Introduction
Circadian rhythms are biological rhythms with a
periodicity close to 24 h. In mammals, these rhythms are generated
by a master biological clock, located in the hypothalamus and by
clocks localized in peripheral cells (1). At the molecular level, circadian clocks
are operated by transcriptional/translational feedback loops
involving a set of genes called ‘clock genes’ (2,3). Clock
genes operate like oscillators, modulating rhythmic transcription
of a large number of genes in all cells by a mechanism that relies
on coordinated chromatin remodeling events (4,5).
Situations that alter the normal expression pattern of the clock
genes have important repercussions at the systemic, cellular and
molecular levels (6).
The maintenance of the molecular mechanism of the
circadian clockwork has a profound influence on human health, and
its alteration has frequently been associated with multiple
diseases, including cancer (7–12). Several
in vivo and in vitro studies suggest that, in
addition to their main role within the molecular mechanism of the
circadian clock, Period circadian regulator (Per)1 and Per2 genes
can also function as tumor suppressors due to their involvement in
cell proliferation, apoptosis, cell cycle control, and DNA damage
response (13–22). Targeted ablation of Per2 leads to the
development of malignant lymphomas (13), whereas its ectopic expression in
cancer cell lines results in growth inhibition, cell cycle arrest,
apoptosis, and loss of clonogenic ability (15,18).
Accumulating evidence suggests that deregulation or
significantly decreased expression of Per1 and Per2 genes in humans
is associated with increased risk of breast, prostate, ovarian,
endometrial, pancreatic, colorectal, gastric, liver, skin, lung,
and head and neck cancers, leukemia, lymphomas, and glioma
(23–41). Decreased expression of Per1 or Per2
genes has been associated with promoter hypermethylation in breast,
endometrial, and non-small lung cancer cells (23,27,35). On
the other hand, treatment of non-small cell lung cancer cells and
other cancer cell lines with SAHA, an HDACi, induce the expression
of Per1 gene (35), whereas TSA
induced the expression of Per3 in myeloid leukemia cells (41); however, the role of HDACi on Per2
expression has not been tested, neither their role on Per1 and Per2
expression in gastric cancer cells. Studies in rodents have shown
that histone H3 acetylation is of great relevance to maintain the
activation and rhythmic expression of clock genes in liver cells
(42).
Despite this evidence, the transcriptional
regulation of Per1 and Per2 genes by epigenetic modifications is
not fully understood, and the role of HDACi on Per1 and Per2
expression in gastric cancer cells has not been explored.
Therefore, the aim of this study was to investigate whether HDACi
regulate the expression of Per1 and Per2 genes in two human gastric
cancer cell lines, and to determine histone-specific modifications
in response to the HDACi treatment.
Materials and methods
Cell culture and treatments with
HDACi
KATO III and NCI-N87 human gastric carcinoma cells
were acquired from ATCC (Manassas, VA, USA). KATO III cells were
grown in Iscove's modified Dulbecco's medium (IMDM) supplemented
with 20% fetal bovine serum, 0.5% penicillin-streptomycin, and 70
mg/l kanamycin. NCI-N87 cells were grown in RPMI-1640 supplemented
with 10% fetal bovine serum, 0.5% penicillin-streptomycin and 70
mg/l kanamycin. Both cell lines were grown at 37°C in a humidified
5% CO2/95% air atmosphere. Exponentially growing cells
were trypsinized and seeded in 6-well plates; when cells reached
70–80% confluence by microscopic examination (day 2 or 3
post-plating), the medium was changed, and sodium butyrate (NaB)
(1, 2 or 3 mM) or trichostatin A (TSA) (50, 100 or 150 nM) were
added. Cells were treated during 48 or 96 h with these reagents,
replacing the medium with inhibitors every 24 h.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
KATO III and NCI-N87 cells treated during 48 or 96 h
as described above, were washed twice with 1× PBS, then 1 ml of
Trizol reagent was added to isolate total cellular RNA, according
to the manufacturer's recommendations (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). RNA concentration was
determined with a NanoDrop 1000 (Thermo Fisher Scientific, Inc.),
and the RNA integrity by agarose gel electrophoresis.
Total RNA (1 µg) was reverse-transcribed using 200 U
of M-MLV reverse transcriptase and 50 pmoles of random hexamers in
a final volume of 20 µl, according to the instructions provided by
the manufacturer (Invitrogen; Thermo Fisher Scientific, Inc.).
RT-qPCR reactions were performed in triplicate,
containing 6 µl of 2× SYBR Green qPCR reaction mix (Invitrogen;
Thermo Fisher Scientific, Inc.), 1 µl of cDNA from each sample, and
5 pmol of each primer (forward and reverse) for Per1 or Per2
(Table I), in a final volume of 12
µl. RT-qPCR reactions were run as follow: 2 min at 50°C, 8.5 min at
95°C, 40 cycles of 95°C for 15 sec, 60°C for 1 min, followed by a
dissociation analysis, in a 7500 thermal cycler (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Reactions with primers
for GAPDH and ACTB were used as internal control (Table I). The efficiencies of RT-qPCR
reactions were determined with the LinReg program (43), and the relative mRNA expression levels
were calculated according to the method described by Pfaffl
(44). The expression of Per1 and
Per2 genes in control cells, without treatment, was considered as
one.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction and promoter
analysis of Per1 and Per2 genes. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction and promoter
analysis of Per1 and Per2 genes.
| Gene | Genbank accession
no. | Forward
(5′-3′) | Reverse
(5′-3′) | Position |
|---|
| PER1 | NM_002616.2 |
GGACATGACCTCTGTGCTGA |
CATCAGGGTGACCAGGATCT | 3,688–3,887 |
| PER2 | NM_022817.2 |
ACAGCTTTGGCTTCTGGTGT |
TATTGGCCATCATGGTCTGA | 4,574–4,774 |
| GAPDH | NM_002046.5 |
GTCAGTGGTGGACCTGACCT |
TGAGGAGGGGAGATTCAGTG | 908-1,307 |
| ACTB | NM_001101.3 |
TCCCTGGAGAAGAGCTACGA |
AGCACTGTGTTGGCGTACAG | 787–980 |
| PER1p | – |
TGTCTCTCCCCTCCTCTCAAa |
AGATACGCTGCGCCTCTTTAa | −437 to −241 |
| PER2p | – |
AGGAACCGACGAGGTGAAC |
CCGCTGTCACATAGTGGAAA | −410 to −217 |
Chromatin immunoprecipitation (ChIP)
assays
Chromatin immunoprecipitation (ChIP) assays were
performed as previously described (45). After cross-linking, cell lysates were
sonicated on ice, soluble chromatin (equivalent to 50 µg of DNA)
was pre-cleared with protein A/G plus-agarose beads (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and then incubated overnight
at 4°C on a Nutator platform with 2 µg of antibodies to H3
modifications (Abcam, Cambridge, MA, USA), or with antibodies to
Sp1 and Sp1 (Santa Cruz Biotechnology, Inc.). The
immunoprecipitates were recovered by incubation with protein A/G
agarose beads and washed sequentially for 5 min with low-salt
buffer, high-salt buffer, LiCl wash buffer; and twice with TE
buffer. Immunoprecipitated DNA was eluted, then incubated for 30
min at 37°C with RNase A, followed by overnight incubation at 55°C
with proteinase K. DNA was reverse cross-linked and extracted with
phenol, phenol-chloroform, and chloroform-isoamyl alcohol, then
precipitated with ethanol and resuspended in 20 µl of
H2O. Input samples (equivalent to 5 µg of DNA) were
resuspended in 20 µl of H2O after reversal cross-linking
and ethanol precipitation. PCR was conducted with 1 µl of
immunoprecipitates or input samples, 10 µl of 2× PCR reaction mix
(Promega Corporation, Madison, WI, USA), 5 pmoles of each primer
for the Per1 or Per2 promoter (Table
I), 1 µl of DMSO, and water up to 20 µl; with the following
program: 3 min at 95°C, 30 cycles of 30 sec at 95°C, 30 sec at
60°C, 1 min at 72°C, and a final extension of 7 min at 72°C. PCR
products were separated on 2% agarose gels and visualized by
ethidium bromide staining with a XR 170–8170 photo documentation
station (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Methylation specific PCR (MS-PCR)
KATO III cells were treated with 3 mM NaB or 100 nM
TSA for 48 or 96 h with 5 µM of 5-Aza-2′-deoxycitydine (Aza). Cells
were then lysed and treated with sodium bisulfite, followed by the
DNA purification with the kit EZ-DNA Methylation-Direct (Zymo
Research Corp., Irvine, CA, USA), following the directions from the
manufacturer. Bisulfite-modified DNA was used to perform
methylation-specific PCR (MS-PCR) using specific primers to
distinguish methylated and unmethylated forms of Per1 and Per2
promoters, as reported (41). PCR
reactions were conducted with 1 µl of modified DNA, 10 µl of 2× PCR
mix (Promega Corporation), 5 pmoles of sense and antisense primers
in a final volume of 20 µl. Cycling parameters were as described
above, except that the annealing temperature was 56–62°C, according
to the Tm of each primer set (41).
