Despite improvements in early detection and
treatment method in recent years, colorectal cancer (CRC) remains
the third most frequent and the fourth leading cause of
cancer-associated mortalities worldwide (1,2).
Approximately 65% of CRC cases are sporadic with no family history
or apparent genetic predisposition (3). The remaining cases are familial, arising
from moderately penetrant inherited susceptibility, possibly
interacting with environmental factors (3,4).
CRC, like numerous other solid tumors, is a
heterogeneous disease in which different subtypes may be
distinguished by their specific clinical and/or molecular features.
The majority of sporadic CRCs (~85%) exhibit chromosomal
instability (CIN), with changes in chromosome number and structure
(5–8).
These changes include gains or losses of chromosomal segments,
chromosomal rearrangements, and loss of heterozygosity (LOH), which
results in gene copy number variations (CNVs) (5–8). These
alterations affect the expression of tumor-associated genes, and/or
genes that regulate cell proliferation or cell cycle checkpoints,
which, in turn, may activate pathways essential for CRC initiation
and progression (9,10). The remaining sporadic cases (~15%)
have high-frequency microsatellite instability (MSI) phenotypes.
However, hereditary CRC has two well-described forms: Familial
adenomatous polyposis (FAP) (<1%) patients inherit a mutated
copy of the adenomatous polyposis (APC) gene, whereas
hereditary non-polyposis colorectal cancer (HNPPC, or Lynch
syndrome) (1–3%) is characterized by MSI, a consequence of a
defective DNA mismatch repair (MMR) system (11). The other forms of hereditary CRC
include a rare syndrome called hamartomatous polyposis syndrome
(<1%) and the common inherited cases caused by less penetrant
inherited mutations (32%) (3).
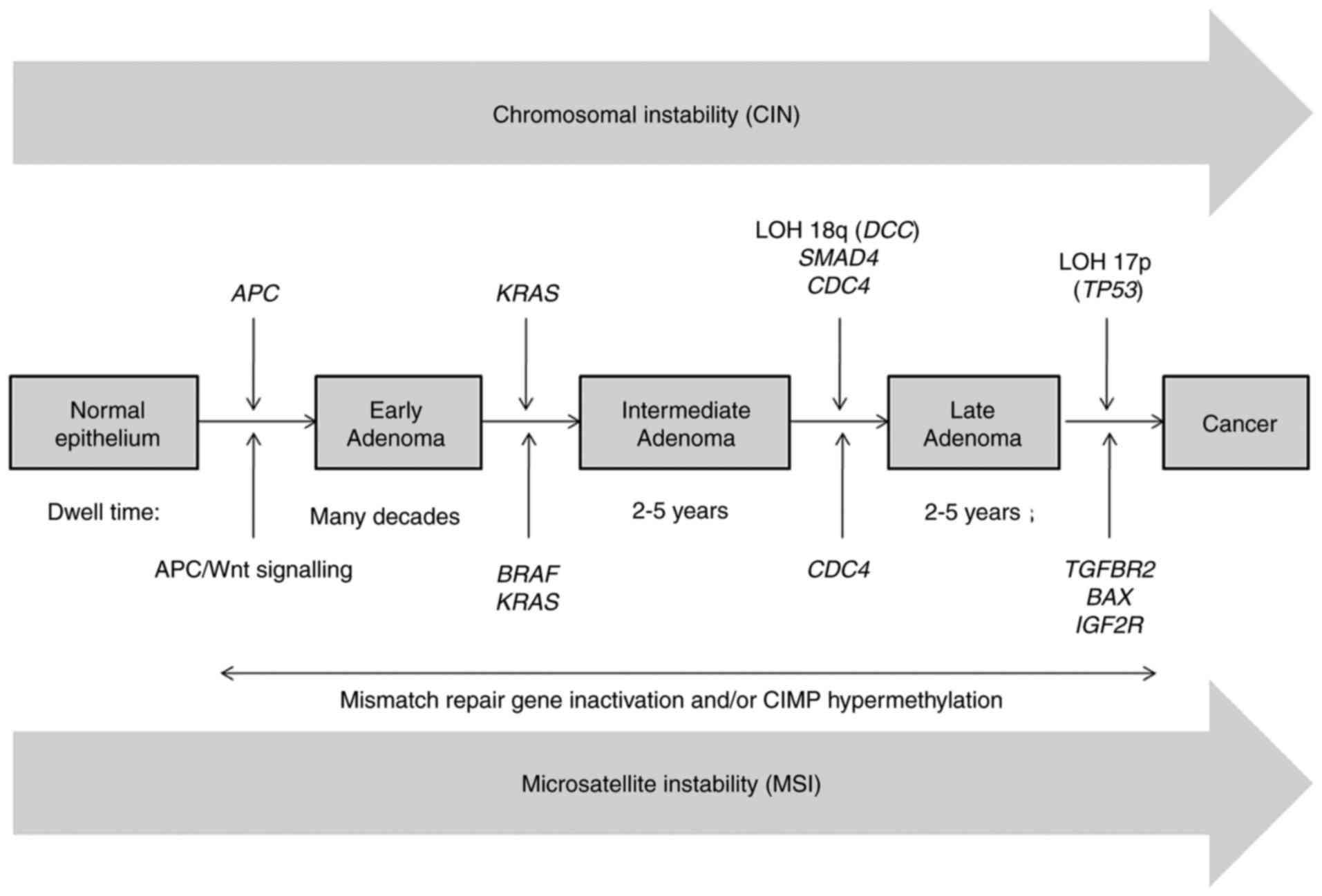
Sequential acquisition of genetic and epigenetic
alterations is well defined, and confirmed to drive the initiation
and progression of adenomas to carcinomas in sporadic and inherited
forms of CRC (12–14). Generally, CRC formation begins with
the transformation of a normal colorectal epithelium to a benign
adenoma, and then progresses through the stepwise accumulation of
multiple genetic and epigenetic aberrations, subsequently leading
to invasive and metastatic tumors (12–14). This
process may take years to decades to escape the multiple regulatory
layers of the cells and to fully develop (Fig. 1) (13,15). There
are three major pathways associated with CRC pathogenesis, namely:
CIN, MSI and CpG island methylator phenotype (CIMP) (16).
The extent to which cancer has spread at the time of
diagnosis is described as its stage. Currently, CRC staging is
primarily based on the tumor-nodes-metastasis (TNM) system proposed
by the American Joint Committee on Cancer (17). The survival rate of patients with CRC
largely depends on the stage at which tumor is first diagnosed and
varies between stages. For example, the 5-year-survival rate for
patients with stage I colon cancer is 93.2%, which drops to 8.1%
for patients with stage IV (17).
Although TNM is currently the most common CRC staging system, and
an important basis to determine the treatment method and assessing
prognosis, it is not a reliable tool for prediction and prognosis.
Particularly, CRC patients with similar histopathology may have
completely different progression and outcome depending on their
genetic and epigenetic background (18). Thus, understanding the molecular
pathways underlying the initiation and development of CRC is
essential to identify novel molecular biomarkers for diagnosis and
prognosis, thereby improving the outcome. The present review
summarized the current knowledge of the genetic and epigenetic
integrity, the consequences of the DNA MMR machinery associated
with CRC, and the role of molecular characterization in early
diagnosis and in the treatment of CRC.
