Introduction
RBEL1A represents one of the four isoforms of RBEL1
and harbors a N-terminus Ras/Rab-like GTPase domain followed by a
GTP-binding regulatory domain, a protein-rich region and a
C-terminus nuclear localization signal (1,2). RBEL1A
functions as a GTPase by binding to GTP. Apart from its GTPase
function, RBEL1A was identified to be upregulated in primary breast
cancer tissues compared with its expression in adjacent normal
tissues (3). Downregulation of RBEL1A
led to a suppression of cell growth through cell cycle arrest and
inhibition of cellular migratory and invasive abilities. Those
results indicated the association of RBEL1A with poor prognosis in
several types of cancer (4).
The p53 tumor suppressor is activated under stress
conditions. Activated p53 functions as a positive transcriptional
regulator for >60 genes, which in turn regulate cell cycle, DNA
repair, apoptosis and senescence (5,6). In
cancer, p53 is involved in the induction of chemosensitivity via
its transcriptional activity to regulate cell cycle and apoptosis
(7,8).
The transcriptional activity of p53 depends on the process of
oligomerization (9). In cancer cells,
a number of strategies to prevent p53-mediated cellular control by
inhibiting the transcriptional activity of p53 via dissociating
tetramers have been revealed (10,11). S100B
protein binds specifically to the tetramerization domain of p53
monomers but rarely with the p53 tetramers and leads to a shift of
equilibrium favoring monomeric conformation (12,13).
Apoptosis repressor with caspase recruitment domain (ARC) has been
identified to interact with the C-terminus domain (amino acids,
301–393) of p53 and interferes with the tetramerization of p53
(14).
RBEL1A has been identified to serve an inhibitory
function in the tetramerization of p53 (15). RBEL1A binds to residues 315–360 and
decreases the oligomerization of the exogenously expressed
C-terminus domain (residues, 301–393) of p53 in vitro.
Depletion of RBEL1A increases the oligomerization of p53 and
induces its transcriptional targets, including p21 and Puma in
breast cancer cells (12). However,
whether upregulation of RBEL1A serves any functions in regulating
chemosensitivity via interaction with p53, remains unresolved.
In the present study, changes in the expression
profile of RBEL1A in response to cisplatin treatment were assessed.
Additionally, whether RBEL1A-p53 interaction is regulated by
chemotreatment was examined. Collectively, the results demonstrated
that chemotreatment induced RBEL1A and negatively regulated the
function of p53 by decreasing the protein level of p53 and blocking
the oligomerization of p53 in MCF-7 cells. This may lead to the
development of a promising therapeutic strategy for cancer through
the targeting of p53.
Materials and methods
Cell lines
Human breast cancer cell line MCF-7 was obtained
from the American Type Culture Collection (ATCC; Manassas, VA,
USA). Cells were incubated in minimum essential medium (MEM; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.). Cells
were maintained at 37°C in a humidified atmosphere containing 5%
CO2. To investigate the molecular mechanism underlying
the effects of RBEL1A on p53 and p21, 10 µM MG132 (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) was incubated with MCF-7 cells for
24 h at 37°C in a 5% CO2 incubator.
Plasmids and antibodies
Plasmids encoding HA-tagged p53 (HA-p53, Addgene,
Inc., Cambridge, MA, USA), FLAG-tagged p53 (FLAG-p53, Addgene,
Inc.) or RBEL1A (Addgene, Inc.) was cloned for mammalian expression
from the cytomegalovirus immediate-early promoter in pcDNA3.1
vector (Thermo Fisher Scientific, Inc.). Antibodies used in
chromatin-immunoprecipitation were as follows: Anti-HA tag antibody
(1:500; cat. no. ab9110; Abcam, Cambridge, UK) and anti-FLAG tag
antibody (1:500; cat. no. ab1162; Abcam). Polyclonal RBEL1A
antibody was generated according to the protocol described by
Montalbano et al (3). For
western blot analysis, the primary antibodies used were as follows:
Anti-p21 antibody (1:2,000; cat. no. ab109520; Abcam), anti-p53
antibody (1:2,000; cat. no. ab1101; Abcam), anti-MDM2 antibody
(1:2,000; cat. no. ab16895; Abcam). The secondary antibody used
were as follows: Goat anti-rabbit IgG H&L (HRP) antibody
(1:5,000; cat. no. ab7090; Abcam), Rabbit anti-mouse IgG H&L
(HRP) antibody (1:5,000; cat. no. ab6728; Abcam).
Transfection
MCF-7 cells were seeded at 3×105
cells/well in a 6-well plate and attached overnight. Cells were
transfected at 1.6 µg plasmid/well using Lipofectamine
2000® (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. After 4 h, medium was refreshed and
cells were incubated in MEM supplemented with 10% FBS for 48 h.
Cytotoxicity assay
MCF-7 cells were plated at a density of
2×104 cells/well in a 96-well plate and were left to
attach overnight. Target cells were incubated with serial
concentrations of cisplatin (1, 5, 10, 20, 40, 60, 80 and 100 µM;
Sigma-Aldrich; Merck KGaA) for 24 h. The medium was then removed
and 200 µl fresh medium supplemented with 20 µl MTT (5 mg/ml
dissolved in PBS; Merck KGaA) was added to each well. Following 4 h
incubation at 37°C, supernatant was removed and 200 µl dimethyl
sulfoxide (Merck KGaA) was added into each well. Absorbance at 570
nm was measured using a microplate reader (Synergy 2 Multi-Mode
Microplate Reader; BioTek Instruments, Inc., Winooski, VT,
USA).