PCR products were separated on 2% agarose gels, visualized and
documented as described above.
In silico analysis
In silico analysis to search for potential
Sp1 and Sp3 binding sites at the Per1 and Per2 promoters was done
using the JASPAR database (46).
Statistical analysis
RT-qPCR data were analyzed by the Kruskal-Wallis
test, except those obtained with Aza treatment that were analyzed
with a U-Mann Whitney test, as they did not fit a normal
distribution, whereas data from ChIP assays were analyzed with
one-way analysis of variance followed by a Tukey post-hoc test,
using the Statistica 7 software (StatSoft, Inc., Tulsa, OK, USA). A
P-value <0.05 was considered significant. Plots were done with
Sigmaplot 10.0 software (Systat Software, San Jose, CA USA).
Results
Treatment of KATO III cells with NaB
or TSA induce Per1 and Per2 mRNA expression
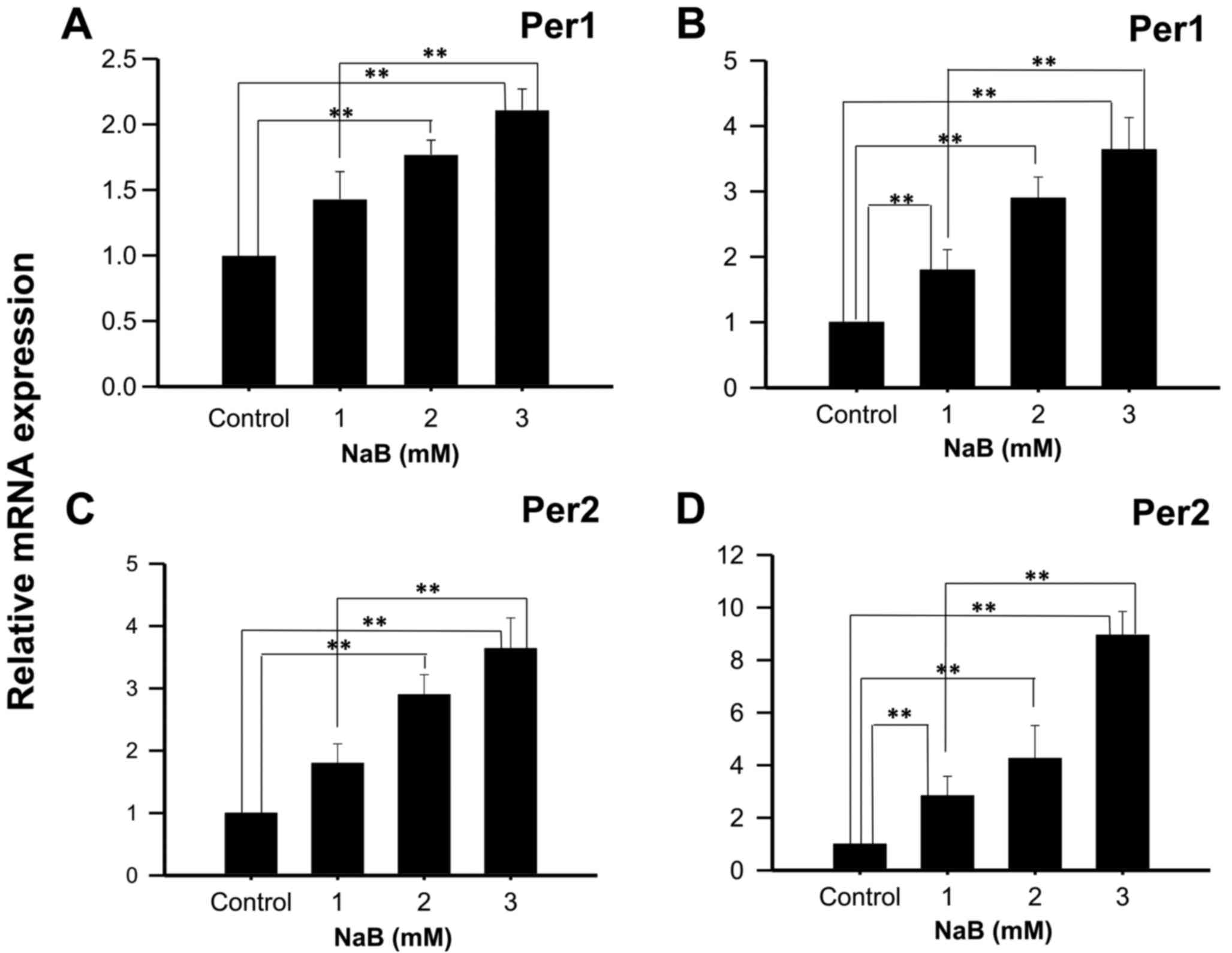
KATO III cells treated with 2 or 3 mM NaB during 48
h showed a significant increase in Per1 mRNA expression, compared
to control cells (1.77±0.11, and 2.11±0.16 fold higher,
respectively; P<0.01; Fig. 1A),
and with 1, 2 and 3 mM NaB during 96 h (2.89±0.60; 3.79±0.73, and
4.84±0.40 fold higher, respectively; P<0.01; Fig. 1B). Similarly, a significant induction
in Per2 mRNA expression was found in KATO III cells treated with 2
or 3 mM NaB for 48 h compared to control cells (2.90±0.32 and
3.64±0.49 fold higher, respectively; P<0.01; Fig. 1C), as well as with 1, 2 or 3 mM NaB
for 96 h (2.84±0.74; 4.36±1.25 and 8.85±1.03 fold higher,
respectively; P<0.01; Fig. 1D).
Significant differences were also found between cells treated with
1 and 3 mM NaB for 48- and 96-h (P<0.01).
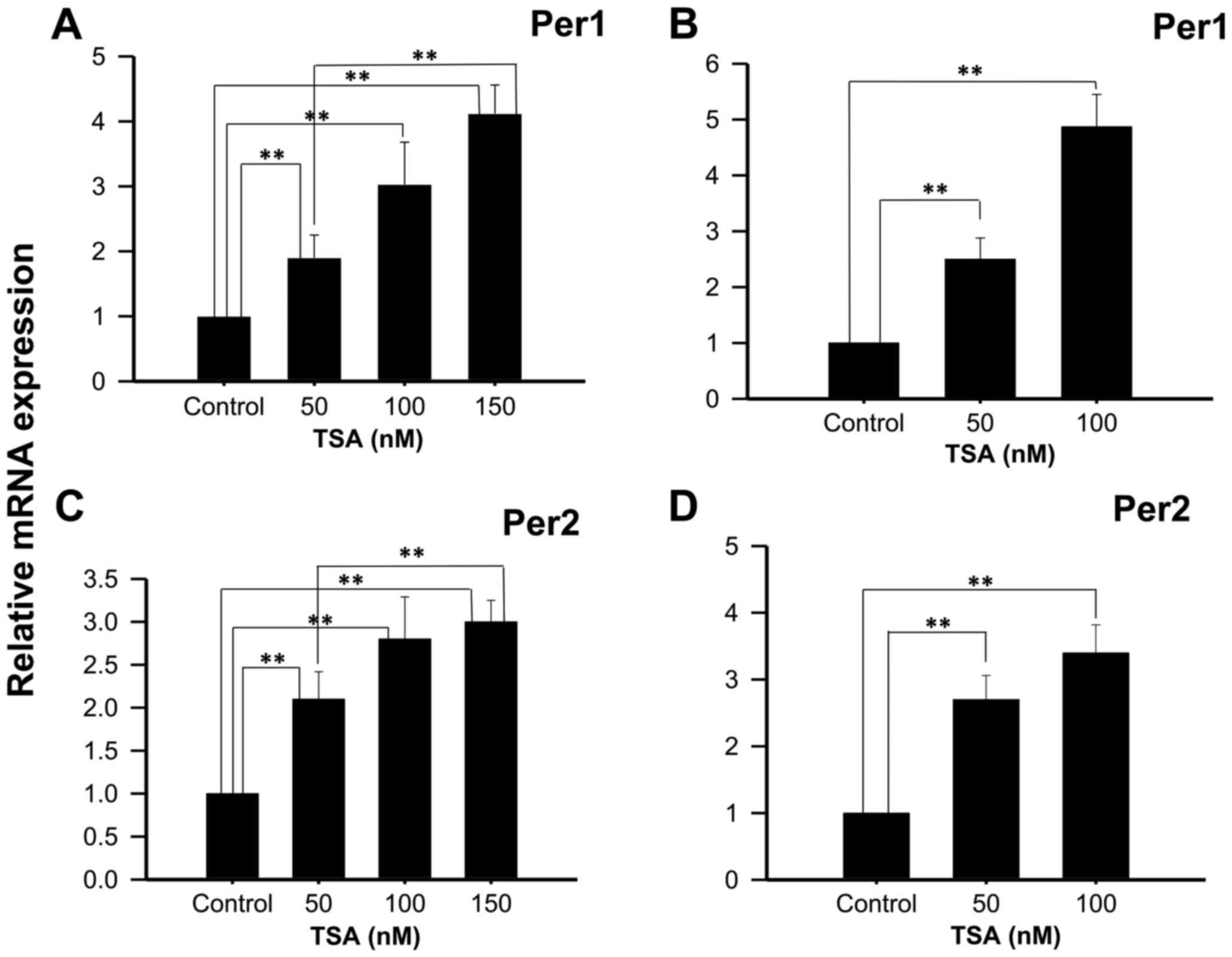
Experiments performed with 50, 100 or 150 nM TSA, a
potent and specific class I and IIa HDACi, show increased Per1 mRNA
expression at 48 h (1.90±0.35; 3.03±0.65 and 4.12±0.54 fold higher
than control untreated cells, respectively; P<0.01; Fig. 2A), as well as with 50 or 100 nM TSA
for 96 h (2.5±0.38 and 4.87±0.58 fold higher than control untreated
cells, respectively; P<0.01; Fig.