The average rate of genomic mutation in normal human
cells is estimated to be ~2.5×10−8
mutations/nucleotide/generation (19,20).
However, this rate is higher in cancer cells due to the sequential
accumulation of multiple mutations during cell divisions forming a
so-called ‘mutator phenotype’ (21).
Accordingly, mutations in MMR genes, genes that regulate cell cycle
checkpoints, and/or cellular responses may elevate mutation rates
to the level commonly observed in human tumors (21). The ‘mutator phenotype’ may have
various manifestations, including point mutations, CIN, MSI, CIMP
and LOH (21).
CIN appears to be the most common type of genetic
instability in CRC, observed in 85% of adenoma-carcinoma
transitions (5–7). CIN refers to a high rate of gains or
losses of whole, or large portions of chromosomes. This leads to
karyotypic variability from cell to cell that consequently forms an
aneuploidy, sub-karyotypic amplification, chromosomal
rearrangement, and a high frequency of LOH at tumor suppressor gene
loci (5,6). In addition, CIN tumors are recognized by
the accumulation of mutations in specific oncogenes, including KRAS
proto-oncogene GTPase (KRAS) and B-Raf proto-oncogene
serine/threonine kinase (BRAF), and tumor suppressor genes,
such as APC and tumor protein p53 (TP53), thereby
contributing to CRC tumorigenesis (6,10). The
multistep genetic model of colorectal carcinogenesis proposed by
Fearon and Vogelstein is now widely accepted, and used as a
paradigm for solid tumor progression (12). According to this model, inactivation
of APC occurs as the first event, followed by oncogenic
KRAS mutations in the adenomatous stage, and eventually,
deletion of chromosome 18q and inactivation of the tumor-suppressor
gene TP53 on chromosome 17p occur during the transition to
malignancy (Fig. 1) (12,22–25).
Array-based comparative genomic hybridization and
single nucleotide polymorphism techniques have enabled scientists
to effectively determine CNVs in the entire human genome with
higher resolution. Although the allelic loss of all chromosomal
arms has been detected in certain tumors, its frequency varies
considerably, and only a few of them are highly recurrent in CRC,
including losses at chromosomal arms 1p, 5q, 8p, 17p, 18p, 18q, 20p
and 22q (26–31). A high-frequency allelic loss at a
specific chromosomal region denotes the presence of a candidate
tumor-suppressor gene, including APC on chromosome 5q,
TP53 on chromosome 17p, DCC netrin 1 receptor (DCC),
SMAD family member (SMAD2 and SMAD4) on chromosome
18q (31). In contrast, a gain of
chromosomal material suggests the presence of the potential
oncogenes or genes that favor cell growth or survival. In CRC,
gains at chromosome 7, and chromosomal arms 1q, 8q, 12q, 13q and
20q have been repeatedly reported by different research groups
(26–31). It was reasoned that these chromosomal
changes are associated with a gain and loss of function of
tumor-associated genes offering mutated cells growth and survival
advantages, leading to progressive conversion of normal cells into
cancer cells (32,33). However, the gains/losses of
chromosomal materials generally span a large region and comprise a
large number of genes making identification of target genes
challenging.
In the field of stem cell research, genetic analysis
of human embryonic stem cell (hESC) lines, a pluripotent cell type
that shares numerous characteristics with cancer cells, has also
revealed multiple CNVs, and few of them are also recurrent,
including losses of chromosomal band 18q21qter, and whole or
partial gains of chromosomes 1, 12, 17 and 20 (34,35).
Notably, 20q11.21 amplification was identified in >20% of the
screened hESC lines (36).
Previously, BCL2 like 1 (BCL2L1), which is located in the
smallest common chromosomal region of gain and regulates the
mitochondrial apoptotic pathway, has been confirmed as the
key-driver gene of this amplification (37,38).
Accordingly, the overexpression of Bcl-xL, an anti-apoptotic
isoform of BCL2L1 has offered cells a survival advantage by
preventing apoptosis (37,38). Overexpression of this gene may also be
responsible for the gain of 20q in various human cancer types
(39).
Allelic loss at chromosome 18q is detected in ~70%
of primary CRC in the late carcinogenic process (29,31,40,41),
and is considered as a poor prognosis marker for survival in
patients with CRC (42,43). The high frequency of allelic deletions
involving chromosome 18q suggests the presence of candidate
tumor-suppressor genes whose inactivation may serve a significant
role in CRC, including DCC, SMAD2 and SMAD4 (12,25,44).
DCC, located in the chromosome band 18q21.2, encoding a
component of the neutrin-1 receptor, was proposed as a putative
tumor-suppressor gene (45). However,
much of the reported data on the loss and inactivation of
DCC is circumstantial and fails to provide conclusive
evidence that DCC functions as a tumor-suppressor gene
(46). Furthermore, to the best of
our knowledge, there is no evidence that germline mutations of
DCC serve a role in heritable cancer; and few somatic
mutations in DCC have been reported in CRC (46). The presence of two other
well-established tumor suppressor genes, SMAD2 and
SMAD4 in the region of loss also challenges the function of
DCC as a tumor-suppressor gene (47,48). In
fact, SMAD2 and SMAD4 genes are localized in 18q21.1,
the common region of loss of 18q in CRC (25). These SMAD genes encode
downstream signal transducers for transforming growth factor-β
(TGF-β), and their alterations may confer resistance to
TGF-β and contribute to tumorigenesis (49). SMAD4 was identified to be
inactivated in ~60% of pancreatic cancer (50). However, the frequency of SMAD4
and SMAD2 somatic mutations is relatively low in CRC
(51–53). Nevertheless, smaller regions of loss,
which exclude SMAD2 and SMAD4, have been reported in
head and neck squamous cancer (54).
In addition, their gene expression is retained in CRC with LOH of
18q (46). Taken together, these
observations suggest that SMAD2 and SMAD4 are
unlikely to constitute the major chromosome 18q target for
inactivation in CRC, and that other tumor suppressor genes besides
the DCC and SMAD genes may be the target for
chromosome 18q loss.
Another type of genomic instability is MSI, a
typical characteristic of cancerous cells, occurring in 15–20% of
sporadic CRC and in >95% of HNPPC. Microsatellites are
repetitive DNA sequences consisting of tandem repeats, usually
between one to five base pairs. Patients with MSI phenotype exhibit
a high frequency of replication errors, particularly in repetitive
DNA sequences, primarily due to the slippage of the DNA polymerase
(94). The progressive
insertion/deletions of nucleotides within the microsatellite
sequences result in the appearance of longer or shorter alleles
compared with those detected in the normal cells of the same
individual (95,96).
To access the MSI status of a cancer, a standard
panel of five microsatellite markers, including two mononucleotide
(BAT26 and BAT25) and three dinucleotide (D2S123, D5S346, and
D17S250) repeats, has been recommended according to the Bethesda
Guidelines (97). Tumors are then
classified based on the number of microsatellites exhibiting
instability. Particularly, tumors are classified as MSI high
(MSI-H) when ≥30% of the markers exhibit instability; those with
<30% markers exhibiting instability are defined as MSI low, and
those with no apparent instability are microsatellite stable (MSS)
(97,98).
