Introduction
Malignant melanoma is one of the most aggressive
malignancies in humans, and is responsible for 60-80% of skin
cancer-associated mortalities (1). As
revealed in 2013, the 5-year survival rate of patients with
metastatic malignant melanoma is ~14%, and its incidence has been
increasing in the white population over the previous 2 decades
(1). However, the mechanisms leading
to the malignant transformation of melanocytes remains poorly
understood. As a non-receptor tyrosine kinase, focal adhesion
kinase (FAK) is widely expressed in various human tissues and cell
types, including mesenchymal cells, neuronal cells, platelets,
lymphocytes and erythrocytes (2,3).
Overexpressed in a wide range of types of cancer (4), FAK promotes tumor cell migration and
invasion (5). FAK is also involved in
various signaling pathways that promote tumor growth and metastasis
(6). However, the precise mechanisms
underlying the regulatory role of FAK in cell migration remain to
be elucidated.
Coiled-coil domain containing 80 (CCDC80) has been
suggested to be a multifunctional protein among vertebrates
(7). Previous studies have
demonstrated that the expression of CCDC80 was downregulated in
thyroid carcinomas, papillary carcinomas and colorectal and
pancreatic cell lines (8–10). Ectopic expression of CCDC80 inhibits
cancer cells growth in vitro, indicating that CCDC80 may be
a candidate target for tumor therapy (10). In addition, CCDC80 also modulates the
apoptotic pathways (10–12). However, the role of CCDC80 as a
possible tumor suppressor remains unclear.
In the present study, the mechanism underlying the
regulation of B16F10 cell migration by FAK was investigated. The
knockdown of FAK promoted the expressions of CCDC80 and E-cadherin,
and suppressed the migration of B16F10 cells. Meanwhile, CCDC80 was
demonstrated to inhibit the migration of B16F10 cells, and elevate
the level of E-cadherin. Clinical data for patients with melanoma
from the Oncomine Cancer Microarray database were also analyzed and
compared with the experimental data.
Materials and methods
Biological chemicals and
antibodies
Blasticidin was obtained from Invitrogen; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). The primary antibody
against E-cadherin (cat. no.: 610181) was purchased from BD
Biosciences (Franklin Lakes, NJ, USA), and the primary antibodies
against extracellular-signal-regulated kinase (ERK; cat. no.:
4695S) and phosphorylated ERK (p-ERK; cat. no.: 4377S) were
obtained from Cell Signaling Technology Inc. (Danvers, MA, USA).
The primary antibody against tubulin (cat. no.: sc-32293) was
obtained from Santa Cruz Biotechnology, Inc., (Dallas, TX,
USA).
Cells and cell culture
Using small interfering (si)RNA, the stable
interference of FAK in B16F10 cells [American Type Culture
Collection (ATCC), Manassas, VA, USA] (siFAK) and siFAK negative
control (siNC) cell lines were constructed by the State Key
Laboratory of Pharmaceutical Biotechnology, School of Life
Sciences, Nanjing University (Nanjing, Jiangsu). These cells,
alongside B16F1 cells (ATCC), were cultured at 37°C and 5%
CO2 in Dulbecco's modified Eagle's medium (DMEM; Wisent,
Inc., St. Bruno, QC, Canada) supplemented with 10% fetal bovine
serum (FBS; Life Technologies; Thermo Fisher Scientific, Inc.) and
3 mg/l−1 blasticidin.
Gene chip analysis
siFAK and siNC cells were lysed in 1 ml
TRIzol® reagent (Life Technologies; Thermo Fisher
Scientific, Inc.), and gene chip analysis was performed by
CapitalBio Corporation (Beijing, China).
Migration assays
Cell migration was monitored by wound healing and
transwell assays. For the wound healing assay, B16F10 cells that
were transfected with pcDNA3.1-CCDC80 or pcDNA3.1 plasmid or
siCCDC80 RNA were seeded at an initial density of 2×105
cells/well, and cultured (37°C, 5% CO2) in DMEM
supplemented with 10% FBS overnight. Following culture (37°C, 5%
CO2) of the cells in serum-free DMEM for an additional
24 h, a micro-pipette tip was used to create a wound in the
monolayer of cells. Wound closures were observed by phase-contrast
microscopy (magnification, ×40; Olympus Corporation, Tokyo, Japan),
and digital images were captured at the interval times of 0 and 24
h (Photoshop CS6, Adobe Systems, Inc., San Jose, CA, USA). The
percent migration rate was calculated using the formula: (1-T/C)
×100%, where T and C represent the scrape distance at the indicated
treatment time (24 h) and at the previous treatment time (0 h),
respectively.
