Introduction
Bladder cancer (BLCA) is among the most malignant
types of cancer; 76,790 new cases and 16,390 mortalities were
reported in the United States in 2016 (1). Based on a recent study on cancer in
China, there were 80,500 new cases and 32,900 mortalities from BRCA
reported in 2015 (2). Metastasis and
early relapse are common in BLCA, thus determining the prognosis is
important for patients with BLCA (3).
However, the current clinical staging system is insufficient to
predict the outcome of patients with BLCA (4). Therefore, novel molecular biomarkers for
prediction of BLCA prognosis are urgently required.
According to a previous study, single biomarkers
often fail to accurately predict the prognosis of patients in
datasets, whereas multiple biomarkers perform more effectively
(5). In the present study, random
forest variable hunting coupled with Cox multivariate regression
were used to produce a model based on gene expression levels to
evaluate the prognosis of patients with BLCA from The Cancer Genome
Atlas (TCGA) dataset. The patients with high risk scores had a
significantly shorter survival time than those with low risk
scores, which was validated in 3 further independent cohorts.
Furthermore, the association between risk score and other clinical
information demonstrated that the risk score was associated with
the pathological stage, while a nomogram based on risk score and
clinical information indicated that the risk score corresponded the
most with the outcome of bladder cancer.
Materials and methods
Data processing
mRNA expression levels from the ‘TCGA Bladder Cancer
(BLCA)’ dataset (n=407) were downloaded from UCSC Xena (http://xena.ucsc.edu/) and converted to RNAseq by
expectation-maximization (RSEM) values using the Xena website.
Genes not expressed in any of the samples were filtered from the
dataset. log 2-transformed RSEM values were retained for model
development.
Raw data from the expression profiles GSE31684,
GSE48075 and E-MTAB-4321 were downloaded from Gene Expression
Omnibus (https://www.ncbi.nlm.nih.gov/geo) and Array Express
(www.ebi.ac.uk/arrayexpress/) in CEL
format. Background correction and normalization with Robust
Multiarray Averaging were performed on the raw data (6,7). Probes
were matched to the HUGO Gene (https://www.genenames.org/) Nomenclature
Committee-approved gene names. Probes without annotation were
discarded, genes matching more than one probe were merged and mean
values were used to represent gene expression. The Z-score was
calculated in each dataset for each gene across samples and used
for further analysis (8).
Gene selection and model
construction
Univariate Cox regression analysis was performed on
the training (TGCA) dataset. Gene expression significantly
associated with overall survival (OS) in the training dataset was
selected for further analysis, with a threshold of P<0.001.
Random forest variable hunting was performed using 100 replications
and 100 steps to select the most significant candidate genes,
including zinc finger protein 230 (ZNF230), BCL2-like 14 (BCL2L14),
AHNAK, transmembrane protein 109 (TMEM109), apolipoprotein L2
(APOL2), advanced glycation end-product specific receptor (AGER)
and amine oxidase, copper containing 2 (AOC2).
Multivariate Cox regression analysis was implemented
to calculate the risk score using the candidate genes and overall
survival information. Risk score was calculated using the following
formula; where βi indicates the coefficients evaluated
with gene expression and xi refers to gene relative
expression level.
Risk score=∑inβi*xi
Coefficients were locked to calculate the risk
scores of the three test datasets.
Statistical analysis
All statistical analysis in this study was performed
with R (version 3.0.1; https://www.r-project.org/) and R packages.
Normalization of raw data was performed using the ‘affy’ package
(v1.56.0) (9), the survival analysis
and Cox probability hazard analyses were performed using the
‘survival’ (v1.4–8) package, random forest variable hunting was
performed using the ‘randomForestSRC’ package (v2.0.5) (10) and the receiving operating
characteristic (ROC) curves were drawn using the ‘pROC’ package
(v1.11.0) (11). The gene set
enrichment analysis (GSEA) was performed using Java GSEA software
(http://software.broadinstitute.org/gsea/index.jsp)
(v5.2) (12).