It is now accepted that MSI is associated with
post-replicative DNA MMR deficiency, primarily involving mutL
homolog 1 (MLH1) and mutS homolog 2 (MSH2) (94,99–101).
Impairment of MMR genes can occur by either mutational inactivation
or by epigenetic inactivation through CpG island methylation of the
promoter of the genes. Loss or insufficiency of MMR activity leads
to replication errors with an increased mutation rate and a higher
potential for malignancy. In MSI-H gastric cancer, for example,
hypermethylation of MLH1 promoter is responsible for the
development of >50% of cases, whereas mutations in MLH1
and MSH2 account for ~15% of cases (102,103).
Small insertions/deletions may create frame-shift
mutations within repetitive tracts present in the coding region of
essential tumor-suppressor or tumor-associated genes, resulting in
an inactive protein and contributing to tumorigenesis in cancers
with MSI-H (104). Using a
large-scale genomic screen of coding region microsatellites, Mori
et al (105) identified nine
loci that were mutated in >20% of tumors, namely: Transforming
growth factor-β receptor (TGFBR2) (79.1%), BCL2 associated X
apoptosis regulator (BAX) (37.5%), human mutS homolog 3
(26.2%), activin A receptor, type II (58.1%), SEC63 homolog protein
translocation regulator (48.8%), absent in melanoma 2 (47.6%),
NADH-ubiquinone oxidoreductase (27.9%), cordon-bleu WH2 repeat
protein like 1 (23.8%) and proliferation-associated
2G4/ErbB3-binding protein 1 (20.9%). TGFBR2, encoding a
kinase receptor involved in transduction of the TGFB1/2/3
signal from the cell surface to the cytoplasm to inhibit cellular
proliferation, is the most commonly affected gene. Particularly,
instability in the poly-adenine tract of this gene has been
detected in ~85% of MSI-H colorectal tumors, rendering an inactive
receptor and thus eliminating the growth-suppressive effects of
TGFB1 (106). Another
commonly mutated gene in CRC is BAX, a pro-apoptotic gene
belonging to the BCL2 family. Frame-shift mutations within
the poly-guanine sequence have been detected in 50% of MSI-H
colorectal tumors, causing silencing of this gene and suppressing
apoptosis (107). These alterations
in the gene functions represent a possible mechanism for MSI
carcinogenesis.
Transcription inactivation by DNA hypermethylation
at promoter CpG islands of tumor-suppressor genes, causing gene
silencing, is now recognized as an important mechanism in human
carcinogenesis (108–111). The CpG island methylator phenotype
has been identified in 30–35% colorectal adenoma cases, and is
considered as an early event and a characteristic for the serrated
pathway of colorectal tumorigenesis (108,112,113).
However, the quantitative DNA methylation study performed by Ogino
et al (114) reported that
CIMP accounts for 17% of CRC, which is less frequent compared with
previously reported and that clinical features of CIMP are similar
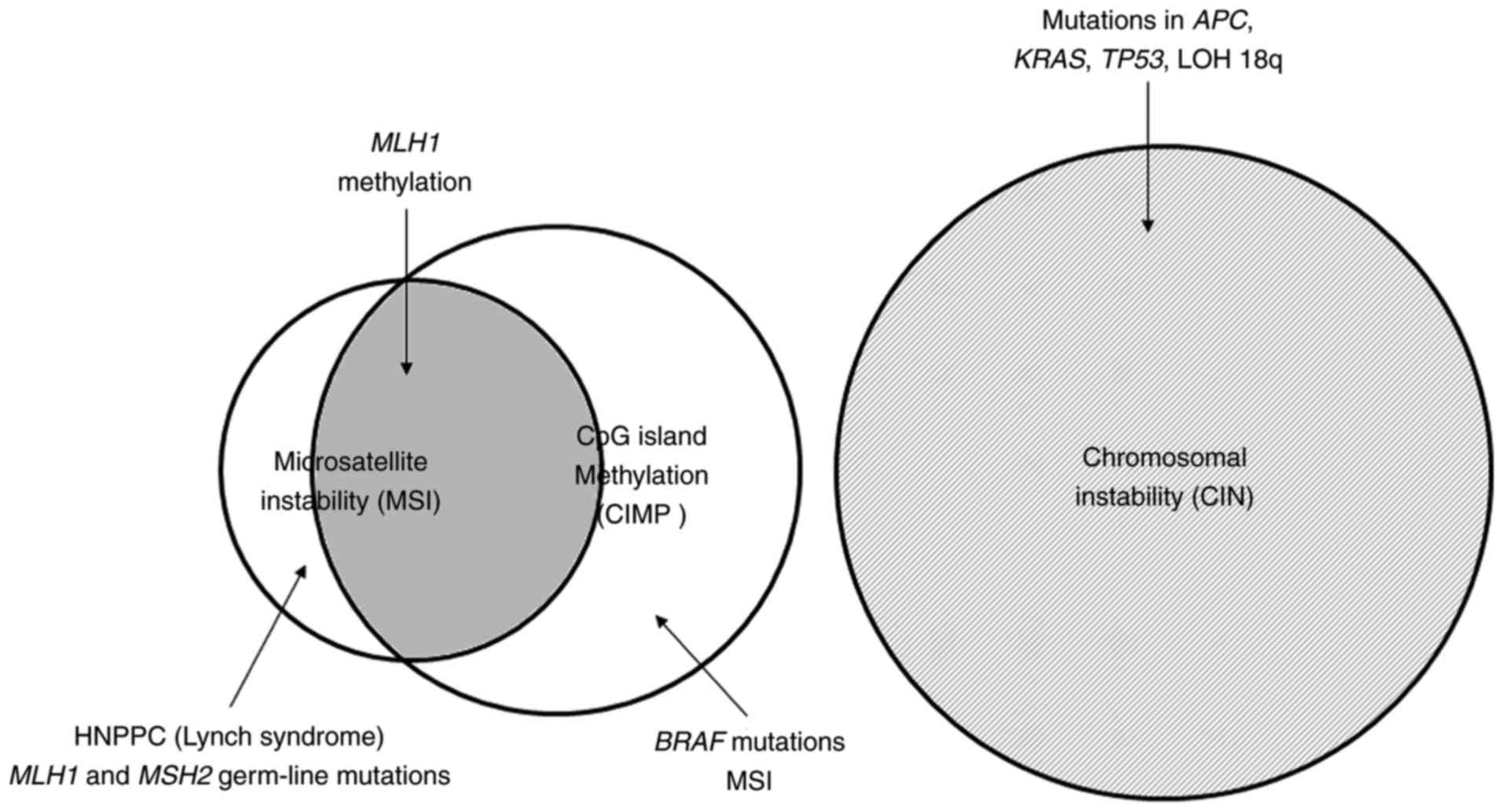
to those of MSI-associated CRC (114). Notably, sporadic MSI colorectal
tumors are almost exclusively associated with CIMP-associated
methylation of MLH1 leading to inactivation of this gene
(107,115). In contrast, the familial MSI cases
(Lynch syndrome) are generally caused by germline mutations in the
MMR genes, primarily including MLH1 and MSH2, and
accounts for <5% of all CRC cases (Fig. 2) (107,116).