RNA interference (RNAi)
Knockdown of RBEL1A was performed by transfecting
RBEL1A-specific small hairpin (sh)RNA construct in a pLKO.1
lentiviral vector. The structure of the primers for shRNA consisted
of the following elements: Sense, loop (underlined), and
antisense). The primers were as follows: shRBEL1A,
5′-CCGCCAGTGTTTCTCAGGGATCTCGAGATCCCTGAGAAACACTGGCGG-3′; shScramble,
5′-AGGTTCCATGTGCGGTTCACCCTCGAGGGTGAACCGCACATGGAACCT-3′; shp53,
5′-CCGACTCCAGTGGTAATCTACTTCAAGAGAGTAGATTACCACTGGAGTCTTTTT-3′;
shScramble,
5′-CCAAGTCCTGGTTCAGCACATTTCAAGAGAATGTGCTGAACCAGGACTTTTTT-3′. 293T
cells were transfected with a target vector along with packaging
plasmid psPAX2 and envelope plasmid pMD2.G (Addgene, Inc.) at the
ratio of 1:1.5:1. At 4 h post-transfection, the medium was replaced
with fresh medium supplemented with 10% FBS. Following 3 days of
culture, the supernatants containing viral particles were collected
and the titer was determined. Briefly, on day 1, 1×105
MCF-7 cells were plated in a 12-well plate and were left to attach
overnight. On day 2, cells were infected with 3-fold serial
dilutions of the viruses in MEM containing 10 µg/ml polybrene
(Sigma-Aldrich; Merck KGaA). GFP-positive cells were observed under
microscopy and the optimal dilution was determined.
Dual-Luciferase® reporter
assay
pG13-Luciferase (pG13-luc; a gift from Dr Jianjun
Chen, Sichuan University, Chengdu, China) was stored in the
laboratory. Cells were seeded into a 12-well plate for conducting
the luciferase assays. Transfection of cells was performed using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. At 24 h post-transfection
at 37°C, cell lysates were subjected to the luciferase assay. To
detect luciferase and β- galactosidase activity, a luciferase
substrate (Promega Corporation, Madison, WI, USA) and the
Galacto-Star™ β-galactosidase Reporter Gene Assay System
for Mammalian cells (Cat. no.: T1012; Thermo Fisher Scientific,
Inc.) were employed according to the manufacturer's protocol.
Relative values of luciferase activity were calculated using
β-galactosidase activity as an internal control for
transfection.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA isolated from MCF-7 cells using Trizol
(Thermo Fisher Scientific, Inc) was performed for first strand cDNA
synthesis using Superscript III RT-qPCR kit (Thermo Fisher
Scientific, Inc.). The following primers were used for the qPCR:
RBEL1A, 5′-CCGATGTGACTGACGAGGATGAG-3′ (forward) and
5′-GTGTTTGCTCTTCTTCTTGGCAGC-3′ (reverse); β-actin,
5′-CATGTACGTTGCTATCCAGGC-3′ (forward) and
5′-CTCCTTAATGTCACGCACGAT-3′ (reverse); p53,
5′-CAGCACATGACGGAGGTTGT-3′ (forward) and
5′-TCATCCAAATACTCCACACGC-3′ (reverse); p21,
5′-TGTCCGTCAGAACCCATGC-3′ (forward) and 5′-AAAGTCGAAGTTCCATCGCTC-3′
(reverse). For chromatin immunoprecipitation analysis, the primers
were as follows: p21 promoter region, 5′-CTGGACTGGGCACTCTTGTC-3′
(forward) and 5′-CTCCTACCATCCCCTTCCTC-3′ (reverse); and DHFR 5′UTR,
5′-TGTAAAACGACGGCCAGTC-3′ (forward) and 5′-CCAGGAAACAGCTATGACC-3′
(reverse). The PCR program was as follows: 5 min 95°C hot start, 40
cycles of 10 sec 94°C, 10 sec 60°C and 1 min 72°C; 10 min 72°C
incubation. Purified PCR products were cloned into a pCR2.1 vector
(Thermo Fisher Scientific, Inc.) followed by DNA sequencing. In
order to quantify gene expression, the 2−∆∆Cq method was
used (16). RT-qPCR was performed in
triplicate.
Chromatin immunoprecipitation and
western blot analysis
MCF-7 cells were plated in 100 mm tissue culture
dishes and co-transfected with Flag-p53 and HA-p53 constructs for
24 h. Cells were trypsinized and placed in 6-well plates and left
to attach overnight. Then, cells were treated with cisplatin
[IC30 concentration, evaluated by the isobolographic
method, as described previously (17)] for 24 h. Cells treated with equal
volume of dimethyl sulfoxide were used as the control group. Whole
cells were lysed using a lysis buffer [50 mM Tris/HCl (pH 8.0), 1
mM EDTA, 120 mM NaCl, 10% glycerol, 0.5% NP40, 1 mM dithiothreitol
(DTT) and 0.5 mM phenylmethylsulfonyl fluoride (PMSF)], sonicated
for 30 cycles (for each cycle, 30 sec on/30 sec off) using
Diagenode Bioruptor Standard (Model UCD200), and centrifuged at
21,000 × g and 4°C for 10 min. Supernatant was diluted 10 times
with lysis buffer, and incubated with 20 µl protein A/G-agarose
beads (Sigma-Aldrich; Merck KGaA) and anti-FLAG/anti-HA antibody.