Results
Risk score staging system
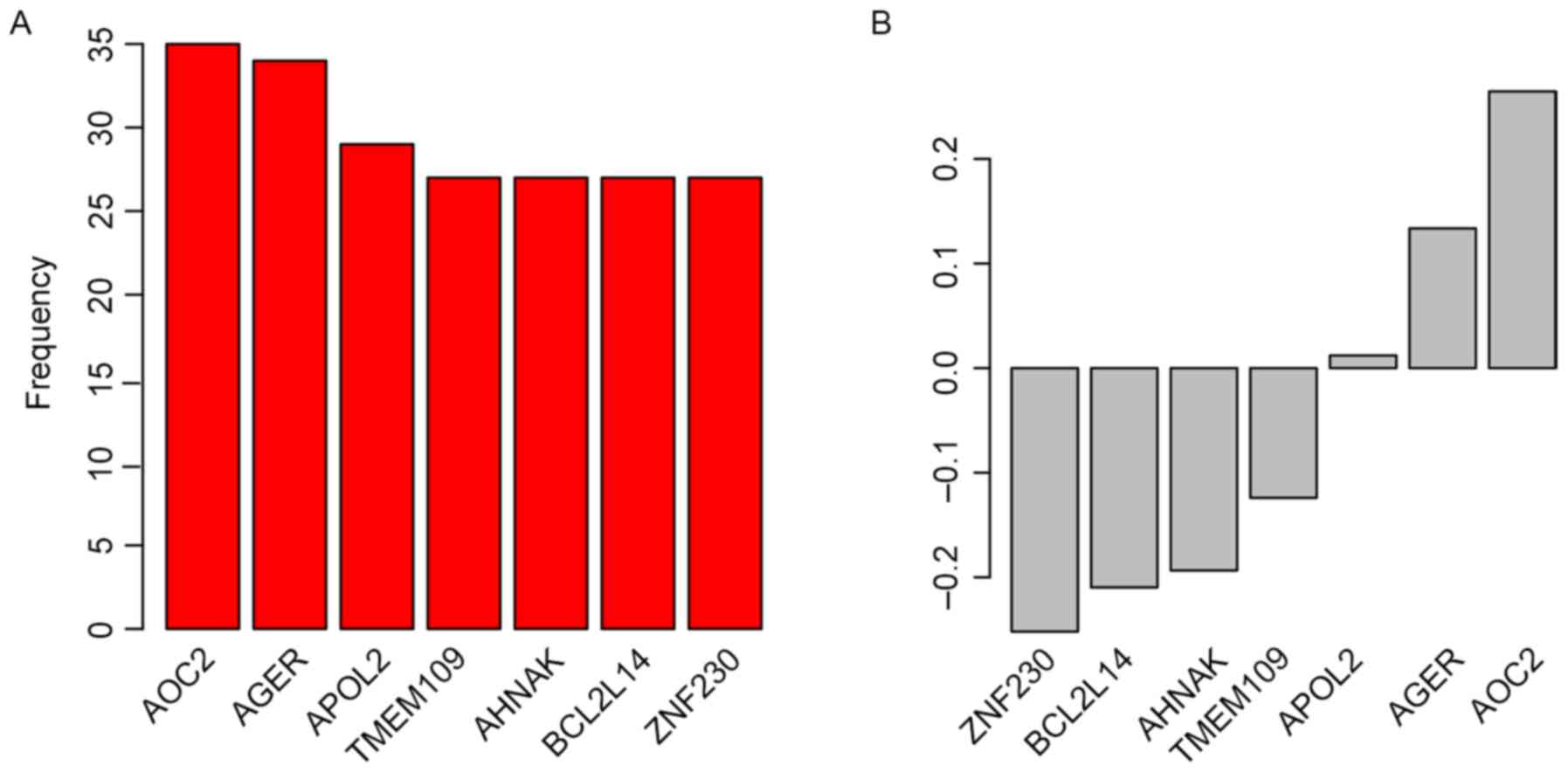
Candidate genes for the staging system were selected
by Univariate Cox regression analysis between gene expression and
OS in the ‘TCGA Bladder Cancer (BLCA)’ dataset. Random forest
variable hunting was implemented to select the most suitable
combination of candidate genes; 7 genes were identified (Fig. 1A). Multivariate Cox regression
analysis was performed and coefficients were calculated. The risk
score of each patient was calculated using the following formula:
Risk score=(0.012050982× ZNF230) + (−0.124027149× BCL2L14) +
(−0.251893959× AHNAK) + (0.264530911× TMEM109) + (0.133540278×
APOL2) + (−0.19351212× AGER) + (−0.209706035× AOC2); where gene
name represents the Z-score for that gene. Parameters for each gene
are detailed in Table I. Genes with
positive coefficients indicate genes identified as cancer drivers,
whereas genes with negative coefficients were identified as tumor
suppressor genes (Fig. 1B).