The CIMP status of CRC is currently assessed by a
panel of methylation markers categorizing CRC as exhibiting or not
exhibiting DNA methylation on the basis of certain thresholds
(114,115,117,118).
CIMP+ colorectal tumors appear to have a distinct
profile, including associations with the proximal colon, poor
differentiation, MSI status, BRAF mutation and wild-type
KRAS (113–115,119–121).
Particularly, the frequency of BRAF mutations in
CIMP+ tumors is significantly higher compared with their
CIMP− counterparts (114,115).
Shen et al (122) analyzed
the genetic and epigenetic alterations in 97 primary CRC samples,
and demonstrated that CIMP-high tumors are associated with MSI
status (80%) and BRAF mutation (53%); CIMP-low tumors are
associated with KRAS mutations (92%); and CIMP−
tumors typically have a high rate of p53 mutations (71%) (122). Furthermore, CIMP status has also
been indicated to be negatively associated with 18q LOH status in
colorectal tumors (117).
Particularly, CIMP-0 was associated with 18q LOH-positive tumors
and vice versa (117).
The prognosis and therapeutic options for patients
with CRC are associated with the stage at which they are first
diagnosed. While early stage CRC is often cured with surgery alone,
more advanced or metastatic CRC generally require additional
adjuvant chemotherapy or targeted therapy, either alone or as a
combined treatment. Early detection of CRC thus becomes important
to reduce the incidence and mortality of the disease. Furthermore,
due to their heterogeneity, the benefits from adjuvant chemotherapy
for stage II and III CRC patients may vary largely. Thus,
identifying molecular prognostic markers that are capable of
recognizing patients with CRC more likely to recur or benefit from
adjuvant chemotherapy may improve the prognosis and assist in the
selection of appropriate therapy, and subsequently the general
outcomes.
It is now widely known that certain alterations at
the molecular level favor CRC onset, progression and metastasis
(60). Several known mutations are
considered to be associated with a poorer patient outcome and/or
failure of response to a certain therapy (18). Patients with inactive TP53
mutations, for example, are at an increased risk of mortality
compared with their counterparts, but this mutation does not appear
to affect the outcome of chemotherapy (123). However, the presence of somatic
KRAS mutations has been considered as a predictor of
resistance to anti-EGFR therapy (83–86,124).
Thus, KRAS mutation status is currently used in clinical
settings to predict the therapeutic effectiveness of CRC prior to
chemotherapy to avoid any undesired effects and medical costs
(125). APC is another
commonly affected gene whose mutations generally appear in the
early stage of CRC development (55,60).
Notably, the risk of CRC for a patient with FAP, which begins with
a germline mutation in one allele of the APC gene is ~100%
by the age of 40 years (6,7). Therefore, APC mutations are being
considered as good diagnostic markers for identifying individuals
at risk of CRC.
The majority (~75%) of CRC with MSI are sporadic
cases caused by the loss of DNA MMR activity due to methylation of
the promoter of the MLH1 gene, while the other 25% of cases
are classified as Lynch syndrome caused by germline mutations in
the MMR genes (Fig. 2). Generally,
MSI is detected earlier in life in patients with Lynch syndrome
(<50 years old) as compared with the sporadic cases (>65
years old) (126). Particularly, CRC
with MSI are more likely to occur in the proximal colon (126). Evidence has suggested that MSI is a
favorable prognostic biomarker for CRC (127–129).
However, its predictive role for the response to chemotherapeutic
agents, including 5-fluorouracil (5-FU) is conflicting. Several
studies demonstrated a lack of benefit of 5-FU-based adjuvant
chemotherapy in patients with CRC with MSI tumors (130–133),
while others reported the beneficial effects (127,134).
Des Guetz et al (135)
performed a meta-analysis involving 3,690 patients from seven
different studies, and reported that chemotherapy had a beneficial
effect among MSS, but not MSI-H patients (135). In addition, the more improved
survival rate of MSI-H patients was due to a better prognosis
rather than the benefit of chemotherapy (135). These findings suggested that MSI may
be considered as a predictive marker of chemoresistance and that
patients with CRC with MSI may be spared from adjuvant treatment.
The MSI status among patients with CRC, thus, is highly valuable in
prognosis and therapy of CRC, and should be thoroughly evaluated by
performing polymerase chain reaction analysis using the Bethesda
panel and/or immunohistochemistry staining for DNA MMR proteins,
including MLH1 and MSH2, in order to contribute to treatment
decision-making regarding chemotherapy administration.
Several groups have used gene expression profiling
to classify CRC, and to identify genes associated with prognosis
and prediction of disease outcome. De Sousa et al (136) used an unsupervised classification
strategy involving >1,100 individuals with colon cancer and
defined three main colon cancer subtypes. Two subtypes are
associated with two well-characterized subsets of colon cancer,
namely the CIN and the MSI group. The third subtype was largely MSS
and overlaps partly with the CIMP group, and is associated with
poor prognosis and resistance to anti-EGFR therapy (136). Using a similar approach, Sadanandam
et al (137) defined six
clinically relevant CRC subtypes by associating their gene
expression profiles with corresponding clinical response to
cetuximab. Patients with stem-like subtype and inflammatory subtype
tumors, with poor and intermediate disease-free survival, exhibited
an improved response to the combination chemotherapy regimen
FOLFIRI (5-FU with irinotecan) in adjuvant or metastatic settings,
whereas transit-amplifying- and goblet-like-subtype tumors, with
markedly better prognosis, did not appear to benefit from these
treatments. However, cetuximab-sensitive transit-amplifying and
cetuximab-resistant transit-amplifying subtypes may be efficiently
treated with cetuximab or a cMET inhibitor, respectively, in the
metastatic setting (137). Although
there are significant associations between MSI status and specific
subtypes, the transcriptional signatures-based subtypes allow
better refinement and provide insights for the development of
subtype-specific therapies, which, in turn, may contribute to the
more effective management of this disease.
Despite the great advancement in CRC research, the
role of the molecular characterization in diagnostic tests and
therapeutic decisions remains limited due to the fact that the
function of the majority of mutations remains unclear and rarely
provides any valuable diagnostic information. Further research is
required to develop more easily applicable molecular tests for
early detection of CRC, which is essential to improving the
prognosis and treatment efficiency. Furthermore, it is essential to
identify novel therapeutic targets as the majority of CRC cases are
insensitive to EGFR inhibitor therapy.
Recent studies have provided a better understanding
of CRC and assist in the development of novel treatment regimens.
Particularly, the implementation of targeted next-generation
sequencing (NGS) in clinical settings allows a reliable
identification of the most common mutations, and is able to guide
therapeutic decisions for patients with CRC based on personalized
medicine (138). NGS is currently
the most important technology for early diagnosis and prognosis, as
well as identification of novel predictive biomarkers for available
treatments with targeted therapy and immunotherapy for patients
with CRC (138). Specifically, the
combination of CRISPR/Cas9 technology and immunotherapy would
significantly improve patient care by reducing side effects
(139,140).
