. Necroptosis is characterized
by necrotic cell death morphology and activation of autophagy
related to inflammatory response (10).
Here we found that JY-1-106-treated cells did not
show any features of apoptosis, since DAPI staining do not
highlighted chromatin condensation, indicative of apoptotic bodies'
formation (Fig. 2B, blue staining).
Instead, in the cellular model used, we observed vacuoles formation
after 48 h incubation upon JY-1-106, with a maximum between 72 and
96 h (data not shown). The combination with RARγ SR11253 inhibitor
massively amplified this process (Fig.
2B, green staining). To confirm the hypothesis that autophagy
promotes necroptosis in apoptosis-deficient cells (18) we evidenced an increase in lactate
dehydrogenase LDH release from damaged cells. LDH activity was
enhanced after 96 h for both compounds, alone and or in combination
(Fig. 2C) the result, corroborates
the DAPI staining that show nuclear swelling (indicate by white
arrow) but not chromatin condensation (Fig. 2B, blue staining).
In the complexity of the biochemical mechanisms
governing TNBC cells, the negative modulation of RARα in favor to
RARγ shown cell proliferation, cancer survival and tumor growth in
MMTV-Myc onco-mice (26).
Experimental evidence established that the resistance to vitamin-A
derivatives of the ER-negative breast cancer cells has been linked
to a down-regulation of RARα levels (26,33). In
MDA-MB-231 cells, it was reported a very low expression of RARα
while RARγ is strongly overexpressed (27). The activation of the RARα pathway is
related to tumor growth inhibition, differentiation and cell death,
whereas RARγ was functionally linked to the promotion of tumor
growth. The pharmacologic activation of RARα or inhibition of RARγ
activity reduces cancer cell growth and the enhancing of the
RARα/RARγ ratio is favorable to cell death (33). In this context, the pharmacologic
inhibition of RARγ controlled by SR11253 reduces cancer cell
growth, enhancing the RARα/RARγ ratio, favorable to cell death
(26). RARγ antagonist SR11253 alone
inhibited the proliferation of MDA-MB-231 cells which was not
further inhibited by RA or the RARα agonist Am580 individually, as
was expected due to the low expression of RARα. It is notable that
both compounds, alone or in combination, had no effect on the
modulation of RARγ (Fig. 3B).
JY-1-106 by itself induced expression of RARα (Fig. 3A) with a favorable ratio of RARα/RARγ
which helps cell death (Fig. 3C). In
HL60 cells, a model of pro-mielocitic leukemia hematologic
malignant, we demonstrated that RARα expression was unaffected by
JY-1-106 and only RARγ was downregulated with the combined
treatment (34). The results obtained
here and the others in HL60 leukemia cells indicate that the impact
of JY-1-106 and retinoid receptor compounds is dependent upon cell
type and exist a cross talk between Bcl-xL/Mcl-1
activity and RARs expression profile, since the BH3 mimetic is able
to modulate RARs. In any case, the finality of this cross talking
is to induce cell death following either apoptosis (in leukemia) or
authophagy/necrosis (in TNBC). The p53 protein is a critical
transcriptional activator promoting apoptosis, autophagy and
therefore necrosis (35,36). Whereas the functions of p53 in
promoting apoptosis and autophagy are well established, recently it
was also identified to have a role in activating necrosis. Vaseva
et al (36) show that p53
stimulates necrotic cell death in tumor cells genetically deficient
to undergo apoptosis triggering mitochondrial permeability
transition pore. In MDA-MB-231 cells, the p53 harbor a
tumor-derived mutation (Arg280 to Lys280) in the DNA Binding Domain
which still maintain the positive charge to interact with the
phosphate backbone of the DNA consensus sequence (37,38). In
our set of experiments we have shown a strong upregulation of the
p53 mRNA using the Bcl-xL inhibitor with no additional effect of
the RARγ antagonist after 96 h incubation. These findings suggest
p53 involvement during the autophagic/necrotic by JY-1-106
(Fig. 3D) highlighting that the
action of this molecule on p53 is independent by the co-treatment.
Therefore, JY-1-106 assume an interesting profile for preclinical
TNBC treatment, considering that also promote RARα expression
without altering RARγ (39).
Therapy-induced autophagy and necrosis in cancer treatments has
been investigated and may be therapeutically useful since they are
in early phase clinical trials (40).
It worth to note that, last year the molecule ABT-199 was approved
by US-FDA as Venetoclax (April 2016). Venetoclax represents the
first pharmacological agent today in therapy, which targets
specifically Bcl-2 pathway, for the treatment of Chronic
Lymphocytic Leukemia, in patients harboring specific genetic
characteristic (8). Our results
showed a synergistic effect in reducing cell viability and inducing
autophagy and necrosis by the combination of the
Bcl-xL/Mcl-1 inhibitor JY-1-106 with a specific RARγ
antagonist. The combined treatment seems to be an attractive
strategy for controlling cancer progression in basal-like tumor
cell model.