2B). Similarly, significant increases in Per2 mRNA expression
were found in KATO III cells treated with 50, 100 or 150 nM TSA for
48 h compared to control untreated cells (2.1±0.32±0.49 and
3±2.3±0.25, respectively; P<0.01; Fig.
2C), as well as with 50 or 100 nM TSA for 96 h compared to
control untreated cells (2.7±0.28; 3.4±0.33, respectively; Fig. 2D).
Treatment of NCI-N87 cells with NaB or
TSA induce the expression of Per1 and Per2 mRNA
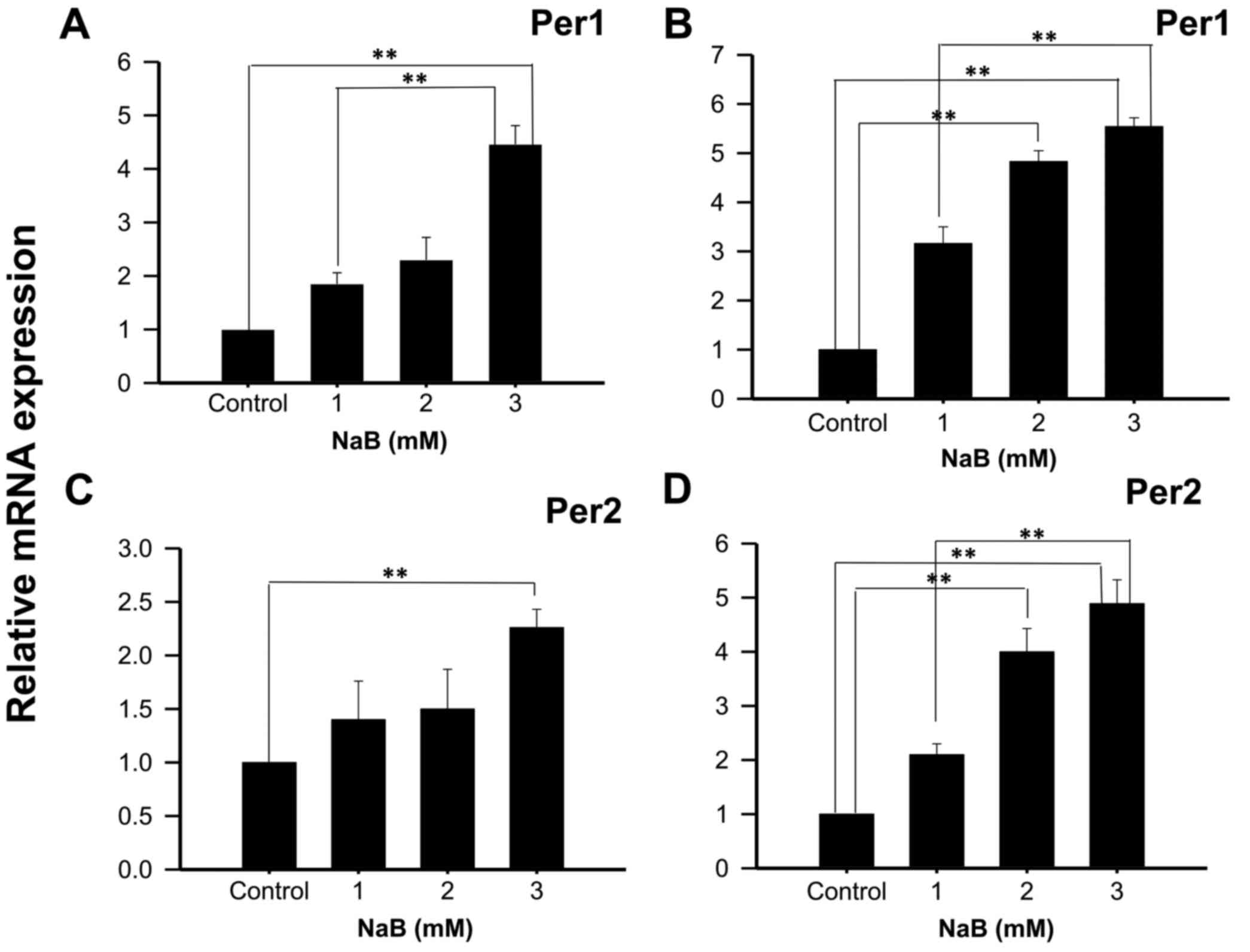
With the intention to show that the effect observed
in KATO III cells was not cell line-specific, Per1 and Per2 mRNA
expression was also analyzed in NCI-N87 cells treated with NaB or
TSA during 48 or 96 h. The results show a significant increase in
Per1 mRNA expression in cells treated with 3 mM NaB for 48 h
(4.46±0.35 fold higher than control untreated cells; P<0.01;
Fig. 3A). Extending the exposure time
to 96 h with 2 and 3 mM NaB induced Per1 expression (4.83±0.22 and
5.54±0.18 fold higher than control untreated cells, respectively;
P<0.01; Fig. 3B). Significant
differences were also found in Per2 mRNA expression with 3 mM NaB
at 48 h (2.26±0.17 fold higher than control untreated cells;
P<0.01; Fig. 3C), and with 2 and 3
mM at 96 h (4.00±0.38 and 4.89±0.36 fold higher than control
untreated cells, respectively; P<0.01; Fig. 3D).