The authors would like to thank Dr. Adam F. Johnson
(Center for Molecular Biology, Institute of Research and
Development, Duy Tan University, Danang, Vietnam) for the critical
reading of the manuscript.
This study was supported by the National Foundation
for Science and Technology Development (NAFOSTED; grant no.
106-YS.01-2015.12).
Not applicable.
HTN conceptualized the article, critically
discussed the findings and co-wrote the article. HQD co-wrote the
article. Both authors revised the article and approved the final
version.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Torre LA, Siegel RL, Ward EM and Jemal A:
Global cancer incidence and mortality rates and trends-an update.
Cancer Epidemiol Biomarkers Prev. 25:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burt R: Inheritance of colorectal cancer.
Drug Discov Today Dis Mech. 4:293–300. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hendon SE and DiPalma JA: U.S. practices
for colon cancer screening. Keio J Med. 54:179–183. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markowitz SD and Bertagnolli MM: Molecular
origins of cancer: Molecular basis of colorectal cancer. N Engl J
Med. 361:2449–2460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsang AH, Cheng KH, Wong AS, Ng SS, Ma BB,
Chan CM, Tsui NB, Chan LW, Yung BY and Wong SC: Current and future
molecular diagnostics in colorectal cancer and colorectal adenoma.
World J Gastroenterol. 20:3847–3857. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grady WM and Pritchard CC: Molecular
alterations and biomarkers in colorectal cancer. Toxicol Pathol.
42:124–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fransén K, Klintenäs M, Österström A,
Dimberg J, Monstein HJ and Söderkvist P: Mutation analysis of the
BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas.
Carcinogenesis. 25:527–533. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pino MS and Chung DC: The chromosomal
instability pathway in colon cancer. Gastroenterology.
138:2059–2072. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lynch HT and de la Chapelle A: Hereditary
colorectal cancer. N Engl J Med. 348:919–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fearon ER and Vogelstein B: A genetic
model for colorectal tumorigenesis. Cell. 61:759–767. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grady WM: Epigenetic events in the
colorectum and in colon cancer. Biochem Soc Trans. 33:684–688.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kinzler KW and Vogelstein B: Lessons from
hereditary colorectal cancer. Cell. 87:159–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tejpar S and Van Cutsem E: Molecular and
genetic defects in colorectal tumorigenesis. Best Pract Res Clin
Gastroenterol. 16:171–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
O'Connell JB, Maggard MA and Ko CY: Colon
cancer survival rates with the new American Joint Committee on
Cancer sixth edition staging. J Natl Cancer Inst. 96:1420–1425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reimers MS, Zeestraten EC, Kuppen PJ,
Liefers GJ and van de Velde CJ: Biomarkers in precision therapy in
colorectal cancer. Gastroenterol Rep (Oxf). 1:166–183. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nachman MW and Crowell SL: Estimate of the
mutation rate per nucleotide in humans. Genetics. 156:297–304.
2000.PubMed/NCBI
|
|
20
|
Roach JC, Glusman G, Smit AF, Huff CD,
Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et
al: Analysis of genetic inheritance in a family quartet by
whole-genome sequencing. Science. 328:636–639. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loeb LA, Loeb KR and Anderson JP: Multiple
mutations and cancer. Proc Natl Acad Sci USA. 100:776–781. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Markowitz S, Wang J, Myeroff L, Parsons R,
Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein
B, et al: Inactivation of the type II TGF-beta receptor in colon
cancer cells with microsatellite instability. Science.
268:1336–1338. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Samuels Y, Wang Z, Bardelli A, Silliman N,
Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riqqins GJ, et al:
High frequency of mutations of the PIK3CA gene in human cancers.
Science. 304:5542004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baker SJ, Fearon ER, Nigro JM, Hamilton
SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF,
Nakamura Y, et al: Chromosome 17 deletions and p53 gene mutations
in colorectal carcinomas. Science. 244:217–221. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thiagalingam S, Lengauer C, Leach FS,
Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton
SR, Kern SE, et al: Evaluation of candidate tumour suppressor genes
on chromosome 18 in colorectal cancers. Nat Genet. 13:343–346.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Diep CB, Kleivi K, Ribeiro FR, Teixeira
MR, Lindgjærde OC and Lothe RA: The order of genetic events
associated with colorectal cancer progression inferred from
meta-analysis of copy number changes. Genes Chromosomes Cancer.
45:31–41. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jasmine F, Rahaman R, Dodsworth C, Roy S,
Paul R, Raza M, Paul-Brutus R, Kamal M, Ahsan H and Kibriya MG: A
genome-wide study of cytogenetic changes in colorectal cancer using
SNP microarrays: Opportunities for future personalized treatment.
PLoS One. 7:e319682012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baudis M: Genomic imbalances in 5918
malignant epithelial tumors: An explorative meta-analysis of
chromosomal CGH data. BMC Cancer. 7:2262007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones AM, Douglas EJ, Halford SE, Fiegler
H, Gorman PA, Roylance RR, Carter NP and Tomlinson IP: Array-CGH
analysis of microsatellite-stable, near-diploid bowel cancers and
comparison with other types of colorectal carcinoma. Oncogene.
24:118–129. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zarzour P, Boelen L, Luciani F, Beck D,
Sakthianandeswaren A, Mouradov D, Sieber OM, Hawkins NJ, Hesson LB,
Ward RL and Wong JW: Single nucleotide polymorphism array profiling
identifies distinct chromosomal aberration patterns across
colorectal adenomas and carcinomas. Genes Chromosomes Cancer.
54:303–314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Foulds L: The natural history of cancer. J
Chronic Dis. 8:2–37. 1958. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nowell PC: The clonal evolution of tumor
cell populations. Science. 194:23–28. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nguyen HT, Geens M and Spits C: Genetic
and epigenetic instability in human pluripotent stem cells. Hum
Reprod Update. 19:187–205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lund RJ, Närvä E and Lahesmaa R: Genetic
and epigenetic stability of human pluripotent stem cells. Nat Rev
Genet. 13:732–744. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
International Stem Cell Initiative, . Amps
K, Andrews PW, Anyfantis G, Armstrong L, Avery S, Baharvand H,
Baker J, Barker D, Munoz MB, et al: Screening ethnically diverse
human embryonic stem cells identifies a chromosome 20 minimal
amplicon conferring growth advantage. Nat Biotechnol. 29:1132–1144.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nguyen HT, Geens M, Mertzanidou A, Jacobs
K, Heirman C, Breckpot K and Spits C: Gain of 20q11.21 in human
embryonic stem cells improves cell survival by increased expression
of Bcl-xL. Mol Hum Reprod. 20:168–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Avery S, Hirst AJ, Baker D, Lim CY,
Alagaratnam S, Skotheim RI, Lothe RA, Pera MF, Colman A, Robson P,
et al: BCL-XL Mediates the strong selective advantage of a 20q11.21
amplification commonly found in human embryonic stem cell cultures.