The beads were washed three times with buffer containing 20 mM
HEPES (pH 7.9), 120 mM NaCl, 1 mM EDTA, 1 mM PMSF, and 1 mM DTT
followed by centrifugation and boiled with 200 ul elution buffer
(10 mM Tris-HCl, pH 8.0, 0.5 mM EDTA). Eluted sample was analyzed
by RT-qPCR, with the aforementioned protocols. Then, western blot
analysis was performed as aforementioned. Total proteins were
isolated using radioimmunoprecipitation lysis and extraction buffer
(Thermo Fisher Scientific, Inc.) and quantified using a
bicinchoninic acid protein assay kit (Sigma-Aldrich; Merck KGaA)
according to the manufacturer's instructions. Proteins (20 µg/lane)
from total cell lysates were fractionated using SDS-PAGE and a
10–15% gel and transferred onto polyvinylidene fluoride membranes
(Thermo Fisher Scientific, Inc.). Membranes were blocked with 5%
skimmed milk in Tris-buffered saline with 0.2% Tween-20 (TBST) at
room temperature for 1 h. Membranes were then incubated with
anti-p21 antibody, anti-p53 antibody or anti-MDM2 antibody
(dilution, 1:2,000) overnight at 4°C, respectively. After three
washes with TBST, secondary antibody (cat. no. ab7090; dilution,
1:5,000; Abcam) was incubated with the membrane at room temperature
for 1 h. Imaging was performed using X-ray films (Kodak, Rochester,
NY, USA) as described previously (3).
Cell cycle analysis
Cells were harvested by trypsinization and fixed for
4 h with 70% ice-cold ethanol at −20°C. Fixed cells were washed
with ice-cold PBS for three times and stained with 1 ml propidium
iodide (50 µg/ml; Sigma-Aldrich; Merck KGaA) containing 0.1% Triton
X-100 and 0.1 mg/ml RNase in darkness at room temperature for 30
min and analyzed by flow cytometry with ModFit LT software (Verify
Software, Topsham, MN, USA).
Cell Counting kit-8 (CCK-8)
MCF-7 cells were seeded at a density of
1×104 cells/well in 96-well plates and incubated
overnight. Following cisplatin treatment, 10 µl CCK-8 solution was
added to each well and incubated for 4 h at 37°C. Cell
proliferation was determined by measuring the absorbance at a
wavelength of 450 and 620 nm. Cell viability was calculated as
(OD450-OD620 in treatment
group)/(OD450-OD620 in control group) ×100.
Experiments were performed in triplicate. Two individual
experiments were performed.
EdU incorporation assay
The apollo DNA labeling kit (Guangzhou RiboBio Co.,
Ltd., Guangzhou, China) was used to analyze cell proliferation.
MCF-7 cells were seeded in a 12-well plate (2×105
cells/well), treated with 50 mM EdU for 2 h and fixed with 4%
paraformaldehyde for 15 min at room temperature. The cells were
incubated with 2 mg/ml glycine for 10 min to reverse fixation and
washed with PBS three times. The cells were permeated with 100
µl/well permeabilization buffer containing 0.5% Triton X-100 and
incubated with 100 µl of 1X apollo solution for 30 min in the dark.
Following this, cells were observed under fluorescence microscope
(magnification, ×100; Olympus Corporation, Tokyo, Japan).
Invasion assay
Transwell membranes were precoated with 100 µl