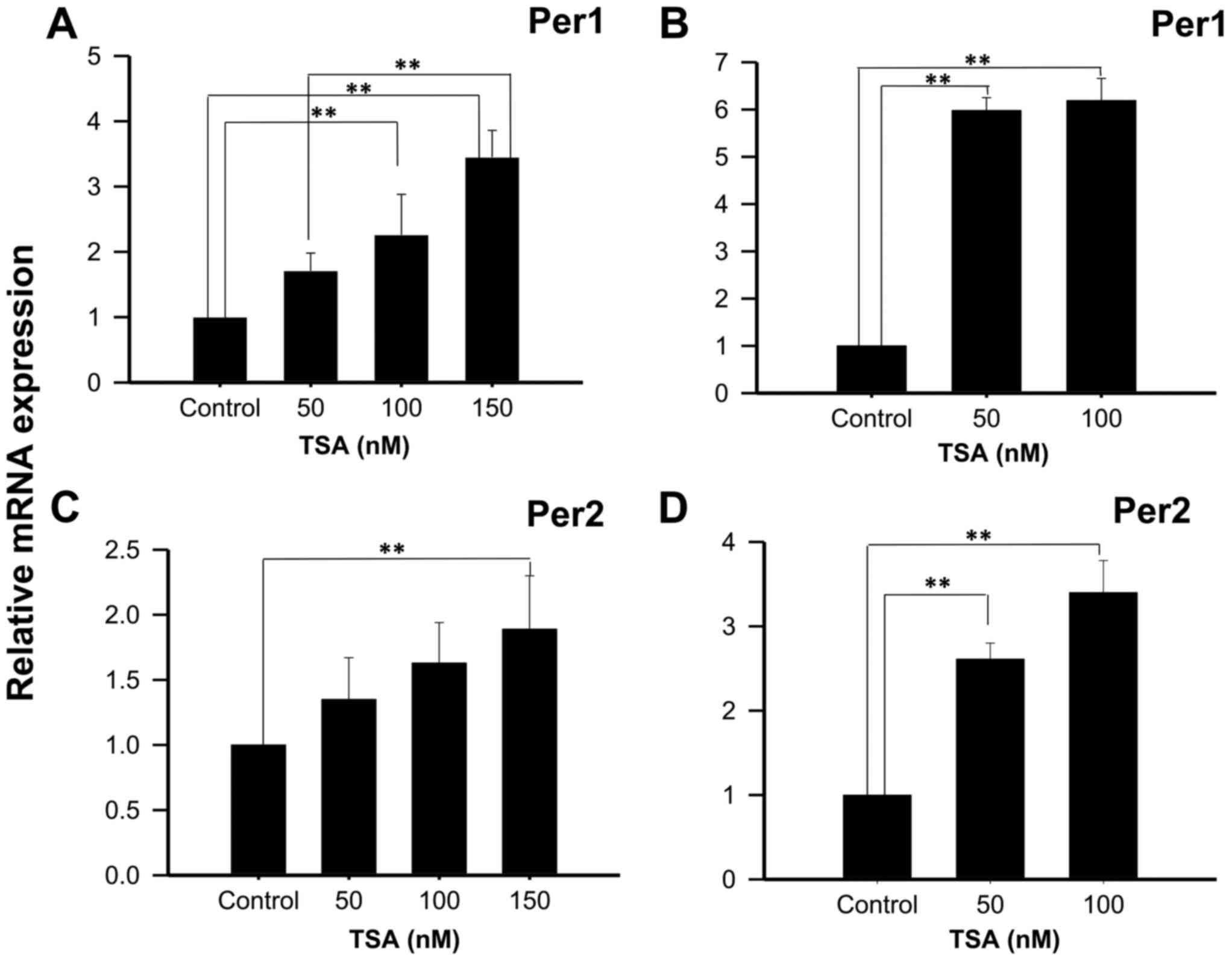
On the other hand, treatment of NCI-N87 cells with
50, 100 or 150 nM TSA for 48 h increases Per1 mRNA expression
(1.71±0.27; 2.26±0.62, and 3.45±0.41 fold higher than control
untreated cells, respectively; P<0.01; Fig. 4A); whereas treatment for 96 h show
significant increases at 50 and 100 nM TSA (5.98±0.27 and 6.19±0.47
fold higher than control untreated cells, respectively; P<0.01;
Fig. 4B). Similar to the results
found for Per1, treatment of NCI-N87 cells with 150 nM TSA during
48 h induced Per2 mRNA expression (1.89±0.41 fold higher than
control untreated cells, respectively; P<0.01; Fig. 4C). This effect was more evident after
96 h of treatment with 50 or 100 nM of TSA (2.61±0.19 and 3.4±0.38
fold higher than control untreated cells, respectively; P<0.01;
Fig. 4D).
Effect of NaB and TSA on histone
modifications and Sp1 and Sp3 binding to the Per1 and Per2
promoters
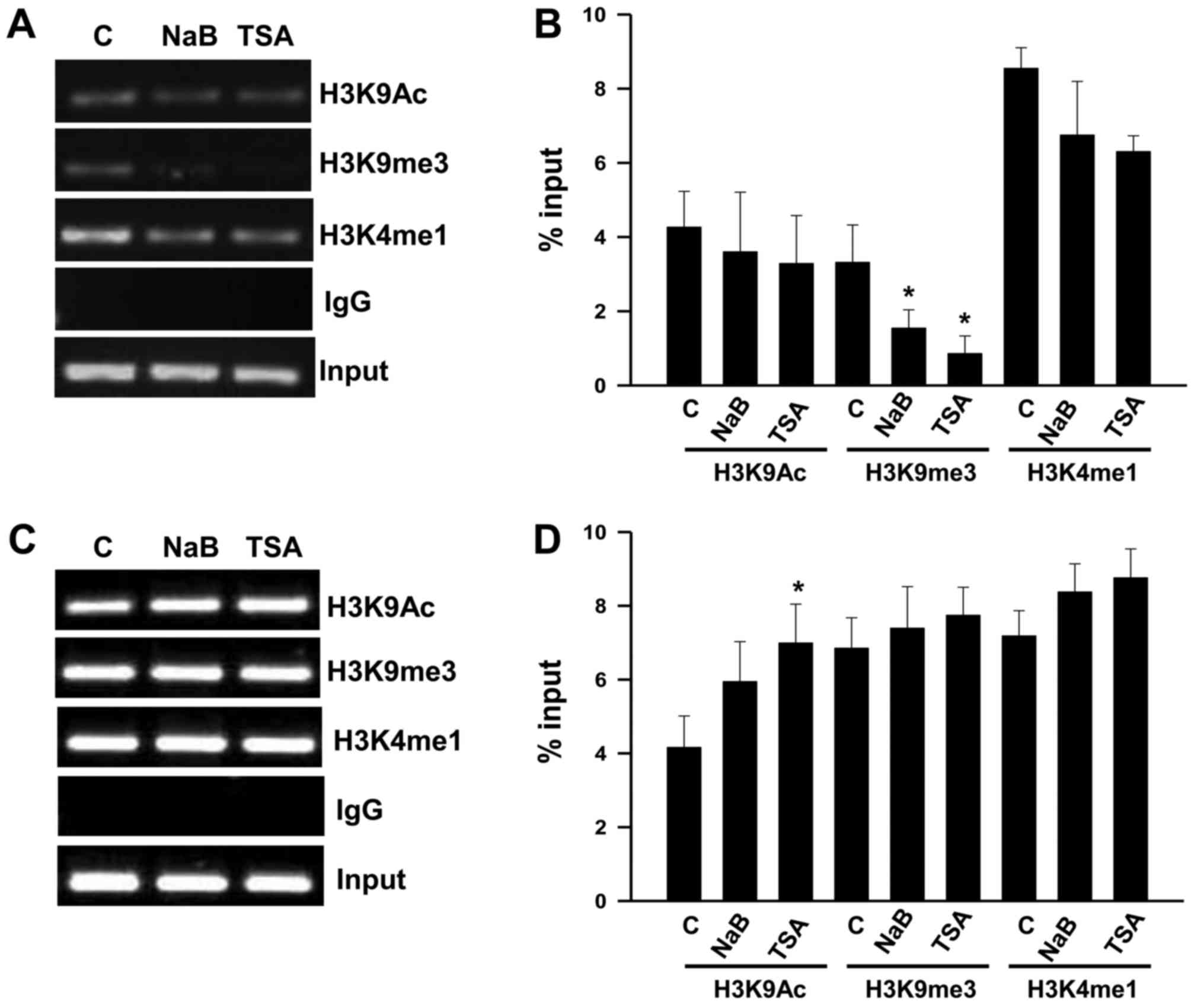
We conducted ChIP assays with KATO III cells to
explore changes in histone modifications, in order to understand
the mechanism for the upregulation of Per1 and Per2 mRNA by NaB and
TSA. We found that treatment with NaB and TSA decreases H3K9me3
(P<0.05) at the Per1 promoter, whereas H3K9Ac and H3K4me1 show
no significant changes (Fig. 5A and
B). In contrast, Per2 promoter shows an increase of H3K9Ac in
response to TSA (P<0.05), while H3K9m3 and H3K4me1 did not
change compared to untreated cells (Fig.
5C and D). Treatment with NaB does not modify the analyzed
chromatin marks at the Per2 promoter (Fig. 5C and D).