Acknowledgements
This work was supported by Regional Operative
Programme (ROP) Calabria ESF 2007/2013-IV Axis Human
Capital-Operative Objective M2-Action d.5 Postdoctoral Fellowship,
in collaboration with University of Calabria (Cs), Italy to M.P..
Thanks to this, M.P. was able to work in Kane's Lab and to
collaborate with Dr Fletcher at University of Maryland, School of
Pharmacy.
Glossary
Abbreviations
Abbreviations:
|
Bcl-2
|
B-cell lymphoma-2
|
|
Bcl-xL
|
B-cell lymphoma-extra large
|
|
Mcl-1
|
myeloid cell leukemia-1
|
|
RAR
|
retinoic acid receptor
|
|
Am580
|
4
[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)carboxamido]benzoic
acid
|
|
SR11253
|
6-[2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1,3-dithiolan-2-yl]2-naphthalene
carboxylic acid
|
References
|
1
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Oakes SR, Vaillant F, Lim E, Lee L,
Breslin K, Feleppa F, Deb S, Ritchie ME, Takano E, Ward T, et al:
Sensitization of BCL-2-expressing breast tumors to chemotherapy by
the BH3 mimetic ABT-737. Proc Natl Acad Sci USA. 109:2766–2771.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Emi M, Kim R, Tanabe K, Uchida Y and Toge
T: Targeted therapy against Bcl-2-related proteins in breast cancer
cells. Breast Cancer Res. 7:R940–R952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Young AI, Law AM, Castillo L, Chong S,
Cullen HD, Koehler M, Herzog S, Brummer T, Lee EF, Fairlie WD, et
al: MCL-1 inhibition provides a new way to suppress breast cancer
metastasis and increase sensitivity to dasatinib. Breast Cancer
Res. 18:1252016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bah N, Maillet L, Ryan J, Dubreil S,
Gautier F, Letai A, Juin P and Barillé-Nion S: Bcl-xL controls a
switch between cell death modes during mitotic arrest. Cell Death
Dis. 5:e12912014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oltersdorf T, Elmore SW, Shoemaker AR,
Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges
J, Hajduk PJ, et al: An inhibitor of Bcl-2 family proteins induces
regression of solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kaefer A, Yang J, Noertersheuser P,
Mensing S, Humerickhouse R, Awni W and Xiong H: Mechanism-based
pharmacokinetic/pharmacodynamic meta-analysis of navitoclax
(ABT-263) induced thrombocytopenia. Cancer Chemother Pharmac.
74:593–602. 2014. View Article : Google Scholar
|
|
8
|
Roberts AW, Davids MS, Pagel JM, Kahl BS,
Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR,
Gressick L, et al: Targeting BCL2 with venetoclax in relapsed
chronic lymphocytic leukemia. N Engl J Med. 374:311–322. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen L and Fletcher S: Mcl-1 inhibitors: A
patent review. Expert Opin Ther Pat. 27:163–178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goodall ML, Fitzwalter BE, Zahedi S, Wu M,
Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD and
Thorburn A: The autophagy machinery controls cell death switching
between apoptosis and necroptosis. Dev Cell. 37:337–349. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pattingre S and Levine B: Bcl-2 inhibition
of autophagy: A new route to cancer? Cancer Res. 66:2885–2888.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maiuri MC, Le Toumelin G, Criollo A, Rain
JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K,
Tavernarakis N, et al: Functional and physical interaction between
Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 26:2527–2539.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin J, Zheng Z, Li Y, Yu W, Zhong W, Tian
S, Zhao F, Ren X, Xiao J, Wang N, et al: A novel Bcl-XL inhibitor
Z36 that induces autophagic cell death in Hela cells. Autophagy.