For the transwell assay, 1×105 cells per
insert (8-µm pore size) were incubated (37°C) in serum-free DMEM
for 24 h in the upper chamber, with DMEM with 10% FBS in the lower
chamber. Then, cells inside the inserts were removed with cotton
swab, while cells on the underside of the inserts were fixed in 10%
formalin for 20 min (room temperature) and stained at room
temperature for 30 min (1% crystal violet). Cells from 5 random
microscopic fields/insert were counted in triplicate at
magnification, ×200.
Total RNA isolation and reverse
transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from siNC and siFAK cell
lines using TRIzol® reagent (Life Technologies; Thermo
Fisher Scientific, Inc.). RNA quantity (absorbance at wavelength
260 nm, A260) and purity (A260/A280) were determined by
BioPhotometer® (Eppendorf, Hamburg, Germany).
First-strand cDNA was synthesized with 1.5 µg of total RNA using a
PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan). RT-qPCR
was performed using FastStart Universal SYBR Green Master (Rox)
(Roche Diagnostics, Basel, Switzerland). The thermocycler
conditions were as follows: 95°C for 10 min, then 94°C for 10 sec
and 60°C for 1 min, for 40 cycles. Data were normalized using
GAPDH. The gene-specific primers were synthesized by Nanjing
GenScript (Nanjing, China), and were as follows: CCDC80 forward,
5′-GATCCTGGAGCAGCCTCTGG-3′; CCDC80 reverse,
5′-ACATGGCTTCCAGCCTGACC-3′; nuclear receptor coactivator-3 (NCOA3)
forward, 5′-TTCGCCTAGTCCACTCATCC-3′; NCOA3 reverse,
5′-GTGGACTCCGAGATCGGTAA-3; E-cadherin forward,
5′-GTGGGTCAGGAAATCACATC-3′; E-cadherin reverse,
5′-CTCCAAATCCGATACGTGATC-3′; GAPDH forward,
5′-TGAAGCAGGCATCTGAGGG-3′; GAPDH reverse,
5′-CGAAGGTGGAAGAGTGGGAG-3′.
RNA and plasmid transfection
CCDC80 (siCCDC80) and negative control (NC) siRNA
were synthesized by Shanghai GenePharma Co., Ltd., (Shanghai,
China). The PcDNA3.1-CCDC80 plasmid was donated by Professor
Kolligs (University of Munich, Munich, Germany). Cells were
transfected with the siRNA (100 pmol) or plasmid (1.5 µg) using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the protocol of the manufacturer,
and cultured for 48 h prior to harvesting and subsequent
experimentation. The sequences of the siRNA were as follows:
siCCDC80 forward, 5′-GAUGAGUAUGCAGGAUAUGUU-3′; siCCDC80 reverse,
5′-CAUAUCCUGCAUACUCAUCUU-3′; siNC forward,
5′-UUCUCCGAACGUGUCACGUTT-3′; siNC reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. The 2−ΔΔCq method was used
for quantification (13).
Western blot analysis
Cells were lysed in lysis buffer (Beyotime,
Institute of Biotechnology, Haimen, China), and following staining
with 0.1 g/l G-250 at room temperature for 1 min, protein
determination was performed by measuring absorbance at a wavelength
of 595 nm using the BioPhotometer® (Eppendorf, Hamburg,
Germany). Total proteins (30 µg) were fractionated using SDS-PAGE
(8%) and transferred onto nitrocellulose membranes. The membranes
were blocked with 5% non-fat dried milk in 1× PBS Tween-20 buffer
(5% tween-20) at room temperature for 1 h. and then incubated with
the aforementioned primary antibodies (1:1,000) at room temperature
for 1 h. Horseradish peroxidase-conjugated anti-mouse (cat. no.:
7076S; 1:2,000 dilution) or anti-rabbit IgG (cat. no.: 7074S;
1:2,000 dilution) from Cell Signaling Technology Inc. were used as
the secondary antibodies at room temperature, and the protein bands
were detected using an enhanced chemiluminesence detection system
(Tanon Science and Technology Co., Ltd., Shanghai, China).