 | Table I.Analysis of the candidate genes with
univariate and multivariate Cox regression. |
Table I.
Analysis of the candidate genes with
univariate and multivariate Cox regression.
|
| Univariate | Multivariate |
|---|
|
|
|
|
|---|
| Gene | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| TMEM109 | 1.50 | 1.50–1.30 | <0.001 | 1.30 | 1.08–1.57 | 0.005 |
| AHNAK | 1.40 | 1.40–1.20 | <0.001 | 1.14 | 0.95–1.37 | 0.152 |
| BCL2L14 | 0.76 | 0.76–0.68 | <0.001 | 0.82 | 0.73–0.93 | 0.003 |
| AOC2 | 0.79 | 0.79–0.69 | <0.001 | 1.01 | 0.85–1.2 | 0.890 |
| ZNF230 | 0.73 | 0.73–0.62 | <0.001 | 0.81 | 0.69–0.96 | 0.015 |
| AGER | 0.77 | 0.77–0.68 | <0.001 | 0.88 | 0.75–1.04 | 0.133 |
| APOL2 | 0.73 | 0.73–0.63 | <0.001 | 0.78 | 0.67–0.9 | 0.001 |
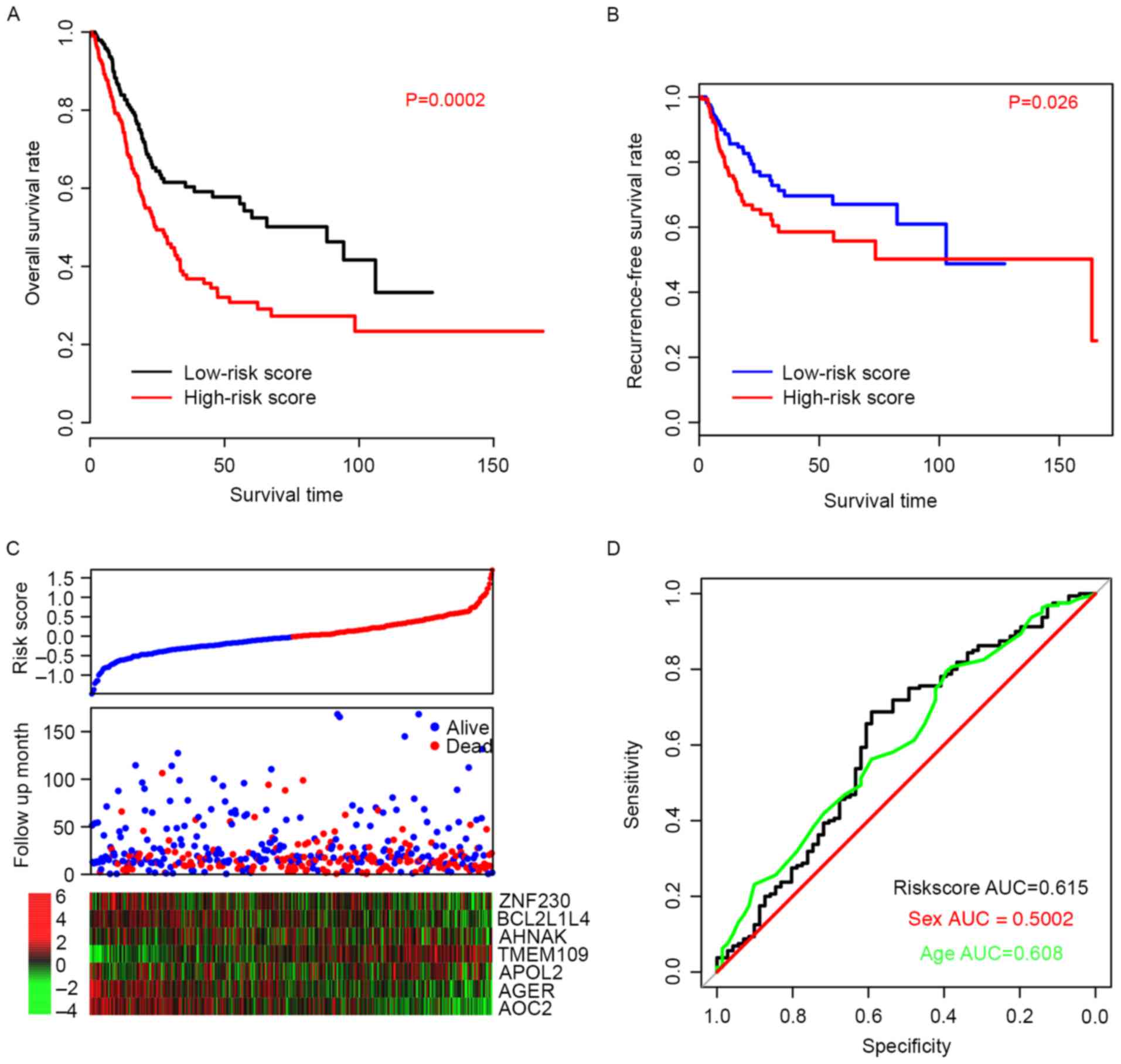
Risk score predicts survival in the
training dataset
The efficiency of the risk score in predicting the
outcome of BLCA patients was evaluated. Using the median risk score
value as a cutoff, patient data from the TCGA dataset was divided
into high-risk and low-risk groups. The OS time of patients in the
high-risk group was significantly longer than patients in the
low-risk group (P=0.0002; Fig. 2A).
The median survival of high-risk patients was 24.6 months (95% CI;
20–33.5 months) whereas the median survival of low-risk patients
was 88 months (95% CI; 45.6-NA months). Furthermore, the
recurrence-free survival (RFS) time was also compared between the
high- and low-risk groups, and the resulting profiles resembled
those of OS (P=0.026; Fig. 2B).
Patients with high-risk scores were more prone to early relapse,
and the expression pattern was consistent with the coefficients of
each gene (Fig. 2C). The ROC curve
for three-year events was also plotted based on age, sex and risk
score (Fig. 2D) and the area under
curve (AUC) was 0.608, 0.500, and 0.615, respectively. These
results suggest that the risk score staging system performed better
in predicting the survival of BLCA patients than other clinical
information.