Matrigel (8%) in MEM and incubated at 37°C for 4 h. A total of
5×103 MCF-7 cells were plated in the upper chambers of
Transwell plates in MEM (200 µl). MEM (600 µl) supplemented with
10% FBS was plated in the lower chambers. Following incubation for
24 h at 37°C, the invasive cells were fixed with 4%
paraformaldehyde at room temperature for 10 min and stained with
0.1% crystal violet stain (in PBS) at room temperature for 10 min.
Stained cells were counted in five randomly-selected fields under a
X71 fluorescence microscope (magnification, ×100; Olympus
Corporation).
Statistical analysis
Data were analyzed using SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA). The relevant data are expressed as the
mean ± standard error of the mean. Statistical significance between
treated and control groups was determined using one-way analysis of
variance followed by Tukey's post hoc test and Student-Neuman-Keuls
method. Statistical significance between two groups was determined
using Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Regulation of mRNA and protein levels
of RBELIA and p53 in response to IC30-cisplatin
treatment in MCF7 cells
It has been reported that RBEL1A is overexpressed in
~67% primary breast tumors (3), which
indicates its potential function in regulating chemosensitivity. In
order to explore whether RBEL1A is involved in the molecular
mechanisms underlying chemosensitivity, IC30 cisplatin
(22.4 µM) was employed to detect the expression of RBEL1A and p53
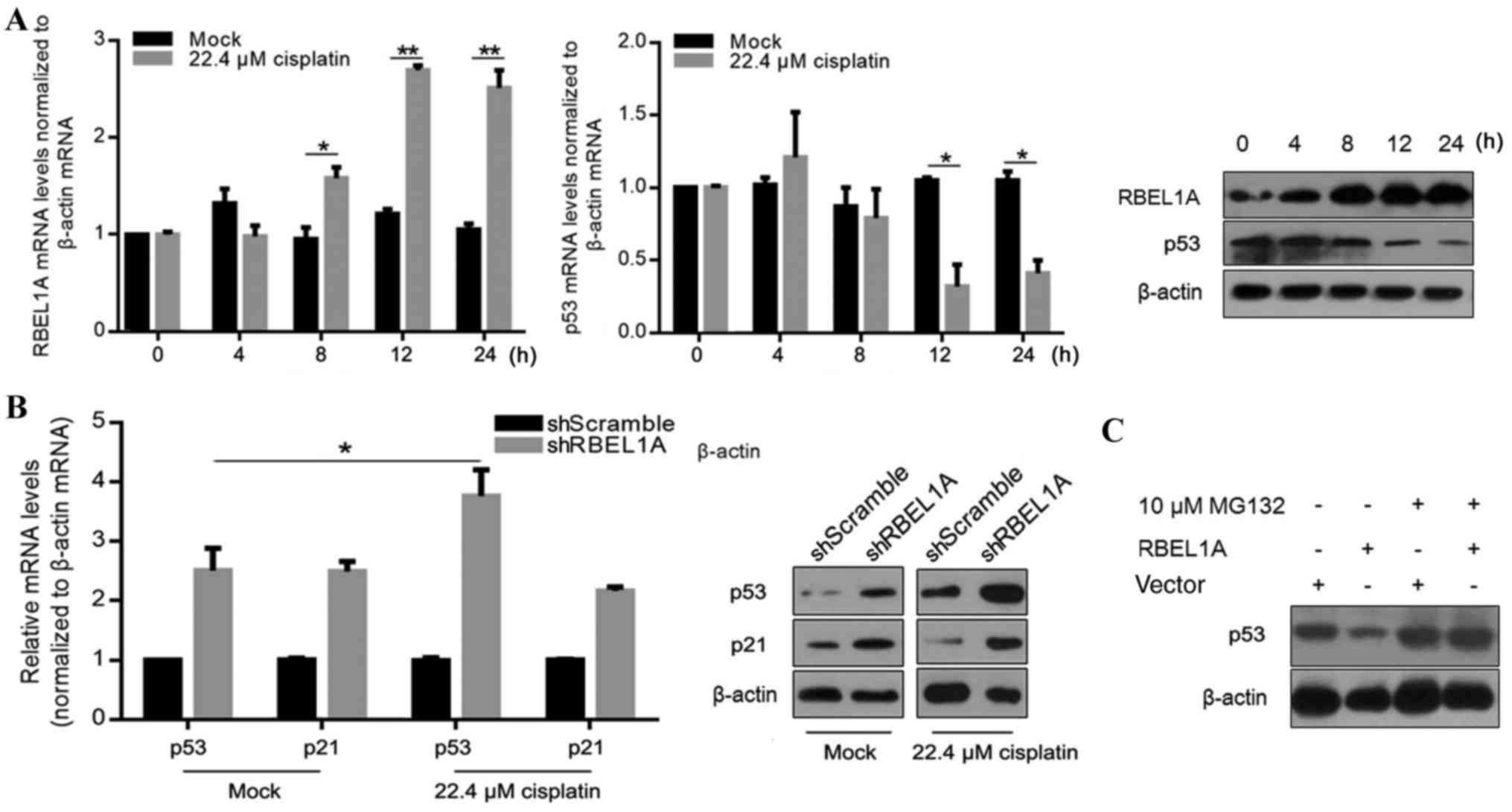
at 0, 4, 8, 12 and 24 h. As presented in Fig. 1A, mRNA levels of RBEL1A were
significantly increased at 8, 12 and 24 h compared with the
control. Consistently, the protein levels were also obviously
increased. Additionally, mRNA and protein levels of p53 were
decreased at 12 and 24 h after cisplatin treatment (Fig. 1A). In breast cancer cells, p53 is
reported to be transcriptionally and post-transcriptionally
regulated following overexpression of RBEL1A (2). Next, the effects of upregulation of
RBEL1A on the target gene of p53, p21, were investigated in
cisplatin-treated MCF-7 cells. The results demonstrated that
downregulation of RBEL1A (using a shRNA target to RBEL1A, shRB3L1A)
let to an upregulation of p53 and p21 in response to cisplatin
treatment in MCF-7 cells (Fig. 1B).
Knockdown of RBEL1A also resulted in an upregulation of p53 and p21
in untreated MCF-7 cells (Fig. 1B),
indicating a regulatory effect of RBEL1A on p53 and p21 under
normal conditions. Next, MCF-7 cells were treated with cisplatin
and with 10 µM MG132, which mediates proteasome inhibition after
ubiquitination, in order to investigate the molecular mechanism
underlying the effects of RBEL1A on p53 and p21. The expression of
p53 was examined using western blot analysis. Fig. 1C demonstrated that RBEL1A-mediated
decrease in p53 protein levels was abrogated in cells treated with
MG132. These results suggest that upregulation of RBEL1A following
cisplatin treatment potentially decreased the expression levels of
p53 by accelerating ubiquitination.