Analysis gene expression by
Oncomine
The Oncomine Platform provides robust, peer-reviewed
analysis methods that compute gene expression signatures, clusters
and gene-set modules, extracting biological observations from the
data. Following the successful registration of an Oncomine
username, Oncomine (www.oncomine.org, last access date August 2016) was
used to analyze the genes, terms searched were ‘CCDC80’, ‘FAK’ and
‘E-cadherin’.
Statistical analysis
The data are presented as the mean ± standard
deviation. Analysis of two groups was performed using Student's
t-test and comparisons of multiple groups was conducted using
one-way analysis of variance with Tukey's post-hoc test. P<0.05
was considered to indicate a statistically significant difference.
Software GraphPad Prism5 (GraphPad Software, Inc., La Jolla, CA,
USA) was used for statistical analysis. A total of 3 replicates
were performed for each experiment.
Results
Knockdown of FAK increases the
expression levels of CCDC80 and E-cadherin in B16F10 cells
To explore the role of FAK in B16F10 melanoma
progression, siFAK and siNC cell lines were constructed. It has
been established previously that compared with those in the siNC
cells, the mRNA and protein levels of FAK in siFAK cells were
decreased markedly (14). In the
present study, the gene chip assay revealed that the mRNA level of
FAK was decreased, while the mRNA levels of CCDC80 and E-cadherin
were increased in the siFAK cells compared with the siNC cells
(Table I). The RT-qPCR assay also
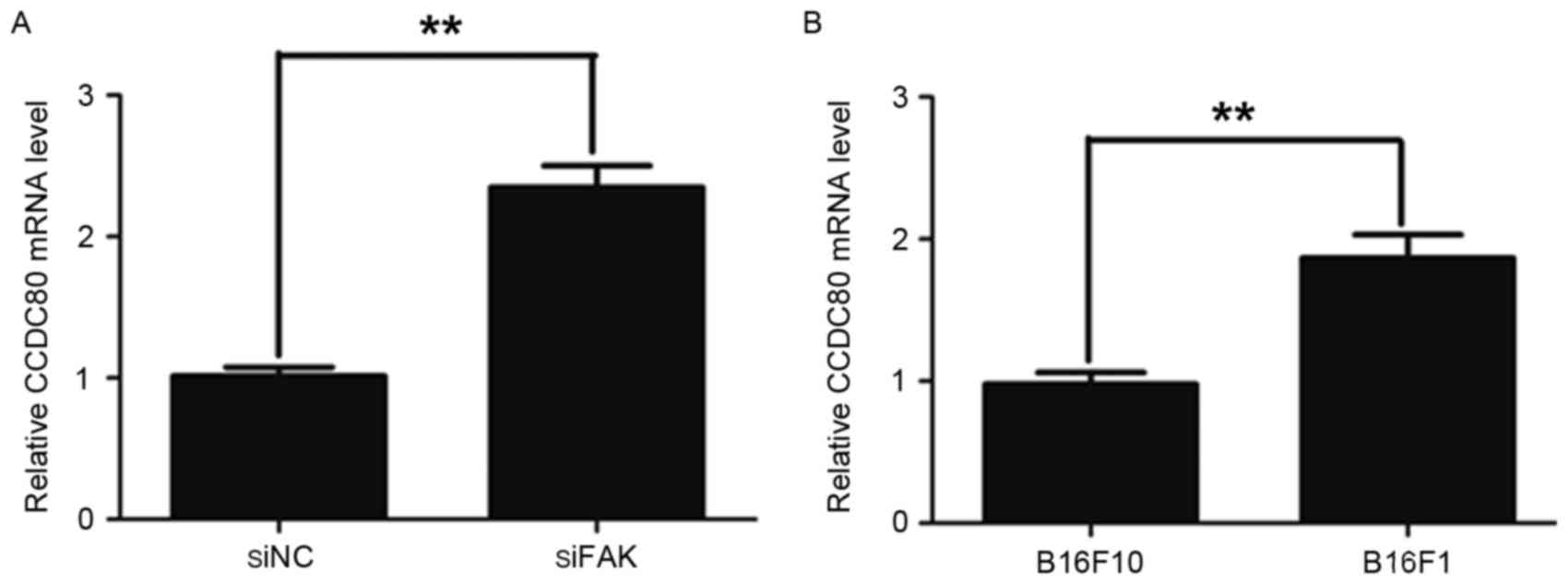
indicated that the CCDC80 mRNA level was significantly increased in
the siFAK cells compared with that in the siNC cells (Fig. 1A). Previously, it was demonstrated
that the expression level of FAK was lower in B16F1 cells, which
exhibit low rates of metastasis, as compared with that in highly
metastatic B16F10 cells (14).