It is known that NaB not only inhibits HDACs, it
also acts on non-histone targets, such as transcription factors, in
particular, those of the Sp family. Sp1 and Sp3 have been
implicated in the mechanism of action of NaB on several promoters
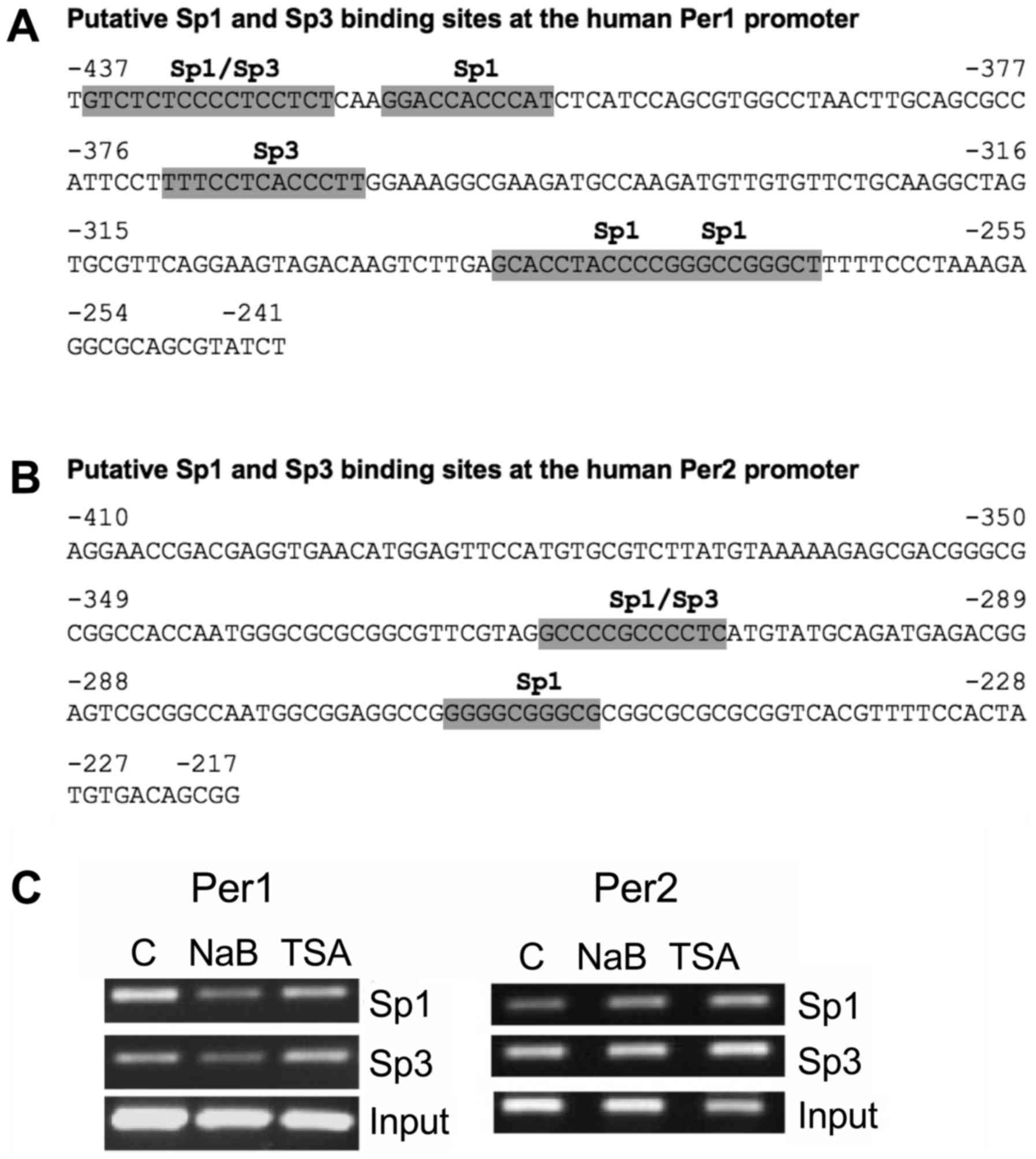
(47–51). We then conducted an in silico
analysis to search for potential Sp1 and Sp3 binding sites at the
Per1 and Per2 promoters. We found four Sp1 and two Sp3 putative
binding sites at the −437 to −241 region of the Per1 promoter
(Fig. 6A), as well as two Sp1 and one
Sp3 putative binding sites at the −410 to −217 region of the Per2
promoter (Fig. 6B). Then we tested
for changes of Sp1 and Sp3 binding to these promoters in response
to NaB and TSA treatment. ChIP analysis shows that binding of Sp1
and Sp3 decrease at the Per1 promoter in NaB treated cells, whereas
TSA has no effect. NaB and TSA treatment increases Sp1 binding to
the Per2 promoter, without affecting Sp3 binding (Fig. 6C).
Effect of NaB, TSA, and Aza on the CpG
methylation of Per1 and Per2 promoters
To further understand the transcriptional activation
of Per1 and Per2 by NaB and TSA we conducted MS-PCR to explore
changes on CpG methylation at the promoters of these genes, as
HDACi treatment may decrease DNA methylation (52,53). We
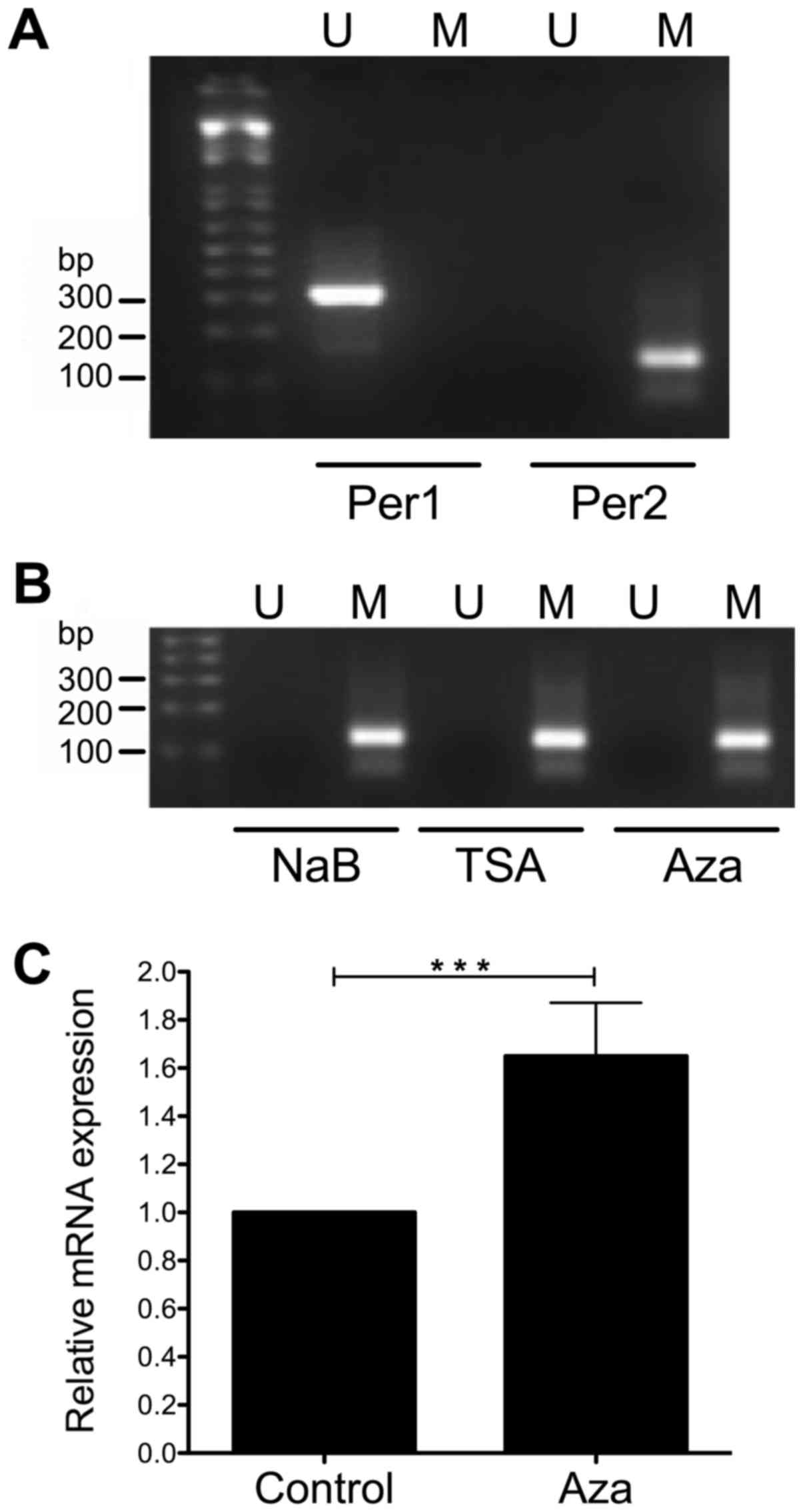
found that Per1 promoter was not methylated at the CpGs detected by
the primers, whereas Per2 promoter was methylated (Fig. 7A). Treatment of KATO III cells with
NaB, TSA and even Aza does not change the methylation status of the
CpGs analyzed by the primers at the Per2 promoter (Fig. 7B). Interestingly, KATO III cells
treated with Aza show a modest but significant increase in Per2
mRNA (1.65±0.22 fold above untreated cells; P<0.001; Fig. 7C).