Stem Cell Reports. 1:379–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Beroukhim R, Mermel CH, Porter D, Wei G,
Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J,
Urashima M, et al: The landscape of somatic copy-number alteration
across human cancers. Nature. 463:899–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ogino S, Nosho K, Irahara N, Shima K, Baba
Y, Kirkner GJ, Meyerhardt JA and Fuchs CS: Prognostic significance
and molecular associations of 18q loss of heterozygosity: A cohort
study of microsatellite stable colorectal cancers. J Clin Oncol.
27:4591–4598. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sheffer M, Bacolod MD, Zuk O, Giardina SF,
Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA and Domany E:
Association of survival and disease progression with chromosomal
instability: A genomic exploration of colorectal cancer. Proc Natl
Acad Sci USA. 106:7131–7136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jen J, Kim H, Piantadosi S, Liu ZF, Levitt
RC, Sistonen P, Kinzler KW, Vogelstein B and Hamilton SR: Allelic
loss of chromosome 18q and prognosis in colorectal cancer. N Engl J
Med. 331:213–221. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zauber P, Sabbath-solitare M, Marotta SP
and Bishop T: Loss of heterozygosity for chromosome 18q and
microsatellite instability are highly consistent across the region
of the DCC and SMAD4 genes in colorectal carcinomas and adenomas. J
Appl Res. 8:14–23. 2008.
|
|
45
|
Fearon ER, Cho KR, Nigro JM, Kern SE,
Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G,
Kinzler KW, et al: Identification of a chromosome 18q gene that is
altered in colorectal cancers. Science. 247:49–56. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mehlen P and Fearon ER: Role of the
dependence receptor DCC in colorectal cancer pathogenesis. J Clin
Oncol. 22:3420–3428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Alazzouzi H, Alhopuro P, Salovaara R,
Sammalkorpi H, Järvinen H, Mecklin JP, Hemminki A, Schwartz S Jr,
Aaltonen LA and Arango D: SMAD4 as a prognostic marker in
colorectal cancer. Clin Cancer Res. 11:2606–2611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Grady WM: Genomic instability and colon
cancer. Cancer Metastasis Rev. 23:11–27. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shi Y, Hata A, Lo RS, Massagué J and
Pavletich NP: A structural basis for mutational inactivation of the
tumour suppressor Smad4. Nature. 388:87–93. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hahn SA, Schutte M, Hoque AT, Moskaluk CA,
da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hurban
RH and Kern SE: DPC4, a candidate tumor suppressor gene at human
chromosome 18q21.1. Science. 271:350–353. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Takagi Y, Kohmura H, Futamura M, Kida H,
Tanemura H, Shimokawa K and Saji S: Somatic alterations of the DPC4
gene in human colorectal cancers in vivo. Gastroenterology.
111:1369–1372. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Takagi Y, Koumura H, Futamura M, Aoki S,
Ymaguchi K, Kida H, Tanemura H, Shimokawa K and Saji S: Somatic
alterations of the SMAD-2 gene in human colorectal cancers. Br J
Cancer. 78:1152–1155. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fleming NI, Jorissen RN, Mouradov D,
Christie M, Sakthianandeswaren A, Palmieri M, Day F, Li S, Tsui C,
Lipton L, et al: SMAD2, SMAD3 and SMAD4 mutations in colorectal
cancer. Cancer Res. 73:725–735. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Takebayashi S, Ogawa T, Jung KY, Muallem
A, Mineta H, Fisher SG, Grenman R and Carey TE: Identification of
new minimally lost regions on 18q in head and neck squamous cell
carcinoma. Cancer Res. 60:3397–3403. 2000.PubMed/NCBI
|
|
55
|
Powell SM, Zilz N, Beazer-Barclay Y, Bryan
TM, Hamilton SR, Thibodeau SN, Vogelstein B and Kinzler KW: APC
mutations occur early during colorectal tumorigenesis. Nature.
359:235–237. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Stanczak A, Stec R, Bodnar L, Olszewski W,
Cichowicz M, Kozlowski W, Szcylik C, Pietrucha T, Wieczorek M and
Lamparska-Pzybysz M: Prognostic significance of Wnt-1, β-catenin
and E-cadherin expression in advanced colorectal carcinoma. Pathol
Oncol Res. 17:955–963. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of β-catenin-Tcf
signaling in colon cancer by mutations in beta-catenin or APC.
Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu W, Dong X, Mai M, Seelan RS, Taniguchi
K, Krishnadath KK, Halling KC, Cunningham JM, Boardman LA, Qian C,
et al: Mutations in AXIN2 cause colorectal cancer with defective
mismatch repair. Nat Genet. 26:146–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Coppedè F, Lopomo A, Spisni R and Migliore
L: Genetic and epigenetic biomarkers for diagnosis, prognosis and
treatment of colorectal cancer. World J Gastroenterol. 20:943–956.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kapitanović S, Cacev T, Radosević S,
Spaventi S, Spaventi R and Pavelić K: APC gene loss of
heterozygosity, mutations, E1317Q, and I1307K germ-line variants in
sporadic colon cancer in Croatia. Exp Mol Pathol. 77:193–200. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Esteller M: Epigenetic lesions causing
genetic lesions in human cancer: Promoter hypermethylation of DNA
repair genes. Eur J Cancer. 36:2294–2300. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Levine AJ: P53, the cellular gatekeeper
for growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
el-Deiry WS: Regulation of p53 downstream
genes. Semin Cancer Biol. 8:345–357. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li XL, Zhou J, Chen ZR and Chng WJ: P53
mutations in colorectal cancer-molecular pathogenesis and
pharmacological reactivation. World J Gastroenterol. 21:84–93.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Leslie A, Carey FA, Pratt NR and Steele
RJ: The colorectal adenoma-carcinoma sequence. Br J Surg.
89:845–860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Takayama T, Miyanishi K, Hayashi T, Sato Y
and Niitsu Y: Colorectal cancer: Genetics of development and
metastasis. J Gastroenterol. 41:185–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sigal A and Rotter V: Oncogenic mutations
of the p53 tumor suppressor: The demons of the guardian of the
genome. Cancer Res. 60:6788–6793. 2000.PubMed/NCBI
|
|
69
|
Liu Y and Bodmer WF: Analysis of P53
mutations and their expression in 56 colorectal cancer cell lines.
Proc Natl Acad Sci USA. 103:976–981. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Béroud C and Soussi T: The UMD-p53
database: New mutations and analysis tools. Hum Mutat. 21:176–181.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Vigil D, Cherfils J, Rossman KL and Der
CJ: Ras superfamily GEFs and GAPs: Validated and tractable targets
for cancer therapy? Nat Rev Cancer. 10:842–857. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Adjei AA: Ras signaling pathway proteins
as therapeutic targets. Curr Pharm Des. 7:1581–1594. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the catalogue of somatic
mutations in cancer. Nucleic Acids Res. 39:(Database issue).
D945–D950. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tan C and Du X: KRAS mutation testing in
metastatic colorectal cancer. World J Gastroenterol. 18:5171–5180.