Therefore, the present study examined the CCDC80 mRNA level in
B16F10 and B16F1 cells. It was identified that the CCDC80 mRNA
level was increased in the B16F1 cells compared with that in the
B16F10 cells (Fig. 1B).
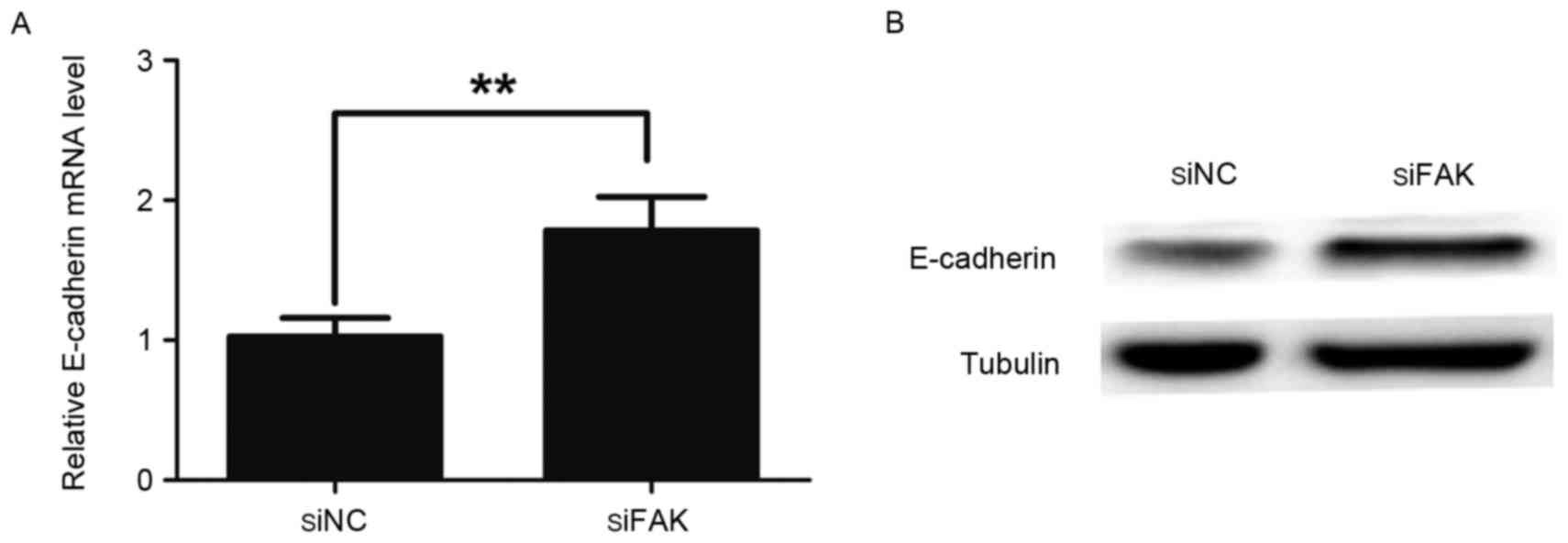
Concomitantly, RT-qPCR and western blotting assays also indicated
that the expression of E-cadherin was significantly increased in
the siFAK cells compared with the siNC cells (Fig. 2A and B). These data suggest that FAK
inhibits the expression levels of CCDC80 and E-cadherin in B16F10
cells.
 | Table I.Gene chip analysis of the FAK, CCDC80
and E-cadherin gene expression. |
Table I.
Gene chip analysis of the FAK, CCDC80
and E-cadherin gene expression.