Discussion
The circadian system may have an important role in
the global epigenetic events occurring during carcinogenesis.
Accumulating evidence indicates that a variety of chromatin
remodelers contribute to various aspects of the circadian epigenome
(4,54). This evidence is further supported by
gene expression and DNA methylation studies at the promoter region
of clock genes silenced in cancer cells (23,27,35,41).
In this study, we found that treatment of KATO III
and NCI-N87 human gastric cancer cells with two HDACi (NaB or TSA)
induce Per1 and Per2 mRNA expression, in a dose-dependent manner.
Our results are consistent with those described by other authors,
who used similar treatments to induce the expression of Per1 and
Per3 genes in cancer cells. It has been shown that TSA and
5-Aza-2′-deoxycytidine increased Per3 mRNA expression in myeloid
leukemia K562 cells (41). Also, HDAC
inhibition by SAHA increased Per1 gene expression in lung cancer
NSCLC cells and other cancer cell lines (35). However, the levels of induction
obtained in our study were higher than those obtained for Per3 in
TSA treated K562 cells (41), but
were lower than those obtained with SAHA in H520, H522, Ishikawa,
MDA-231, and HCT116 cells, which were more than 10-fold higher than
untreated cells (35). This approach
has also been used to induce the expression of epigenetically
silenced tumor suppressor genes in cancer cell lines (55–57).
We also found higher effects of NaB and TSA in cells
treated for 96 h than those treated for 48 h, and larger changes of
Per1 and Per2 expression were found in KATO III than in NCI-N87
cells. It is well established that gene expression levels in
response to HDACi is highly dependent on the cell type (35,45,58). This
could explain the differences observed between KATO III and NCI-N87
cells.
Inhibition of HDAC activity may play a key role in
the expression of Per1 and Per2 genes in gastric cancer cells by
promoting and open chromatin conformation. We found that treatment
with NaB and TSA decreases H3K9me3, a heterochromatin mark, at the
Per1 promoter, whereas TSA increases H3K9Ac, a mark of euchromatin,
at the Per2 promoter, suggesting that these changes may contribute
to their transcriptional activation. It is well established that
H3K9 methylation correlates with transcriptional repression of
multiple genes (59,60).
Enhanced acetylation of H3 and H4 at the Per1
promoter correlate with its transcriptional activation phase in
mouse tissues (4,42,61),
whereas di- and tri-methylation of H3K27 at the promoters of Per1
and Per2 correlate with transcriptional repression (62). A previous study has also shown that
SAHA induced Per1 expression by increasing H3 acetylation at the
Per1 promoter of NSLC cells (35). We
did not find an increase of H3K9Ac at the Per1 promoter in NaB or
TSA treated KATO III cells, but we found a decrease of H3K9
trimethylation. We cannot rule out the possibility that other
residues in H3, such as K27, or in H4 could increase their
acetylation in response to NaB or TSA.
Our data also demonstrate that NaB induced higher
mRNA levels than TSA in both cell lines. NaB not only inhibits
HDACs, it also acts on non-histone targets, such as Sp1 and Sp3
transcriptional factors, who have been implicated in the mechanism
of action of NaB on several promoters (47–51). Our
in silico analysis predicted Sp1 and Sp3 binding sites at
the Per1 and Per2 promoters, thus we tested for changes in binding
of these factors by ChIP assays. We found decreased Sp1 and Sp3
binding to the Per1 promoter in response to NaB, but not to TSA,
whereas Sp1 binding increased at the Per2 promoter in response to
NaB and TSA. This suggests that, Sp1 and Sp3 could negatively
regulate Per1 expression, while Sp1 recruitment could contribute to
the upregulation of Per2 in KATO III cells. Most studies have shown
that NaB induce transcriptional activation by enhancing Sp1 and Sp3
binding to gene promoters (47–50);
however, others have shown the opposite effect (51). NaB down-regulates neuropilin 1 (NRP1)
expression and was associated with decreased binding of Sp1 to the
NRP1 promoter (51). On the other
hand, Sp1 down-regulates STC1 gene expression in HT-29 cells
treated with TSA, by forming a complex with the retinoblastoma
protein (63). Sp1 and Sp3 also
interact with c-Myc to repress p21 promoter (64). Additionally, Sp1 was able to activate
and to repress the thymidine kinase promoter in mouse 3T3
fibroblasts. This ambivalence of Sp1 as a transcriptional modulator
lies on the interaction with other proteins, when it interacts with
HDAC1 acts as a transcriptional suppressor, whereas when interacts
with E2F1-3 promotes transcriptional activation (65). However, further experiments are needed
to fully understand the possible role of Sp1 and Sp3 on Per1
transcriptional regulation, mediated by NaB or TSA.
Some studies have shown that HDACi treatment may
reverse CpG methylation at gene promoters, leading to
transcriptional activation (52,53), thus
we tested this possibility. Our results show that Per2 promoter was
methylated, whereas Per1 promoter was not methylated. The
methylation of the CpGs recognized by the primers at the Per2
promoter did not change in response to NaB or TSA, neither in cells
treated with 5-Aza. However, treatment with Aza induced a modest
Per2 mRNA expression. It will be necessary to conduct DNA
sequencing with bisulfite modified DNA, in order to determine if
other CpGs are methylated at the promoter of these genes, and
whether NaB, TSA or Aza may modify the pattern.
We cannot rule out the possibility that other
mechanisms could indirectly mediate Per1 and Per2 upregulation in
response to HDACi. Possibly by inducing transcription factors that
bind to regulatory elements, which in turn could activate Per1 or
Per2 promoters. Promoter analysis and promoter reporter constructs
have shown important transcription factor binding sites that
activate Per1 or Per2 expression, like E-boxes, CRE-elements,
CAAT-boxes, C/EBP, and others (15,66–68).
Acetylation of BMAL1, mediated by the intrinsic HAT activity of
CLOCK could be involved in the induction of expression of Per1 and
Per2 genes (69,70), by recruiting co-activating proteins
such as P300 to the promoters of Per1 and Per2 genes, as previously
described (42). However, further
experiments are needed to fully understand the transcriptional
regulation of these genes in gastric cancer cells.
In conclusion, the results presented in this work
demonstrate that inhibition of HDACs with NaB or TSA induce the
expression of Per1 and Per2 genes in KATO III and NCI-N87 gastric
cancer cells, through changes in chromatin modifications at Per1
and Per2 proximal promoters, as well as changes of Sp3 and/or Sp1
binding to the Per1 and Per2 promoters. Changes in epigenetic
modifications of Per1 and Per2 could disrupt other clock genes, as
well as a web of genes and cellular pathways under circadian
control. Further experiments will be necessary to address these
issues. The understanding of changes in the epigenome of cancer
cells could contribute to the development of new cancer therapies,
and HDACi offer an excellent alternative.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from
Programa de Mejoramiento del Profesorado, Secretaría de Educación
Pública de México (grant no. PTC-270), Consejo Nacional de Ciencia
y Tecnología, México (grant no. 2015-01-1518), Dirección General de
Asuntos del Personal Académico de la Universidad Nacional Autónoma
de México (grant no. IN217216), and FH-R was supported by a
scholarship (no. 223273) from Consejo Nacional de Ciencia y
Tecnología, México.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contribution
FHR performed, analyzed and discussed the majority
of the experiments, prepared the figures, and wrote the first draft
of the manuscript. AHO performed the ChIP assays, MS-PCR and in
silico analysis, discussed and interpreted the data, and
prepared the figures. LFP performed ChIP assays and critically
revised the manuscript. GR conducted part of the cell culture work
and critically revised the manuscript. MC discussed and interpreted
the data, and critically revised the manuscript. AZH contributed to
the study design, acquired funding, discussed and interpreted the
data, and critically revised and edited the manuscript. JSG
conceived and designed the study, discussed and interpreted the
data, acquired funding, and wrote and edited the manuscript. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lowrey PL and Takahashi JS: Genetics of
circadian rhythms in mammalian model organisms. Adv Genet.