2012.PubMed/NCBI
|
|
77
|
Conlin A, Smith G, Carey FA, Wolf CR and
Steele RJ: The prognostic significance of K-ras, p53, and APC
mutations in colorectal carcinoma. Gut. 54:1283–1286. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Phipps AI, Buchanan DD, Makar KW, Win AK,
Baron JA, Lindor NM, Potter JD and Newcomb PA: KRAS-mutation status
in relation to colorectal cancer survival: The joint impact of
correlated tumour markers. Br J Cancer. 108:1757–1764. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cejas P, López-Gómez M, Aguayo C, Madero
R, de Castro Carpeño J, Belda-Iniesta C, Barriuso J, García Moreno
V, Larrauri J, López R, et al: KRAS mutations in primary colorectal
cancer tumors and related metastases: A potential role in
prediction of lung metastasis. PLoS One. 4:e81992009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kim HS, Heo JS, Lee J, Lee JY, Lee MY, Lim
SH, Lee WY, Kim SH, Park YA, Cho YB, et al: The impact of KRAS
mutations on prognosis in surgically resected colorectal cancer
patients with liver and lung metastases: A retrospective analysis.
BMC Cancer. 16:1202016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Nash GM, Gimbel M, Shia J, Nathanson DR,
Ndubuisi MI, Zeng ZS, Kemeny N and Paty PB: KRAS mutation
correlates with accelerated metastatic progression in patients with
colorectal liver metastases. Ann Surg Oncol. 17:572–578. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Inoue Y, Saigusa S, Iwata T, Okugawa Y,
Toiyama Y, Tanaka K, Uchida K, Mohri Y and Kusunoki M: The
prognostic value of KRAS mutations in patients with colorectal
cancer. Oncol Rep. 28:1579–1584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suqqs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lièvre A, Bachet JB, Boige V, Cayre A, Le
Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, et al: KRAS
mutations as an independent prognostic factor in patients with
advanced colorectal cancer treated with cetuximab. J Clin Oncol.
26:374–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD,
Robitalle S, et al: K-ras mutations and benefit from cetuximab in
advanced colorectal cancer. N Engl J Med. 359:1757–1765. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Siena S, Sartore-Bianchi A, Di
Nicolantonio F, Balfour J and Bardelli A: Biomarkers predicting
clinical outcome of epidermal growth factor receptor-targeted
therapy in metastatic colorectal cancer. J Natl Cancer Inst.
101:1308–1324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Bos JL, Fearon ER, Hamilton SR, Verlaan-de
Vries M, van Boom JH, van der Eb AJ and Vogelstein B: Prevalence of
ras gene mutations in human colorectal cancers. Nature.
327:293–297. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Forrester K, Almoguera C, Han K, Grizzle
WE and Perucho M: Detection of high incidence of K-ras oncogenes
during human colon tumorigenesis. Nature. 327:298–303. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Fernández-Medarde A and Santos E: Ras in
cancer and developmental diseases. Genes Cancer. 2:344–358. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Neumann J, Zeindl-Eberhart E, Kirchner T
and Jung A: Frequency and type of KRAS mutations in routine
diagnostic analysis of metastatic colorectal cancer. Pathol Res
Pract. 205:858–862. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Irahara N, Baba Y, Nosho K, Shima K, Yan
L, Dias-Santagata D, Iafrate AJ, Fuchs CS, Haigis KM and Ogino S:
NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol.
19:157–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Vaughn CP, ZoBell SD, Furtado LV, Baker CL
and Samowitz WS: Frequency of KRAS, BRAF, and NRAS mutations in
colorectal cancer. Genes Chromosomes Cancer. 50:307–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kosmidou V, Oikonomou E, Vlassi M,
Avlonitis S, Katseli A, Tsipras I, Mourtzoukou D, Kontogeorgos G,
Zografos G and Pintzas A: Tumor heterogeneity revealed by KRAS,
BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor
mutation profile differences and their therapeutic implications.
Hum Mutat. 35:329–340. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Abdel-Rahman WM and Peltomäki P: Molecular
basis and diagnostics of hereditary colorectal cancers. Ann Med.
36:379–388. 2014. View Article : Google Scholar
|
|
95
|
Thibodeau SN, Bren G and Schaid D:
Microsatellite instability in cancer of the proximal colon.
Science. 260:816–819. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Boland CR and Goel A: Somatic evolution of
cancer cells. Semin Cancer Biol. 15:436–450. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Boland CR, Thibodeau SN, Hamilton SR,
Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA,
Fodde R, Ranzani GN and Srivastava S: A national cancer institute
workshop on microsatellite instability for cancer detection and
familial predisposition: Development of international criteria for
the determination of microsatellite instability in colorectal
cancer. Cancer Res. 58:5248–5257. 1998.PubMed/NCBI
|
|
98
|
Findeisen P, Kloor M, Merx S, Sutter C,
Woerner SM, Dostmann N, Benner A, Dondog B, Pawlita M, Dippold W,
et al: T25 repeat in the 3′ untranslated region of the CASP2 gene:
A sensitive and specific marker for microsatellite instability in
colorectal cancer. Cancer Res. 65:8072–8078. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Aaltonen LA, Peltomäki P, Leach FS,
Sistonen P, Pylkkänen L, Mecklin JP, Järvinen H, Powell SM, Jen J,
Hamilton SR, et al: Clues to the pathogenesis of familial
colorectal cancer. Science. 260:812–816. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Jiricny J: The multifaceted
mismatch-repair system. Nat Rev Mol Cell Biol. 7:335–346. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Pal T, Permuth-Wey J and Sellers TA: A
review of the clinical relevance of mismatch-repair deficiency in
ovarian cancer. Cancer. 113:733–742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Grady WM and Carethers JM: Genomic and
epigenetic instability in colorectal cancer pathogenesis.
Gastroenterology. 135:1079–1099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Hudler P: Genetic aspects of gastric
cancer instability. Scientific World Journal. 2012:7619092012.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Perucho M: Cancer of the microsatellite
mutator phenotype. Biol Chem. 377:675–684. 1996.PubMed/NCBI
|
|
105
|
Mori Y, Yin J, Rashid A, Leggett BA, Young
J, Simms L, Kuehl PM, Langenberg P, Meltzer SJ and Stine OC:
Instabilotyping: Comprehensive identification of frameshift
mutations caused by coding region microsatellite instability.
Cancer Res. 61:6046–6049. 2001.PubMed/NCBI
|
|
106
|
Parsons R, Myeroff LL, Liu B, Wilison JK
V, Markowitz SD, Kinzler KW and Vogelstein B: Microsatellite
instability and mutations of the transforming growth factor β type
II receptor gene in colorectal cancer. Cancer Res. 55:5548–5550.
1995.PubMed/NCBI
|
|
107
|
Boland CR and Goel A: Microsatellite
instability in colorectal cancer. Gastroenterology.
138:2073–2087.e3. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Toyota M, Ahuja N, Ohe-Toyota M, Herman
JG, Baylin SB and Issa JP: CpG island methylator phenotype in
colorectal cancer. Proc Natl Acad Sci USA. 96:8681–8686. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lao VV and Grady WM: Epigenetics and
colorectal cancer. Nat Rev Gastroenterol Hepatol. 8:686–700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Laird PW: Cancer epigenetics. Hum Mol
Genet. 14:R65–R76. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Jass JR: Serrated adenoma of the
colorectum and the DNA-methylator phenotype. Nat Clin Pract Oncol.