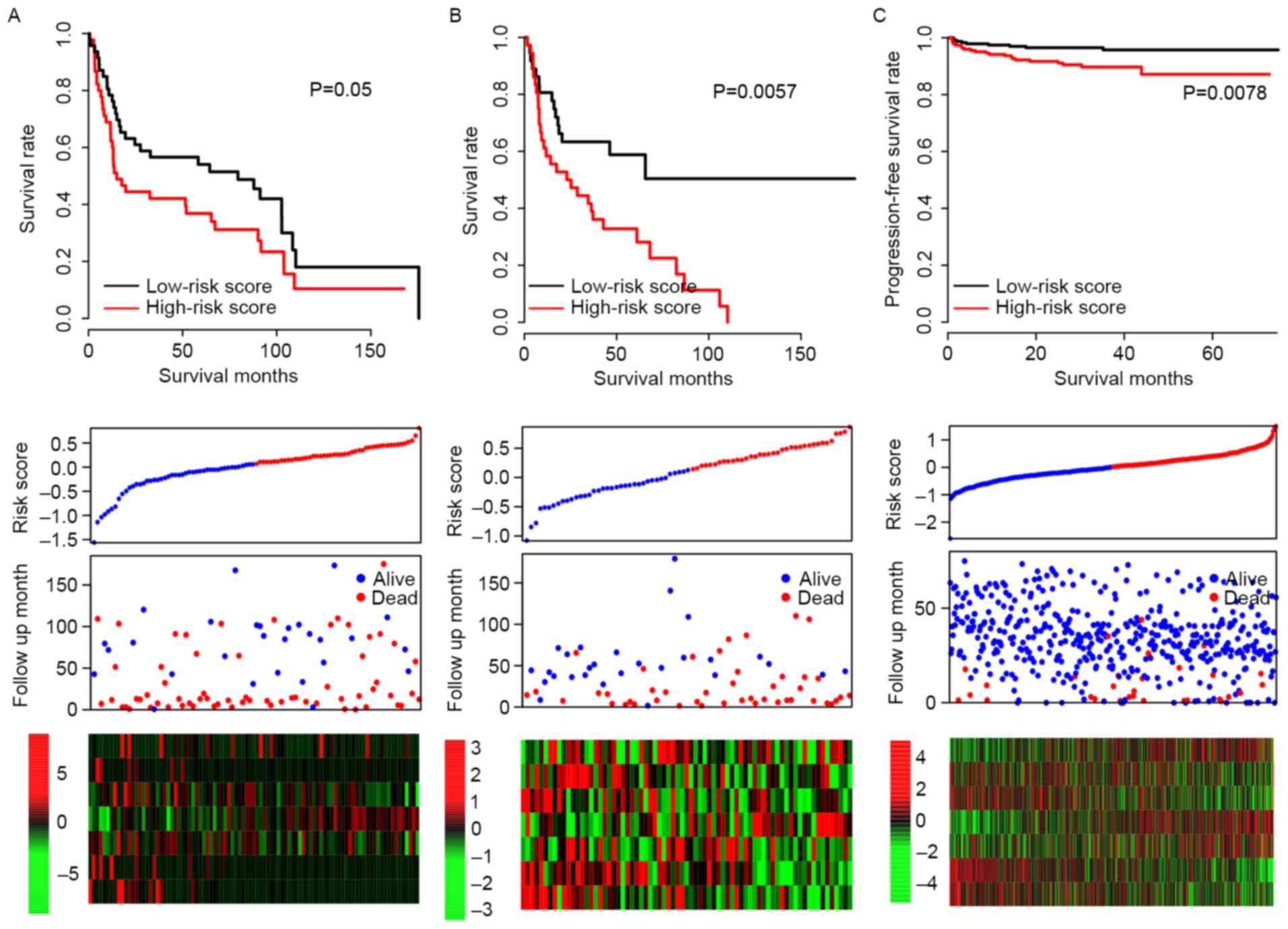
Validation of performance of risk
score in test datasets
It was possible that the model may have overfit to
the training dataset; in order to test the robustness of the model,
subsequent to locking the coefficients for each gene, risk scores
of all patients in three independent test datasets (GSE31684,
GSE48075 and E-MTAB-4321) were evaluated, and the median risk score
value of each dataset was used as a cutoff. Consistent with the OS
profile in the training dataset, the OS rate of the high-risk group
was significantly lower than that of the low-risk group in both
GSE31684 and GSE48075 datasets (P=0.050 and P=0.006, respectively;
Fig. 3A and B). The progression-free
survival curve for E-MTAB4321 resembled the RFS curve for the
training dataset (P=0.0078; Fig. 3C)
and the expression patterns of the 7 genes in the GSE31684,
GSE48075 and E-MTAB-4321 datasets were also similar to the training
dataset. These results indicate that the risk score staging system
is robust across datasets.