| Gene | Fold change (siFAK
vs. siNC) |
|---|
| FAK | −2.12 |
| CCDC80 | 1.72 |
| E-cadherin | 1 |
CCDC80 promotes the expression of
E-cadherin
E-cadherin is a suppressor of tumor invasion and
metastasis (15,16). Loss of E-cadherin expression promotes
metastatic tumor dissemination and predicts poor prognosis
(17). A previous study indicated
that an increase in E-cadherin expression inhibited cancer cell
migration (18). Additionally, CCDC80
was demonstrated to maintain a normal E-cadherin expression rate in
thyroid cancer (8). As demonstrated
in Figs. 1 and 2, knockdown of FAK promoted the expression
levels of CCDC80 and E-cadherin in the B16F10 cells. However, it
was not clear whether CCDC80 regulated E-cadherin expression in
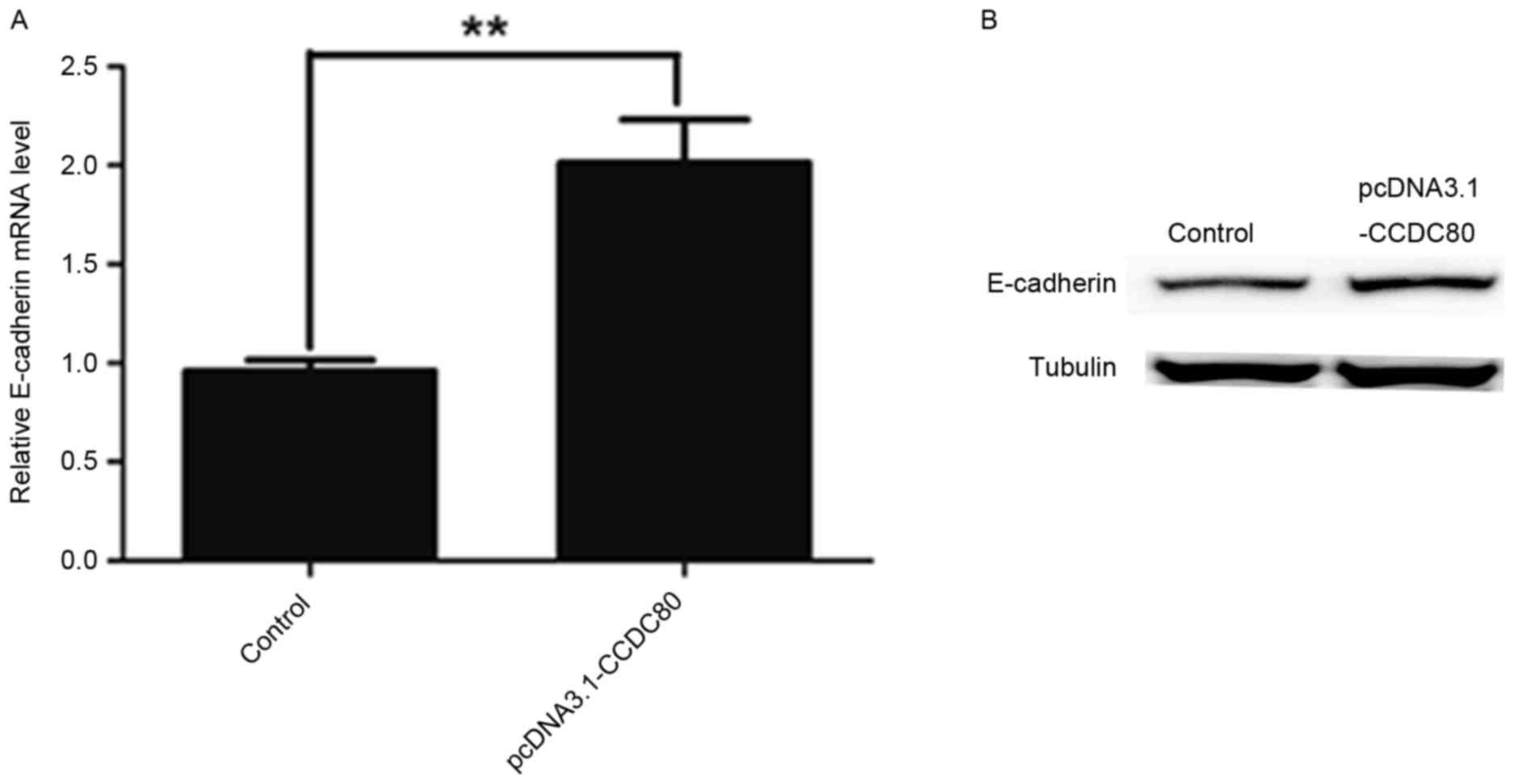
B16F10 cells. In the present study, it was identified that the
overexpression of CCDC80 elevated E-cadherin expression in B16F10
cells at mRNA and protein levels (Fig. 3A
and B). These data indicated that CCDC80 promoted E-cadherin
expression in B16F10 cells.