RBEL1A partially inhibits cisplatin
sensitivity in MCF-7 cells
It has been demonstrated that p53 functions as a
positive regulator of cisplatin-mediated chemotherapy in breast
cancer (13). Variable responses to
cytotoxicity were indicated in response to cisplatin treatment in
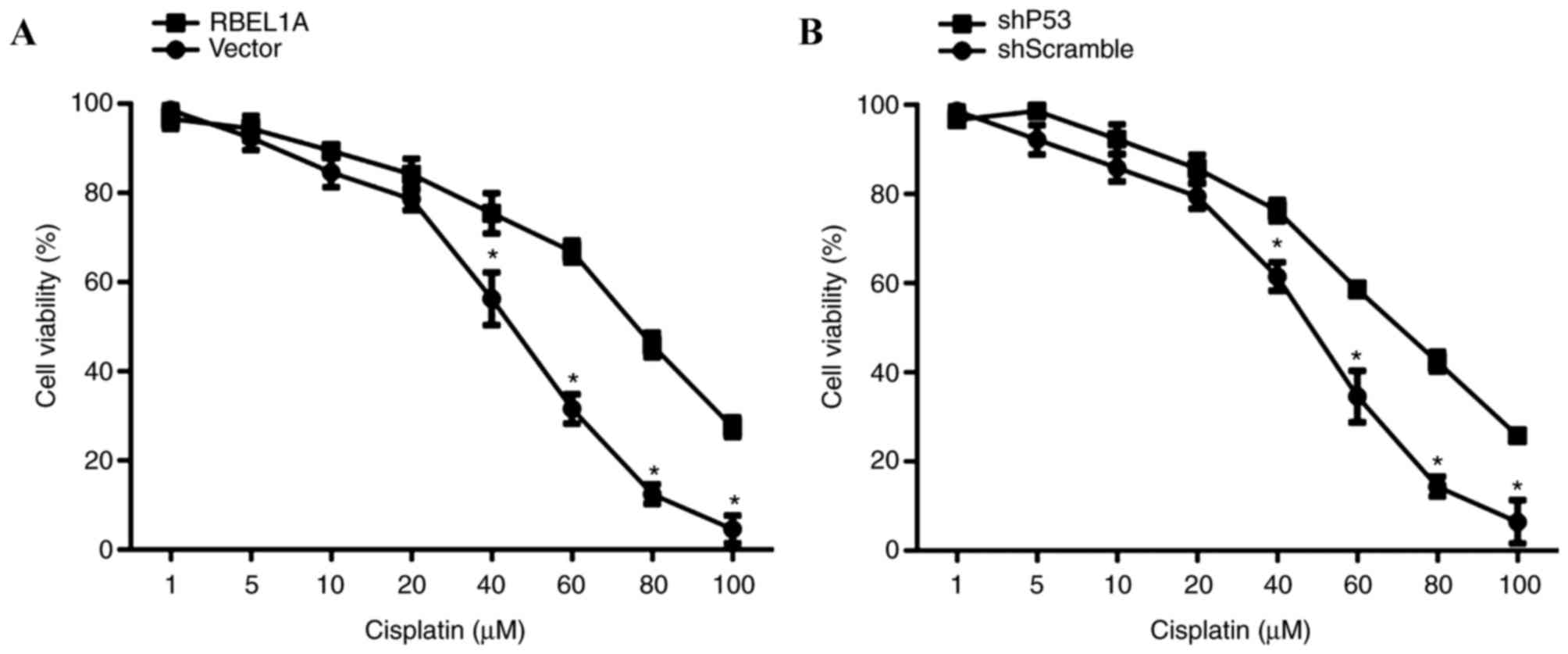
MCF-7-shRBEL1A and MCF-7-shp53 cells. The CCK-8 assay results
illustrated that overexpression of RBEL1A increased cell viability
compared with that of vector-transfected cells, indicating that
RBEL1A led to a significant desensitization of MCF-7 cells to
cisplatin (40, 60, 80 and 100 µM) (Fig.
2A). Additionally, knockdown of p53 (achieved using shp53) also
led to a significant desensitization of MCF-7 cells to cisplatin
(40, 60, 80 and 100 µM) as assessed using a MTT assay (Fig. 2B).
Cisplatin treatment stimulated the
regulatory activity of p53 via upregulating RBEL1
The transcriptional activity of p53 is critical for
inducing chemosensitivity (13),
which may be tightly regulated by cisplatin-induced RBEL1A in MCF-7
cells. Therefore, whether RBEL1A-mediated p53 downregulation in
cisplatin-treated MCF-7 cells may exhibit an effect on the
interaction between p53 and its target DNA sequence was
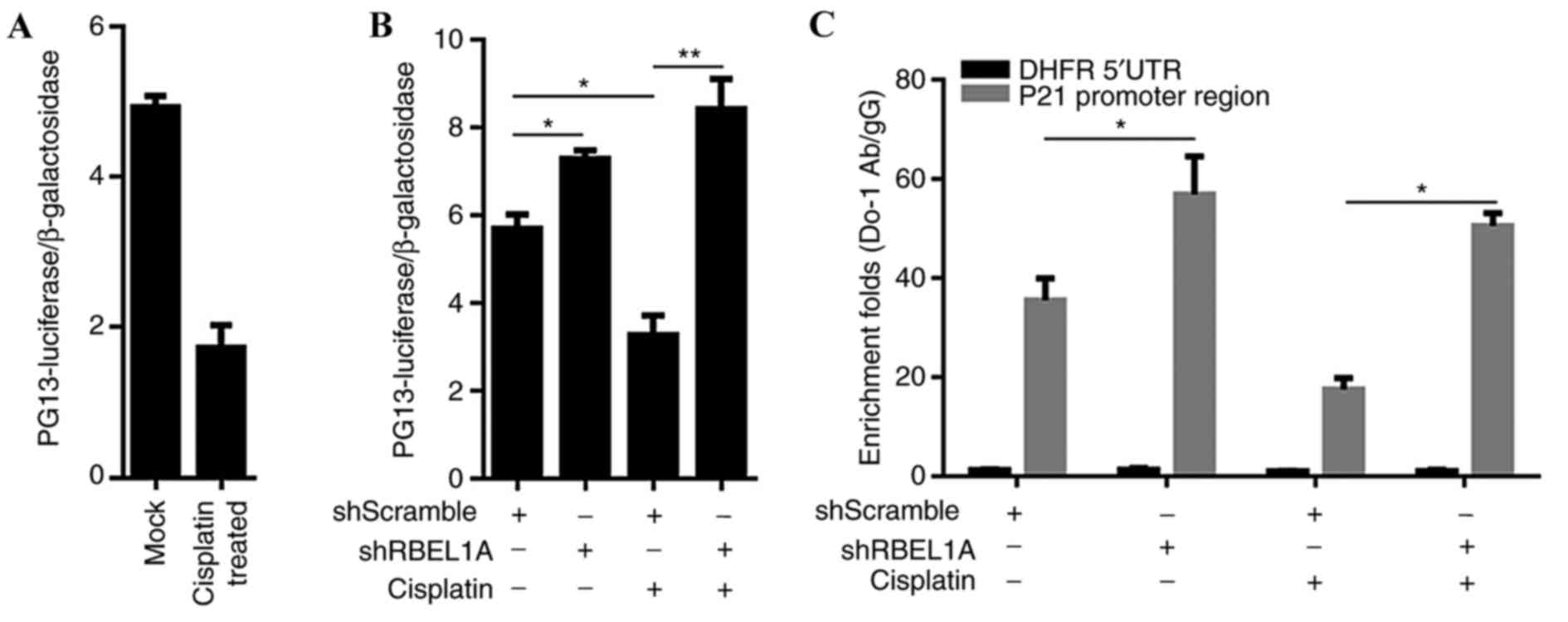
investigated. The transcriptional activity of p53 in
cisplatin-treated MCF-7 cells was confirmed using a pG13L
luciferase reporter assay. Results demonstrated that cisplatin
treatment reduced the transcriptional activity of p53 compared with
mock-treated MCF-7 cells (Fig. 3A).