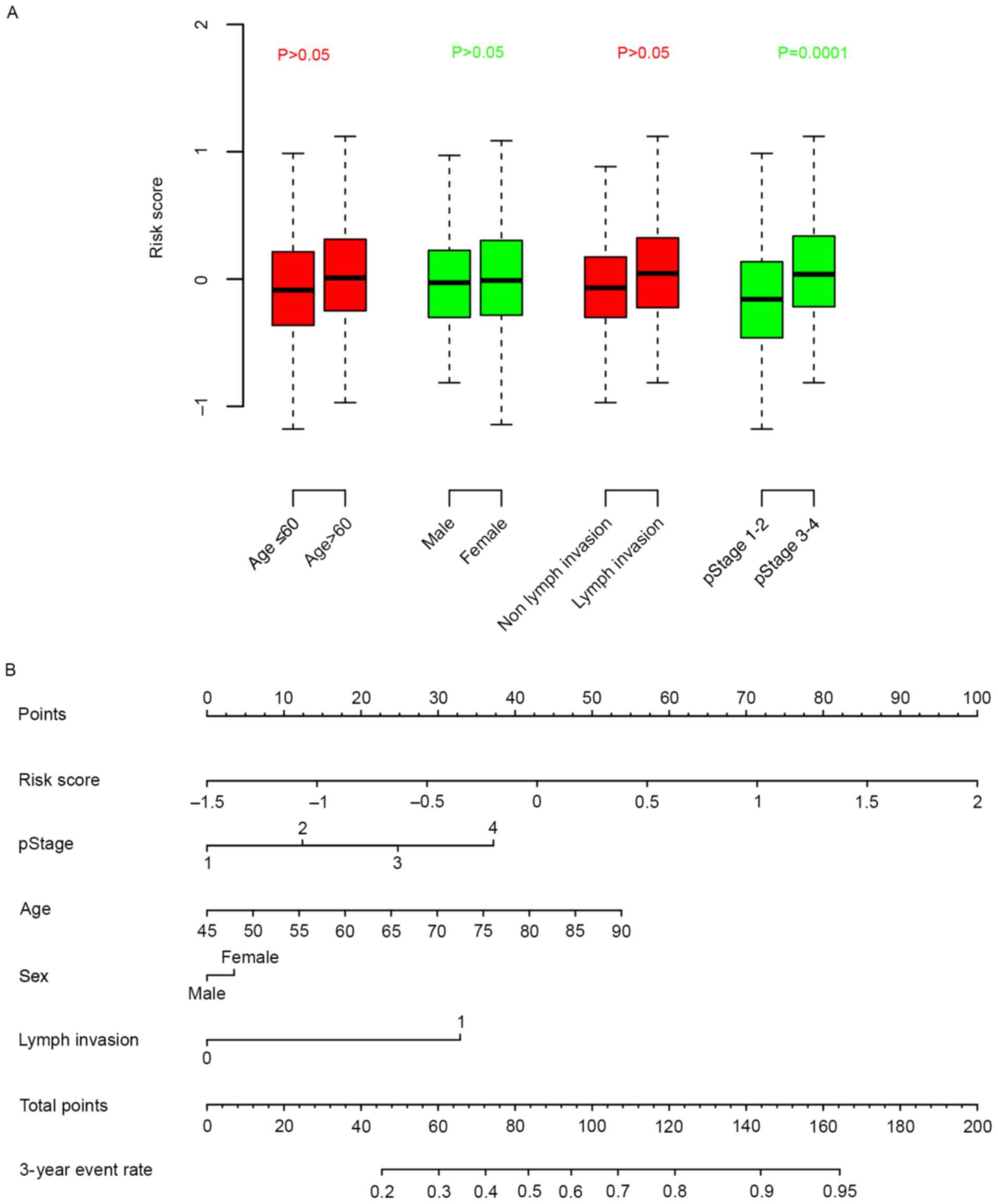
Association between risk score,
clinical information
The association between clinical information and
risk score was calculated. It was observed that the risk score was
independent from age, sex and lymph invasion, but significantly
associated with pathological stage (Fig.
4A). A nomogram for three-year survival, considering
pathological stage, age, sex and lymph invasion status, was plotted
against risk score (Fig. 4B).
According to the nomogram, the risk score ranged the most (from
0–100), indicative of the relative accuracy of the risk score
staging system.