74:175–230. 2011.PubMed/NCBI
|
|
2
|
Partch CL, Green CB and Takahashi JS:
Molecular architecture of the mammalian circadian clock. Trends
Cell Biol. 24:90–99. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Buhr ED and Takahashi JS: Molecular
components of the mammalian circadian clock. Kramer A and Merrow M:
circadian clocks, handbook of experimental pharmacology. 217:3–27.
2013. View Article : Google Scholar
|
|
4
|
Koike N, Yoo SH, Huang HC, Kumar V, Lee C,
Kim TK and Takahashi JS: Transcriptional architecture and chromatin
landscape of the core circadian clock in mammals. Science.
338:349–354. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Katada S and Sassone-Corsi P: The histone
methyltransferase MLL1 permits the oscillation of circadian gene
expression. Nat Struct Mol Biol. 17:1414–1421. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kettner NM, Katchy CA and Fu L: Circadian
gene variants in cancer. Ann Med. 46:208–220. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bechtold DA, Gibbs JE and Loudon AS:
Circadian dysfunction in disease. Trends Pharmacol Sci. 31:191–198.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kiessling S, Beaulieu-Laroche L, Blum ID,
Landgraf D, Welsh DK, Storch KF, Labrecque N and Cermakian N:
Enhancing circadian clock function in cancer cells inhibits tumor
growth. BMC Biol. 15:132017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cash E, Sephton SE, Chagpar AB, Spiegel D,
Rebholz WN, Zimmaro LA, Tillie JM and Dhabhar FS: Circadian
disruption and biomarkers of tumor progression in breast cancer
patients awaiting surgery. Brain Behav Immun. 48:102–114. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Greene MW: Circadian rhythms and tumor
growth. Cancer Lett. 318:115–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yasuniwa Y, Izumi H, Wang KY, Shimajiri S,
Sasaguri Y, Kawai K, Kasai H, Shimada T, Miyake K, Kashiwagi E, et
al: Circadian disruption accelerates tumor growth and
angio/stromagenesis through a Wnt signaling pathway. PLoS One.
5:e153302010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Dycke KC, Rodenburg W, van Oostrom CT,
VanKerkhof LW, Pennings JL, Roenneberg T, van Steeg H and
Vanderhorst GT: Chronically alternating light cycles increase
breast cancer risk in mice. Curr Biol. 25:1932–1937. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fu L, Pelicano H, Liu J, Huang P and Lee
C: The circadian gene Period2 plays an important role in tumor
suppression and DNA damage response in vivo. Cell. 111:41–50. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Papagiannakopoulos T, Bauer MR, Davidson
SM, Heimann M, Subbaraj L, Bhutkar A, Bartlebaugh J, Vander Heiden
MG and Jacks T: Circadian rhythm disruption promotes lung
tumorigenesis. Cell Metab. 24:324–331. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gery S, Gombart AF, Yi WS, Koeffler C,
Hofmann WK and Koeffler HP: Transcription profiling of C/EBP
targets identifies Per2 as a gene implicated in myeloid leukemia.
Blood. 106:2827–2836. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gery S, Komatsu N, Baldjyan L, Yu A, Koo D
and Koeffler HP: The circadian gene per1 plays an important role in
cell growth and DNA damage control in human cancer cells. Mol Cell.
22:375–382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hua H, Wang Y, Wan C, Liu Y, Zhu B, Yang
C, Wang X, Wang Z, Cornelissen-Guillaume G and Halberg F: Circadian
gene mPer2 overexpression induces cancer cell apoptosis. Cancer
Sci. 97:589–596. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiang S, Coffelt SB, Mao L, Yuan L, Cheng
Q and Hill SM: Period-2: A tumor suppressor gene in breast cancer.
J Circadian Rhythms. 6:42008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang X, Wood PA, Oh EY, Du-Quiton J,
Ansell CM and Hrushesky WJ: Down regulation of circadian clock gene
Period 2 accelerates breast cancer growth by altering its daily
growth rhythm. Breast Cancer Res Treat. 117:423–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyazaki K, Wakabayashi M, Hara Y and
Ishida N: Tumor growth suppression in vivo by overexpression of the
circadian component, PER2. Genes Cells. 15:351–358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oda A, Katayose Y, Yabuuchi S, Yamamoto K,
Mizuma M, Shirasou S, Onogawa T, Ohtsuka H, Yoshida H, Yoshida H,
et al: Clock gene mouse period2 overexpression inhibits growth of
human pancreatic cancer cells and has synergistic effect with
cisplatin. Anticancer Res. 29:1201–1210. 2009.PubMed/NCBI
|
|
22
|
Winter SL, Bosnoyan-Collins L, Pinnaduwage
D and Andrulis IL: Expression of the circadian clock genes Pert and
Per2 in sporadic and familial breast tumors. Neoplasia. 9:797–800.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ
and Chang JG: Deregulated expression of the PER1, PER2 and PER3
genes in breast cancers. Carcinogenesis. 26:1241–1246. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dai H, Zhang L, Cao M, Song F, Zheng H,
Zhu X, Wei Q, Zhang W and Chen K: The role of polymorphisms in
circadian pathway genes in breast tumorigenesis. Breast Cancer Res
Treat. 127:531–540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao Q, Gery S, Dashti A, Yin D, Zhou Y, Gu
J and Koeffler HP: A role for the clock gene per1 in prostate
cancer. Cancer Res. 69:7619–7625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tokunaga H, Takebayashi Y, Utsunomiya H,
Akahira J, Higashimoto M, Mashiko M, Ito K, Niikura H, Takenoshita
S and Yaegashi N: Clinicopathological significance of circadian
rhythm-related gene expression levels in patients with epithelial
ovarian cancer. Acta Obstet Gynecol Scand. 87:1060–1070. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yeh KT, Yang MY, Liu TC, Chen JC, Chan WL,
Lin SF and Chang JG: Abnormal expression of period 1 (PER1) in
endometrial carcinoma. J Pathol. 206:111–120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Relles D, Sendecki J, Chipitsyna G, Hyslop
T, Yeo CJ and Arafat HA: Circadian gene expression and
clinicopathologic correlates in pancreatic cancer. J Gastrointest
Surg. 17:443–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mazzoccoli G, Panza A, Valvano MR, Palumbo
O, Carella M, Pazienza V, Biscaglia G, Tavano F, Di Sebastiano P,
Andriulli A and Piepoli A: Clock gene expression levels and
relationship with clinical and pathological features in colorectal
cancer patients. Chronobiol Int. 28:841–851. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mostafaie N, Kallay E, Sauerzapf E, Bonner
E, Kriwanek S, Cross HS, Huber KR and Krugluger W: Correlated
downregulation of estrogen receptor beta and the circadian clock
gene Per1 in human colorectal cancer. Mol Carcinog. 48:642–647.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu ML, Yeh KT, Lin PM, Hsu CM, Hsia HH,
Liu YC, Lin HY, Lin SF and Yang MY: Deregulated expression of
circadian clock genes in gastric cancer. BMC Gastroenterol.