5:314–320. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ullman E, Fan Y, Stawowczyk M, Chen HM,
Yue Z and Zong WX: Autophagy promotes necrosis in
apoptosis-deficient cells in response to ER stress. Cell Death
Differ. 15:422–425. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Samara C, Syntichaki P and Tavernarakis N:
Autophagy is required for necrotic cell death in Caenorhabditis
elegans. Cell Death Differ. 15:105–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao X, Yap JL, Newell-Rogers MK,
Peddaboina C, Jiang W, Papaconstantinou HT, Jupitor D, Rai A, Jung
KY, Tubin RP, et al: The novel BH3 α-helix mimetic JY-1-106 induces
apoptosis in a subset of cancer cells (lung cancer, colon cancer
and mesothelioma) by disrupting Bcl-xL and Mcl-1 protein-protein
interactions with Bak. Mol Cancer. 12:422013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Ikezoe T, Nishioka C and Yokoyama
A: Over-expression of Mcl-1 impairs the ability of ATRA to induce
growth arrest and differentiation in acute promyelocytic leukemia
cells. Apoptosis. 18:1403–1415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kharbanda S, Saxena S, Yoshida K, Pandey
P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, et al:
Translocation of SAPK/JNK to mitochondria and interaction with
Bcl-x(L) in response to DNA damage. J Biol Chem. 275:322–327. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perri M, Pingitore A, Cione E, Vilardi E,
Perrone V and Genchi G: Proliferative and anti-proliferative
effects of retinoic acid at doses similar to endogenous levels in
Leydig MLTC-1/R2C/TM-3 cells. Biochim Biophys Acta. 1800:993–1001.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chambon P: The retinoid signaling pathway:
Molecular and genetic analyses. J Biol Chem. 5:115–125. 1994.
|
|
24
|
Bonofiglio D, Cione E, Qi H, Pingitore A,
Perri M, Catalano S, Vizza D, Panno ML, Genchi G, Fuqua SA and Andò
S: Combined low doses of PPARgamma and RXR ligands trigger an
intrinsic apoptotic pathway in human breast cancer cells. Am J
Pathol. 175:1270–1280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bonofiglio D, Cione E, Vizza D, Perri M,
Pingitore A, Qi H, Catalano S, Rovito D, Genchi G and Andò S: Bid
as a potential target of apoptotic effects exerted by low doses of
PPARγ and RXR ligands in breast cancer cells. Cell Cycle.
10:2344–2354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bosch A, Bertran SP, Lu Y, Garcia A, Jones
AM, Dawson MI and Farias EF: Reversal by RARα agonist Am580 of
c-Myc-induced imbalance in RARα/RARγ expression during MMTV-Myc
tumorigenesis. Breast Cancer Res. 14:R1212012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chavez KJ, Garimella SV and Lipkowitz S:
Triple negative breast cancer cell lines: One tool in the search
for better treatment of triple negative breast cancer. Breast Dis.
32:35–48. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Tang H, Chen J, Song C, Yang L, Liu
P, Wang N and Xie X, Lin X and Xie X: MicroRNA-101 inhibits cell
progression and increases paclitaxel sensitivity by suppressing
MCL-1 expression in human triple-negative breast cancer.
Oncotarget. 6:20070–20083. 2015.PubMed/NCBI
|
|
30
|
Alsabeh R, Wilson CS, Ahn CW, Vasef MA and
Battifora H: Expression of bcl-2 by breast cancer: A possible
diagnostic application. Mod Pathol. 9:439–444. 1996.PubMed/NCBI
|
|
31
|
Edinger AL and Thompson CB: Death by
design: Apoptosis, necrosis and autophagy. Curr Opin Cell Biol.
16:663–669. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kanduc D, Mittelman A, Serpico R,
Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia MG,
Lucchese A, Dini L, et al: Cell death: Apoptosis versus necrosis
(review). Int J Oncol. 21:165–170. 2002.PubMed/NCBI
|
|
33
|
Sheikh MS, Shao ZM, Chen JC, Hussain A,
Jetten AM and Fontana JA: Estrogen receptor-negative breast cancer
cells transfected with the estrogen receptor exhibit increased RAR
alpha gene expression and sensitivity to growth inhibition by
retinoic acid. J Cell Biochem. 53:394–404. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Perri M, Yap JL, Yu J, Cione E, Fletcher S
and Kane MA: BCL-xL/MCL-1 inhibition and RARγ antagonism work
cooperatively in human HL60 leukemia cells. Exp Cell Res.
327:183–191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vaseva AV, Marchenko ND, Ji K, Tsirka SE,
Holzmann S and Moll UM: p53 opens the mitochondrial permeability
transition pore to trigger necrosis. Cell. 149:1536–1548. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bartek J, Iggo R, Gannon J and Lane DP:
Genetic and immunochemical analysis of mutant p53 in human breast
cancer cell lines. Oncogene. 5:893–899. 1990.PubMed/NCBI
|
|
38
|
Wright JD and Lim C: Mechanism of
DNA-binding loss upon single-point mutation in p53. J Biosci.
32:827–839. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sheikh MS, Shao ZM, Li XS, Dawson M,
Jetten AM, Wu S, Conley BA, Garcia M, Rochefort H and Fontana JA:
Retinoid-resistant estrogen receptor-negative human breast
carcinoma cells transfected with retinoic acid receptor-alpha
acquire sensitivity to growth inhibition by retinoids. J Biol Chem.
269:21440–21447. 1994.PubMed/NCBI
|
|
40
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|