Discussion
The prognostic value of clinical information,
including tumor-node-metastasis staging and age, is currently
unreliable for BLCA (13–15). Therefore, an effective molecular
prognostic biomarker is required to guide the therapy and follow up
of patients with BLCA. Various singular molecular markers for
prognosis have been suggested (16–19) but
the clinical power that they have demonstrated across datasets is
unsatisfactory. In contrast, the predictive effect of multiple
genes has been highlighted as a tool of greater potential (11,20–23). In
the present study, a gene expression and multivariate Cox
regression analysis-based model performed well in the prognosis of
1,049 samples in four independent datasets. The risk score
calculated in this model may therefore be suitable for determining
the prognosis of patients with BLCA.
Of the 7 genes in the model, BCL2L14 has previously
been associated with carcinogenesis (24) and a single-nucleotide polymorphism in
this gene has been associated with lung cancer (25). The role of AHNAK is controversial
between different types of cancer (26); AHNAK has been reported to be
downregulated in melanoma and its low expression associated with
reduced survival time (27), whereas
the high expression of AHNAK is reported to be associated with cell
migration and invasion in mesothelioma (28). To the best of our knowledge, the
remaining genes, ZNF230, TMEM109, APOL2, AGER and AOC2, have not
been associated with prognostic value prior to the present
study.
The clinical application of risk score is feasible
as the quantification of gene expression in cancer tissue is
time-efficient, and the risk score model can be applied to data
from various platforms. However, the present study is constrained
by certain limitations. The study is retrospective, thus important
clinical information, including BLCA subtypes and muscle
invasiveness were not included, and other types of survival
information, including progression-free, recurrence-free and
metastasis-free survival, were not directly predicted by the
model.
In summary, the risk score model constructed in this
study is robust and performed effectively in predicting the
survival of BLCA patients. The model has potential to be developed
as a BCLA prognostic tool.
Acknowledgements
Not applicable.
Funding
No funding received.
Availability of data and materials
Raw data was obtained from Gene Expression Omnibus
(https://www.ncbi.nlm.nih.gov/geo) and
Array Express (www.ebi.ac.uk/arrayexpress/). Probes were matched to
the HUGO Gene (https://www.genenames.org/).
Authors' contributions
JC performed data processing and analysis. FL wrote
the manuscript. NL and FL were responsible for the collection of
the relevant literature. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Miller K and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Funt SA and Rosenberg JE: Systemic,
perioperative management of muscle-invasive bladder cancer and
future horizons. Nat Rev Clin Oncol. 14:221–234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Santoni M, Catanzariti F, Minardi D,
Burattini L, Nabissi M, Muzzonigro G, Cascinu S and Santoni G:
Pathogenic and diagnostic potential of BLCA-1 and BLCA-4 nuclear
proteins in urothelial cell carcinoma of human bladder. Adv Urol.
2012:3974122012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Salomaa V, Havulinna A, Saarela O, Zeller
T, Jousilahti P, Jula A, Muenzel T, Aromaa A, Evans A, Kuulasmaa K
and Blankenberg S: Thirty-one novel biomarkers as predictors for
clinically incident diabetes. PLoS One. 5:e101002010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Izadi F, Zarrini HN, Kiani G and Jelodar
NB: A comparative analytical assay of gene regulatory networks
inferred using microarray and RNA-seq datasets. Bioinformation.
12:340–346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deandrés-Galiana EJ, Fernández-Martínez
JL, Saligan LN and Sonis ST: Impact of microarray preprocessing
techniques in unraveling biological pathways. J Comput Biol.
23:957–968. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Colantuoni C, Henry G, Zeger S and Pevsner
J: SNOMAD (Standardization and NOrmalization of MicroArray Data):
web-accessible gene expression data analysis. Bioinformatics.
18:1540–1541. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dazard JE, Choe M, LeBlanc M and Rao JS: R
package PRIMsrc: Bump hunting by patient rule induction method for
survival, regression and classification. Proc Am Stat Assoc.
2015:650–664. 2015.PubMed/NCBI
|
|
11
|
Robin X, Turck N, Hainard A, Tiberti N,
Lisacek F, Sanchez JC and Müller M: pROC: An open-source package
for R and S+ to analyze and compare ROC curves. BMC bioinformatics.