14:672014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao H, Zen ZL, Yang Y, Jin Y, Qiu MZ, Hu
XY, Han J, Liu KY, Liao JW, Xu RH and Zou QF: Prognostic relevance
of Period1 (Per1) and Period2 (Per2) expression in human gastric
cancer. Int J Clin Exp Pathol. 7:619–630. 2014.PubMed/NCBI
|
|
33
|
Lin YM, Chang JH, Yeh KT, Yang MY, Liu TC,
Lin SF, Su W and Chang JG: Disturbance of circadian gene expression
in hepatocellular carcinoma. Mol Carcinog. 47:925–933. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lengyel Z, Lovig C, Kommedal S, Keszthelyi
R, Szekeres G, Battyáni Z, Csernus V and Nagy AD: Altered
expression patterns of clock gene mRNAs and clock proteins in human
skin tumors. Tumor Biol. 34:811–819. 2013. View Article : Google Scholar
|
|
35
|
Gery S, Komatsu N, Kawamata N, Miller CW,
Desmond J, Virk RK, Marchevsky A, Mckenna R, Taguchi H and Koeffler
HP: Epigenetic silencing of the candidate tumor suppressor gene
Per1 in non-small cell lung cancer. Clin Cancer Res. 13:1399–1404.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang MY, Yang WC, Lin PM, Hsu JF, Hsiao
HH, Liu YC, Tsai HJ, Chang CS and Lin SF: Altered expression of
circadian clock genes in human chronic myeloid leukemia. J Biol
Rhythms. 26:136–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hsu CM, Lin SF, Lu CT, Lin PM and Yang MY:
Altered expression of circadian clock genes in head and neck
squamous cell carcinoma. Tumor Biol. 33:149–155. 2012. View Article : Google Scholar
|
|
38
|
Thoennissen NH, Thoennissen GB, Abbassi S,
Nabavi-Nouis S, Sauer T, Doan NB, Gery S, Müller-Tidow C, Said JW
and Koeffler HP: Transcription factor CCAAT/enhancer-binding
protein alpha and critical circadian clock downstream target gene
PER2 are highly deregulated in diffuse large B-cell lymphoma. Leuk
Lymphoma. 53:1577–1585. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xia H, Niu Z, Ma H, Cao SZ, Hao SC, Liu ZT
and Wang F: Deregulated expression of the Per1 and Per2 in human
gliomas. Can J Neurol Sci. 37:365–370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhanfeng N, Yanhui L, Zhou F, Shaocai H,
Guangxing L and Hechun X: Circadian genes Per1 and Per2 increase
radiosensitivity of glioma in vivo. Oncotarget. 6:9951–9958. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang MY, Chang JG, Lin PM, Tang KP, Chen
YH, Lin HY, Liu TC, Hsiao HH, Liu YC and Lin SF: Downregulation of
circadian clock genes in chronic myeloid leukemia: alternative
methylation pattern of hPER3. Cancer Sci. 97:1298–1307. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Etchegaray JP, Lee C, Wade PA and Reppert
SM: Rhythmic histone acetylation underlies transcription in the
mammalian circadian clock. Nature. 421:177–182. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ramakers C, Ruijter JM, Deprez RH and
Moorman AF: Assumption-free analysis of quantitative real-time
polymerase chain reaction (PCR) data. Neurosci Lett. 339:62–66.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Contreras-Leal E, Hernandez-Oliveras A,
Flores-Peredo L, Zarain-Herzberg A and Santiago-Garcia J: Histone
deacetylase inhibitors promote the expression of ATP2A3 gene in
breast cancer cell lines. Mol Carcinog. 55:1477–1485. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Khan A, Fornes O, Stigliani A, Gheorghe M,
Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni
SR, Tan G, et al: JASPAR 2018: Update of the open-access database
of transcription factor binding profiles and its web framework.
Nucleic Acids Res. 46:D260–D266. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Amin MR, Dudeja PK, Ramaswamy K and
Malakooti J: Involvement of Sp1 and Sp3 in differential regulation
of human NHE3 promoter activity by sodium butyrate and
IFN-gamma/TNF-alpha. AJP Gastrointest Liver Physiol. 293:G374–G382.
2007. View Article : Google Scholar
|
|
48
|
Kiela PR, Kuscuoglu N, Midura AJ,
Midura-Kiela MT, Larmonier CB, Lipko M and Ghishan FK: Molecular
mechanism of rat NHE3 gene promoter regulation by sodium butyrate.
Am J Physiol Cell Physiol. 293:C64–C74. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Flores-Peredo L, Rodriguez G and
Zarain-Herzberg A: Induction of cell differentiation activates
transcription of the sarco/endoplasmic reticulum calcium-ATPase 3
gene (ATP2A3) in gastric and colon cancer cells. Mol Carcinog.
56:735–750. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chirakkal H, Leech SH, Brookes KE, Prais
AL, Waby JS and Corfe BM: Upregulation of BAK by butyrate in the
colon is associated with increased Sp3 binding. Oncogene.
25:7192–7200. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu DC, Waby JS, Chirakkal H, Staton CA and
Corfe BM: Butyrate suppresses expression of neuropilin I in
colorectal cell lines through inhibition of Sp1 transactivation.
Mol Cancer. 9:2762010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sarkar S, Abujamra AL, Loew JE, Forman LW,
Perrine SP and Faller DV.: Histone deacetylase inhibitors reverse
CpG methylation by regulating DNMT1 through ERK signaling.
Anticancer Res. 3:2723–2732. 2011.
|
|
53
|
Shin H, Kim JH, Lee YS and Lee YC: Change
in gene expression profiles of secreted frizzled-related proteins
(SFRPs) by sodium butyrate in gastric cancers: Induction of
promoter demethylation and histone modification causing inhibition
of Wnt signaling. Int J Oncol. 40:1533–1542. 2012.PubMed/NCBI
|
|
54
|
Masri S and Sassone-Corsi P: Plasticity
and specificity of the circadian epigenome. Nat Neurosci.
13:1324–1329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Meng CF, Zhu XJ, Peng G and Dai DQ:
Promoter histone H3 lysine 9 di-methylation is associated with DNA
methylation and aberrant expression of p16 in gastric cancer cells.
Oncol Rep. 22:1221–1227. 2009.PubMed/NCBI
|
|
56
|
Chen MY, Liao WS, Lu Z, Bornmann WG,
Hennessey V, Washington MN, Rosner GL, Yu Y, Ahmed AA and Bast RC
Jr: Decitabine and suberoylanilide hydroxamic acid (saha) inhibit
growth of ovarian cancer cell lines and xenografts while inducing
expression of imprinted tumor suppressor genes, apoptosis, G2/M
arrest and autophagy. Cancer. 117:4424–4438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Vibhakar R, Foltz G, Yoon JG, Field L, Lee
H, Ryu GY, Pierson J, Davidson B and Madan A: Dickkopf-1 is an
epigenetically silenced candidate tumor suppressor gene in
medulloblastoma. Neuro Oncol. 9:135–144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chang J, Varghese DS, Gillam MC, Peyton M,
Modi B, Schiltz RL, Girard L and Martinez ED: Differential response
of cancer cells to HDAC inhibitors trichostatin A and depsipeptide.
Br J Cancer. 106:116–125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hublitz P, Albert M and Peters AH:
Mechanisms of transcriptional repression by histone lysine
methylation. Int J Dev Biol. 53:335–354. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Varier RA and MarcTimmers HT: Histone
lysine methylation and demethylation pathways in cancer. Biochim
Biophys Acta. 1815:75–89. 2011.PubMed/NCBI
|
|
61
|
Curtis AM, Seo SB, Westgate EJ, Rudic RD,
Smyth EM, Chakravarti D, Fitzgerald GA and McNamara P: Histone
acetyltransferase-dependent chromatin remodeling and the vascular
clock. J Biol Chem. 279:7091–7097. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Etchegaray JP, Yang X, DeBruyne JP, Peters
AH, Weaver DR, Jenuwein T and Reppert SM: The polycomb group
protein EZH2 is required for mammalian circadian clock function. J
Biol Chem. 281:21209–21215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Law AY, Yeung BH, Ching LY and Wong CK:
Sp1 is a transcriptional repressor to stanniocalcin-1 expression in
TSA-treated human colon cancer cells, HT29. J Cell Biochem.
112:2089–2096. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Gartel AL, Ye X, Goufman E, Shianov P, Hay
N, Najmabandi F and Tyner AL: Myc repress the p21(WAF1/CIP1)
promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA.
98:4510–4515. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Doetzlhofer A, Rotheneder H, Lagger G,
Koranda M, Kurtev V, Brosch G, Wintersberger E and Seiser C:
Histone deacetylase 1 can repress transcription by binding to Sp1.
Mol Cell Biol. 19:5504–5511. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hida A, Koike N, Hirose M, Hattori M,
Sakaki Y and Tei H: The human and mouse Period1 genes: five
well-conserved E-boxes additively contribute to the enhancement of
mPer1 transcription. Genomics. 65:224–233. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Taruscio D, Zoraqi GK, Falchi M, Iosi F,
Paradisi S, DiFiore B, Lavia P and Falbo V: The human Per1 gene:
Genomic organization and promoter analysis of the first human
orthologue of the Drosophila period gene. Gene. 253:161–170. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Koyanagi S, Hamdan AM, Horiguchi M,
Kusunose N, Okamoto A, Matsunaga N and Ohdo S: cAMP-response
element (CRE)-mediated transcription by activating transcription
factor-4 (ATF4) is essential for circadian expression of the
Period2 gene. J Biol Chem. 286:32416–32423. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Doi M, Hirayama J and Sassone-Corsi P:
Circadian regulator CLOCK is a histone acetyltransferase. Cell.
125:497–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Grimaldi B, Nakahata Y, Sahar S, Kaluzova
M, Gauthier D, Pham K, Patel N, Hirayama J and Sassone-Corsi P:
Chromatin remodeling and circadian control: Master regulator CLOCK
is an enzyme. Cold Spring Harb Symp Quant Biol. 72:105–112. 2007.
View Article : Google Scholar : PubMed/NCBI
|