In order to confirm the decrease of the transcriptional activity of
p53, MCF-7 cells were treated with shRBEL1A prior to cisplatin
treatment. According to the results, knockdown of RBEL1A failed to
decrease the transcriptional activity of p53 in cisplatin- or
mock-treated MCF-7 cells (Fig. 3B)
and led to increased transcriptional activity of p53. Additionally,
cisplatin treatment decreased the binding of p53 to p21's promoter
region, as assessed using chromatin-immunoprecipitation (Fig. 3C).
Knockdown of RBEL1A inhibited
proliferation, blocked entry of cell cycle and invasive ability in
cisplatin-treated MCF-7 cells
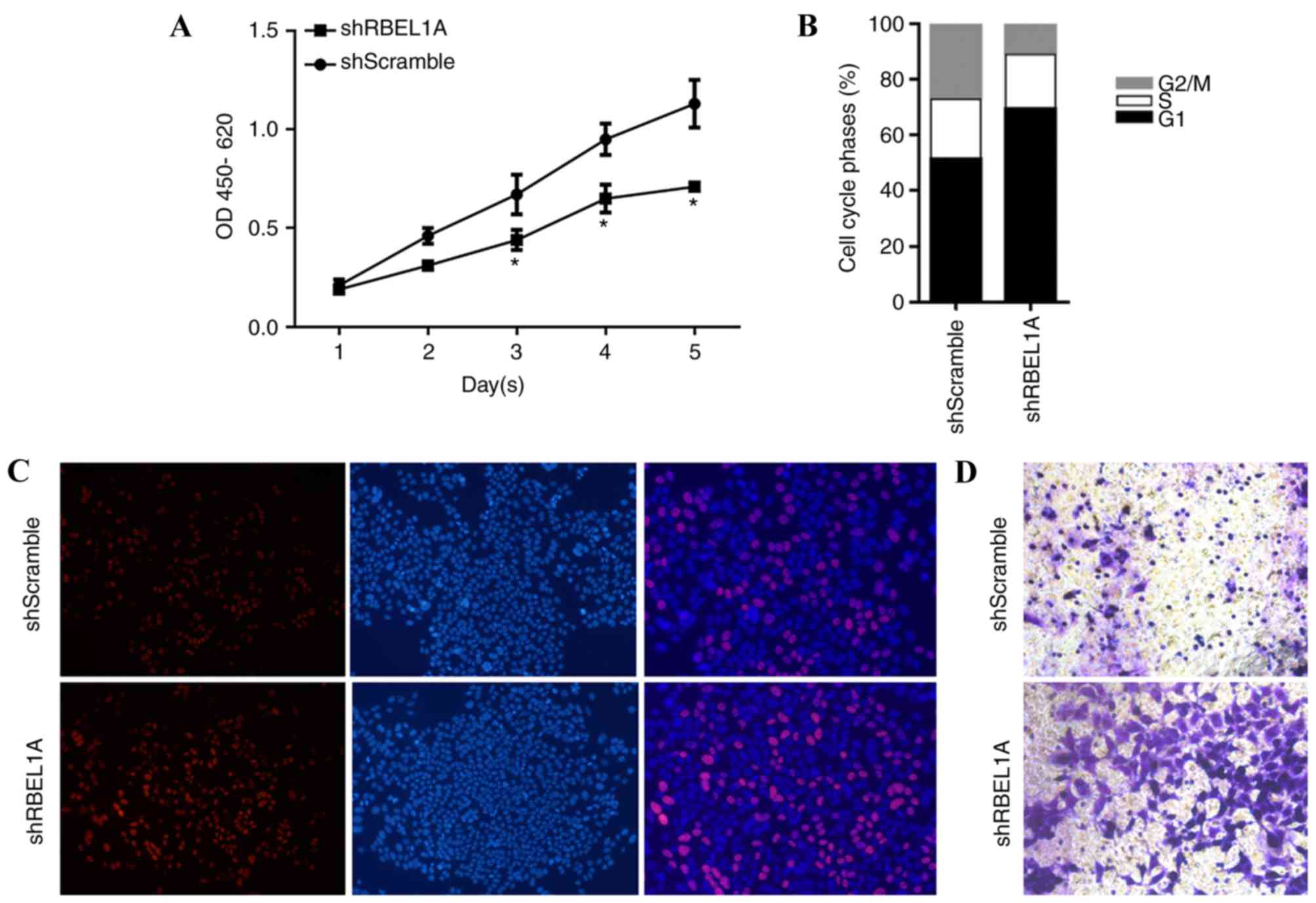
In order to identify the effects of
cisplatin-induced RBEL1A expression on physiological processes,
MCF-7-shScramble and MCF-7-shRBEL1A cells were treated with
IC30 concentration of cisplatin. As presented in
Fig. 4A and B, knockdown of RBEL1A
significantly increased the proliferating rate (at day 3, 4 and 5)
by accelerating cell cycle entry. By performing EdU staining
(red-stained cell represents proliferating cells), it is
demonstrated that treatment with shRBELIA regulated the
proliferation of MCF-7 cells (Fig.