12:772011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao M, He XL and Teng XD: Understanding
the molecular pathogenesis and prognostics of bladder cancer: An
overview. Chin J Cancer Res. 28:92–98. 2016.PubMed/NCBI
|
|
14
|
Dadhania V, Czerniak B and Guo CC:
Adenocarcinoma of the urinary bladder. Am J Clin Exp Urol. 3:51–63.
2015.PubMed/NCBI
|
|
15
|
Boustead GB, Fowler S, Swamy R,
Kocklebergh R and Hounsome L: Stage, grade and pathological
characteristics of bladder cancer in the UK: British association of
urological surgeons (BAUS) urological tumour registry. BJU Int.
113:924–930. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hong Z, Li H, Li L, Wang W and Xu T:
Different expression patterns of histone H3K27 demethylases in
renal cell carcinoma and bladder cancer. Cancer Biomark.
18:125–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nandagopal L and Sonpavde G: Circulating
biomarkers in bladder cancer. Bladder Cancer. 2:369–379. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang J, Platt LT, Maity B, Ahlers KE, Luo
Z, Lin Z, Chakravarti B, Ibeawuchi SR, Askeland RW, Bondaruk J, et
al: RGS6 is an essential tumor suppressor that prevents bladder
carcinogenesis by promoting p53 activation and DNMT1
downregulation. Oncotarget. 7:69159–69172. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Zhang X, Wang L, Dong Z, Du L,
Yang Y, Guo Y and Wang C: Downregulation of urinary cell-free
microRNA-214 as a diagnostic and prognostic biomarker in bladder
cancer. J Surg Oncol. 111:992–999. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gogalic S, Sauer U, Doppler S, Heinzel A,
Perco P, Lukas A, Simpson G, Pandha H, Horvath A and Preininger C:
Validation of a protein panel for the non-invasive detection of
recurrent non-muscle invasive bladder cancer. Biomarkers.
22:674–681. 2017.PubMed/NCBI
|
|
21
|
Urquidi V, Netherton M, Gomes-Giacoia E,
Serie DJ, Eckel-Passow J, Rosser CJ and Goodison S: A microRNA
biomarker panel for the non-invasive detection of bladder cancer.
Oncotarget. 7:86290–86299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Huang J, Sun J, Xiang S, Yang D,
Ying X, Lu M, Li H and Ren G: The transcription levels and
prognostic values of seven proteasome alpha subunits in human
cancers. Oncotarget. 8:4501–4519. 2017.PubMed/NCBI
|
|
23
|
Kavalieris L, O'Sullivan P, Frampton C,
Guilford P, Darling D, Jacobson E, Suttie J, Raman JD, Shariat SF
and Lotan Y: Performance characteristics of a multigene urine
biomarker test for monitoring for recurrent urothelial carcinoma in
a multicenter study. J Urol. 197:1419–1426. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin ML, Park JH, Nishidate T, Nakamura Y
and Katagiri T: Involvement of maternal embryonic leucine zipper
kinase (MELK) in mammary carcinogenesis through interaction with
Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer
Res. 9:R172007. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Spitz MR, Gorlov IP, Dong Q, Wu X, Chen W,
Chang DW, Etzel CJ, Caporaso NE, Zhao Y, Christiani DC, et al:
Multistage analysis of variants in the inflammation pathway and
lung cancer risk in smokers. Cancer Epidemiol Biomarkers Prev.
21:1213–1221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davis TA, Loos B and Engelbrecht AM:
AHNAK: The giant jack of all trades. Cell Signal. 26:2683–2693.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sheppard HM, Feisst V, Chen J, Print C and
Dunbar PR: AHNAK is downregulated in melanoma, predicts poor
outcome, and may be required for the expression of functional
cadherin-1. Melanoma Res. 26:108–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sudo H, Tsuji AB, Sugyo A, Abe M, Hino O
and Saga T: AHNAK is highly expressed and plays a key role in cell
migration and invasion in mesothelioma. Int J Oncol. 44:530–538.
2014. View Article : Google Scholar : PubMed/NCBI
|