2:398–405. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Samowitz WS, Albertsen H, Herrick J, Levin
TR, Sweeney C, Murtaugh MA, Wolff RK and Slattery ML: Evaluation of
a large, population-based sample supports a CpG island methylator
phenotype in colon cancer. Gastroenterology. 129:837–845. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ogino S, Cantor M, Kawasaki T, Brahmandam
M, Kirkner GJ, Weisenberger DJ, Campan M, Laird PW, Loda M and
Fuchs CS: CpG island methylator phenotype (CIMP) of colorectal
cancer is best characterised by quantitative DNA methylation
analysis and prospective cohort studies. Gut. 55:1000–1006. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Weisenberger DJ, Siegmund KD, Campan M,
Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D,
Buchanan D, et al: CpG island methylator phenotype underlies
sporadic microsatellite instability and is tightly associated with
BRAF mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Worthley DL and Leggett BA: Colorectal
cancer: Molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
117
|
Ogino S, Kawasaki T, Kirkner GJ, Ohnishi M
and Fuchs CS: 18q loss of heterozygosity in microsatellite stable
colorectal cancer is correlated with CpG island methylator
phenotype-negative (CIMP-0) and inversely with CIMP-low and
CIMP-high. BMC Cancer. 7:722007. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ogino S, Kawasaki T, Kirkner GJ, Kraft P,
Loda M and Fuchs CS: Evaluation of markers for CpG island
methylator phenotype (CIMP) in colorectal cancer by a large
population-based sample. J Mol Diagn. 9:305–314. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Toyota M, Ohe-Toyota M, Ahuja N and Issa
JP: Distinct genetic profiles in colorectal tumors with or without
the CpG island methylator phenotype. Proc Natl Acad Sci USA.
97:710–715. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Kambara T, Simms LA, Whitehall VLJ, Spring
KJ, Wynter CVA, Walsh MD, Barker MA, Arnold S, McGivern A,
Matsubara N, et al: BRAF mutation is associated with DNA
methylation in serrated polyps and cancers of the colorectum. Gut.
53:1137–1144. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Hawkins N, Norrie M, Cheong K, Mokany E,
Ku SL, Meagher A, OConnor T and Ward R: CpG island methylation in
sporadic colorectal cancers and its relationship to microsatellite
instability. Gastroenterology. 122:1376–1387. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Shen L, Toyota M, Kondo Y, Lin E, Zhang L,
Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, et al: Integrated
genetic and epigenetic analysis identifies three different
subclasses of colon cancer. Proc Natl Acad Sci USA.
104:18654–18659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Munro AJ, Lain S and Lane DP: P53
abnormalities and outcomes in colorectal cancer: A systematic
review. Br J Cancer. 92:434–444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Van Cutsem E, Peeters M, Siena S, Humblet
Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J,
Richardson G, et al: Open-label phase III trial of panitumumab plus
best supportive care compared with best supportive care alone in
patients with chemotherapy-refractory metastatic colorectal cancer.
J Clin Oncol. 25:1658–1664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Heinemann V, Stintzing S, Kirchner T,
Boeck S and Jung A: Clinical relevance of EGFR- and KRAS-status in
colorectal cancer patients treated with monoclonal antibodies
directed against the EGFR. Cancer Treat Rev. 35:262–271. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Boland CR: The molecular biology of
gastrointestinal cancer: Implications for diagnosis and therapy.
Gastrointest Endosc Clin N Am. 18:401–413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Sinicrope FA, Foster NR, Thibodeau SN,
Marsoni S, Monges G, Labianca R, Kim GP, Yothers G, Allegra C,
Moore MJ, et al: DNA mismatch repair status and colon cancer
recurrence and survival in clinical trials of 5-fluorouracil-based
adjuvant therapy. J Natl Cancer Inst. 103:863–875. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Roth AD, Delorenzi M, Tejpar S, Yan P,
Klingbiel D, Fiocca R, d'Ario G, Cisar L, Labianca R, Cunningham D,
et al: Integrated analysis of molecular and clinical prognostic
factors in stage II/III colon cancer. J Natl Cancer Inst.
104:1635–1646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Popat S, Hubner R and Houlston RS:
Systematic review of microsatellite instability and colorectal
cancer prognosis. J Clin Oncol. 23:609–618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Al-Sohaily S, Biankin A, Leong R,
Kohonen-Corish M and Warusavitarne J: Molecular pathways in
colorectal cancer. J Gastroenterol Hepatol. 27:1423–1431. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ribic CM, Sargent DJ, Moore MJ, Thibodeau
SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R,
Shepherd LE, et al: Tumor microsatellite-instability status as a
predictor of benefit from fluorouracil-based adjuvant chemotherapy
for colon cancer. N Engl J Med. 349:247–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Sargent DJ, Marsoni S, Monges G, Thibodeau
SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri
V, et al: Defective mismatch repair as a predictive marker for lack
of efficacy of fluorouracil-based adjuvant therapy in colon cancer.
J Clin Oncol. 28:3219–3226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Yang L, Sun Y, Huang XE, Yu DS, Zhou JN,
Zhou X, Li DZ and Guan X: Carcinoma microsatellite instability
status as a predictor of benefit from fluorouracil-based adjuvant
chemotherapy for stage II rectal cancer. Asian Pacific J Cancer
Prev. 16:1545–1551. 2015. View Article : Google Scholar
|
|
134
|
Tejpar S, Saridaki Z, Delorenzi M, Bosman
F and Roth AD: Microsatellite instability, prognosis and drug
sensitivity of stage II and III colorectal cancer: More complexity
to the puzzle. J Natl Cancer Inst. 103:841–844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Des Guetz G, Schischmanoff O, Nicolas P,
Perret GY, Morere JF and Uzzan B: Does microsatellite instability
predict the efficacy of adjuvant chemotherapy in colorectal cancer?
A systematic review with meta-analysis. Eur J Cancer. 45:1890–1896.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
De Sousa E, Melo F, Wang X, Jansen M,
Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van
Leersum R, Bijlsma MF, et al: Poor-prognosis colon cancer is
defined by a molecularly distinct subtype and develops from
serrated precursor lesions. Nat Med. 19:614–618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sadanandam A, Lyssiotis CA, Homicsko K,
Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA,
Grotzinger C, Del Rio M, et al: A colorectal cancer classification
system that associates cellular phenotype and responses to therapy.
Nat Med. 19:619–625. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
De Rosa M, Pace U, Rega D, Costabile V,
Duraturo F, Izzo P and Delrio P: Genetics, diagnosis and management
of colorectal cancer (Review). Oncol Rep. 34:1087–1096. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Su S, Hu B, Shao J, Shen B, Du J, Du Y,
Zhou J, Yu L, Zhang L, Chen F, et al: CRISPR-Cas9 mediated
efficient PD-1 disruption on human primary T cells from cancer
patients. Sci Rep. 6:200702016. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Liao Y, Chen L, Feng Y, Shen J, Gao Y,
Cote G, Choy E, Harmon D, Mankin H, Hornicek F and Duan Z:
Targeting programmed cell death ligand 1 by CRISPR/Cas9 in
osteosarcoma cells. Oncotarget. 8:30276–30287. 2017. View Article : Google Scholar : PubMed/NCBI
|