4C). Furthermore, invasive activity was also been promoted in
MCF-7-shRBEL1 cells (Fig. 4D). Taken
together, the results demonstrated that knockdown of RBEL1A may
inhibit proliferation, and arrest cell cycle and invasion of MCF-7
cells in response to cisplatin treatment.
Discussion
Oligomerization is critical in p53-mediated
regulation of apoptosis and chemosensitivity (14,15). The
equilibrium of monomer and oligomer shifts under intracellular or
extracellular stress. In normal conditions, p53 may predominantly
exist as latent monomers, the monomers tend to oligomerize to form
dimers, trimers, and dimers of dimer (tetramers) under stress
conditions. Although monomers present slight DNA binding activity,
tetramerized p53 binds tightly and specifically to transactivate
promoters of various target genes that are involved in the
regulation of cellular processes, including cell cycle arrest,
apoptosis, cellular senescence and DNA repair (5). Despite mutations occurring in
oligomerization domain of p53 (residues 301–363), proteins that
block p53 oligomerization are expected to be a novel strategy for
inhibiting p53′ transcriptional activity (8). Several proteins have been reported to be
involved in preventing p53's transcriptional activity via
dissociating p53 oligomers by binding to p53 monomers. The
predominant members of S100 protein family, S100A and S100B bind to
the tetramerization domain of p53 specifically and lead to tetramer
dissociation (10,11). These proteins were upregulated in
various human malignancies, thus indicating their potential
function in tumorigenesis and induction of chemoresistance
(18,19). ARC has been reported to regulate p53's
transcriptional activity via binding directly to p53's
tetramerization domain (11). ARC was
demonstrated to be upregulated in human colon cancer, and thus
inhibited p53 tetramerization and nuclear translocation.
Consequently, the transcriptional regulation of p53 to its target
genes decreased (20). RBEL1A also
interacts with p53 at p53's tetrameric domain and may lead to
dissociation of p53 tetramers (12).
However, whether RBEL1A is involved in the induction of
chemoresistance via regulating p53's transcriptional activity
remains unclear.
In the present study, it was demonstrated that mRNA
and protein levels of RBEL1A were upregulated in response to
cisplatin treatment. Expression of RBEL1B, which is one of the
RBEL1 isoforms was unaffected in response to cisplatin treatment
(data not shown), which is consistent with previous studies,
indicating the positive association between RBEL1A but not RBEL1B
with poor prognosis in breast cancer (3). It has been revealed that upregulation of
RBEL1A inhibited p53 oligomerization in response to cisplatin
treatment in 293 cells (15).
Additionally, upregulation of RBEL1A decreased p53 protein level by
transcriptional inhibition and accelerating protein degradation.
Upregulation of RBEL1A regulated p53's transcriptional activity on
reporter gene and downstream target gene as assessed using
luciferase reporter assay and chromatin-immunoprecipitation.
Although, the protein levels of RBEL1A increased following
cisplatin treatment, its mRNA levels were unchanged, indicating
that the effect of cisplatin on the expression of RBEL1A was on a
post-transcriptional level. Several potential molecular mechanisms
may be involved, including post-transcriptional regulation by
microRNA targeting RBEL1A mRNA or accelerated ubiquitination.
Future studies are required to unravel the molecular mechanisms
underlying the regulation of RBEL1A by chemotreatment.
In breast tumors, nearly half of them contain mutant
p53 and ~70% of mutations in p53 are missense mutations (21). Compared with p53 deficiency, p53
mutants demonstrate increased functional abnormality due to its
multifunction. For example, mutant p53 was reported to positively
regulate signaling pathways involved in cellular proliferation and
metabolism (22). Mutant p53 has been
revealed to promote the expressing level of 15-lypoxygenase, which
is positively associated with tumor progression and survival rate
of breast cancer cells (23). Mutant
p53 was also reported to promote vascular endothelial growth factor
expression in breast cancer (24).
However, the interaction between RBEL1A and mutant p53 remains
unclear.
To conclude, the results of the present study
demonstrated that cisplatin treatment significantly induced the
expression of RBEL1A, thus blocked the transcriptional activity of
p53. This interaction may partially contribute to the induction of
cisplatin-mediated chemosensitization.
Acknowledgements
The authors would like to thank Dr Huimin Shi
(Sichuan University) for language editing.
Funding
The present study was supported by the Sichuan
Provincial Scientific Grant (grant nos. 2016FZ0096 and 2016FZ0093)
and the Luzhou Scientific Grant (grant no. 2014-S-44).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
CC and ZZ designed the experiments. ST performed the
gene expression analysis and cell-related experiments. CZ wrote the
manuscript, provided funding support and performed data
analysis.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Montalbano J, Lui K, Sheikh MS and Huang
Y: Identification and characterization of RBEL1 subfamily of
GTPases in the Ras superfamily involved in cell growth regulation.
J Biol Chem. 284:18129–18142. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lui K, An J, Montalbano J, Shi J, Corcoran
C, He Q, Sun H, Sheikh MS and Huang Y: Negative regulation of p53
by Ras superfamily protein RBEL1A. J Cell Sci. 126:2436–2445. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Montalbano J, Jin W, Sheikh MS and Huang
Y: RBEL1 is a novel gene that encodes a nucleocytoplasmic Ras
superfamily GTP-binding protein and is overexpressed in breast
cancer. J Biol Chem. 282:37640–37649. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Levine AJ, Hu W and Feng Z: The P53
pathway: What questions remain to be explored? Cell Death Differ.
13:1027–1036. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang L, Sheikh MS and Huang Y: Decision
making by p53: Life versus death. Mol Cell Pharmacol. 2:69–77.
2010.PubMed/NCBI
|
|
6
|
Duffy MJ, Synnott NC, McGowan PM, Crown J,
O'Connor D and Gallagher WM: p53 as a target for the treatment of
cancer. Cancer Treat Rev. 40:1153–1160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ozaki T and Nakagawara A: p53: The
attractive tumor suppressor in the cancer research field. J Biomed
Biotechnol. 2011:6039252011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Higashimoto Y, Asanomi Y, Takakusagi S,
Lewis MS, Uosaki K, Durell SR, Anderson CW, Appella E and Sakaguchi
K: Unfolding, aggregation, and amyloid formation by the
tetramerization domain from mutant p53 associated with lung cancer.
Biochemistry. 45:1608–1619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Dieck J, Fernandez-Fernandez MR,
Veprintsev DB and Fersht AR: Modulation of the oligomerization
state of p53 by differential binding of proteins of the S100 family
to p53 monomers and tetramers. J Biol Chem. 284:13804–13811. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin J, Yang Q, Yan Z, Markowitz J, Wilder
PT, Carrier F and Weber DJ: Inhibiting S100B restores p53 levels in
primary malignant melanoma cancer cells. J Biol Chem.
279:34071–34077. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Foo RS, Nam YJ, Ostreicher MJ, Metzl MD,
Whelan RS, Peng CF, Ashton AW, Fu W, Mani K, Chin SF, et al:
Regulation of p53 tetramerization and nuclear export by ARC. Proc
Natl Acad Sci USA. 104:20826–20831. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lui K, Sheikh MS and Huang Y: Regulation
of p53 oligomerization by Ras superfamily protein RBEL1A. Genes
Cancer. 6:307–316. 2015.PubMed/NCBI
|
|
13
|
Boulikas T and Vougiouka M: Cisplatin and
platinum drugs at the molecular level. (Review). Oncol Rep.
10:1663–1682. 2003.PubMed/NCBI
|
|
14
|
Stavridi ES, Chehab NH, Caruso LC and
Halazonetis TD: Change in oliogmerization specificity of the p53
tetramerization domain by hydrophobic amino acid substitutions.
Protein Sci. 8:1773–1779. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fernandez-Fernandez MR, Veprintsev DB and
Fersht AR: Proteins of the S100 family regulate the oligomerization
of p53 tumor suppressor. Proc Natl Acad Sci USA. 102:4735–4740.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tallarida RJ and Murray RB: Manual of
pharmacologic calculations with computer programs. Springer
Publisher; pp. 100–104. 1986
|
|
18
|
Sekine H, Chen N, Sato K, Saiki Y, Yoshino
Y, Umetsu Y, Jin G, Nagase H, Gu Z, Fukushige S, Sunamura M and
Horii A: S100A4, frequently overexpressed in various human cancers,
accelerates cell motility in pancreatic cells. Biochem Biophys Res
Commun. 429:214–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maletzki C, Bodammer P, Breitrück A and
Kerkhoff C: S100 proteins as diagnostic and prognostic markers in
colorectal and hepatocellular carcinoma. Hepat Mon. 12(10 HCC):
e72402012.PubMed/NCBI
|
|
20
|
Mercier I, Vuolo M, Jasmin JF, Medina CM,
Williams M, Mariadason JM, Qian H, Xue X, Pestell RG, Lisanti MP
and Kitsis RN: ARC (apoptosis repressor with caspase recruitment
domain) is a novel marker of human colon cancer. Cell Cycle.
7:1640–1647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Petitjean A, Mathe E, Kato S, Ishioka C,
Tavtigian SV, Hainaut P and Olivier M: Impact of mutant p53
functional properties on TP53 mutation patterns and tumor
phenotype: Lessons from recent developments in the IARC TP53. Hum
Mutat. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kelavkar UP and Badr KF: Effects of mutant
p53 expression on human 15-lipoxygenase-promoter activity and
murine 12/15-lipoxygenase gene expression: Evidence that
15-lipoxygenase is a mutator gene. Proc Natl Acad Sci USA.
96:4378–4383. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reddy N, Everhart A, Eling T and Glasgow
W: Characterization of a 15-lipoxygenase in human breast carcinoma
BT-20 cells: Stimulation of 13-HODE formation by TGF alpha/EGF.
Biochem Biophys Res Commun. 231:111–116. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gallagher EJ and LeRoith D: Minireview:
IGF, insulin, and cancer. Endocrinology. 152:2546–2551. 2011.
View Article : Google Scholar : PubMed/NCBI
|