CCDC80 inhibits B16F10 cell
migration
As CCDC80 promoted E-cadherin expression, it may be
involved in the regulation of B16F10 cell migration. To explore the
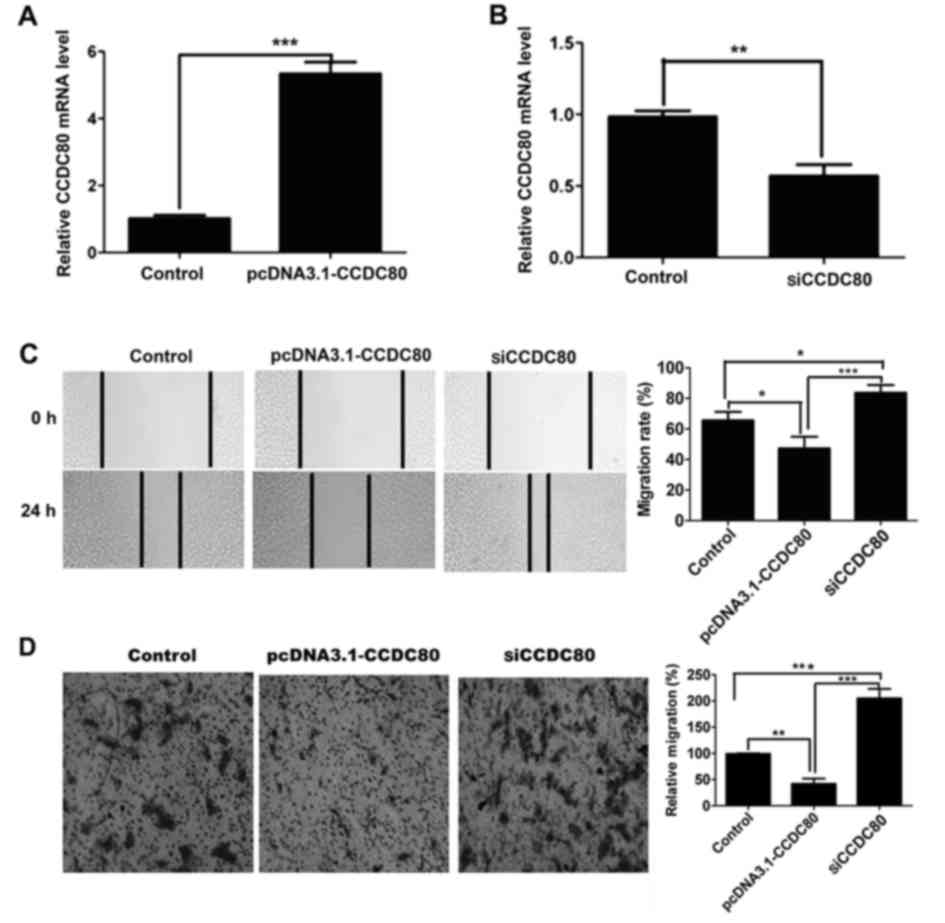
role of CCDC80 in cell migration, the expression level of CCDC80
was upregulated or downregulated in B16F10 cells by transfection of
pcDNA3.1-CCDC80 plasmids or siCCDC80 molecules, respectively
(Fig. 4A and B). The results from the
wound healing assay indicated that the upregulation of CCDC80
inhibited B16F10 cell migration, while the downregulation of CCDC80
promoted B16F10 cell migration compared with the control (Fig. 4C). The transwell assay also suggested
that the upregulation of CCDC80 inhibited B16F10 cell migration
compared with the control (Fig. 4D).
Taken together, these results demonstrated that CCDC80 may inhibit
B16F10 cell migration.
mRNA levels of FAK, CCDC80 and
E-cadherin in patients with melanoma
As aforementioned, it was identified that FAK
regulates E-cadherin expression via CCDC80, and CCDC80 may regulate
B16F10 cell migration. Consequently, the mRNA levels of FAK, CCDC80
and E-cadherin in human melanoma were analyzed using the Oncomine
Cancer Microarray database (www.oncomine.org). As revealed by melanoma datasets
from Riker et al and Haqq et al (data from www.oncomine.org), the expression level of FAK was
upregulated, while the expression levels of CCDC80 and E-cadherin
were downregulated in human melanoma samples compared with the
normal controls (Tables II and
III). In the Critchley-Thorne
melanoma dataset (data from www.oncomine.org), the mRNA level of FAK was
decreased, while the mRNA levels of CCDC80 and E-cadherin were
upregulated (Table IV). The results
from human melanoma datasets are consistent with the data from the
siFAK cells, suggesting that the regulation of B16F10 melanoma cell
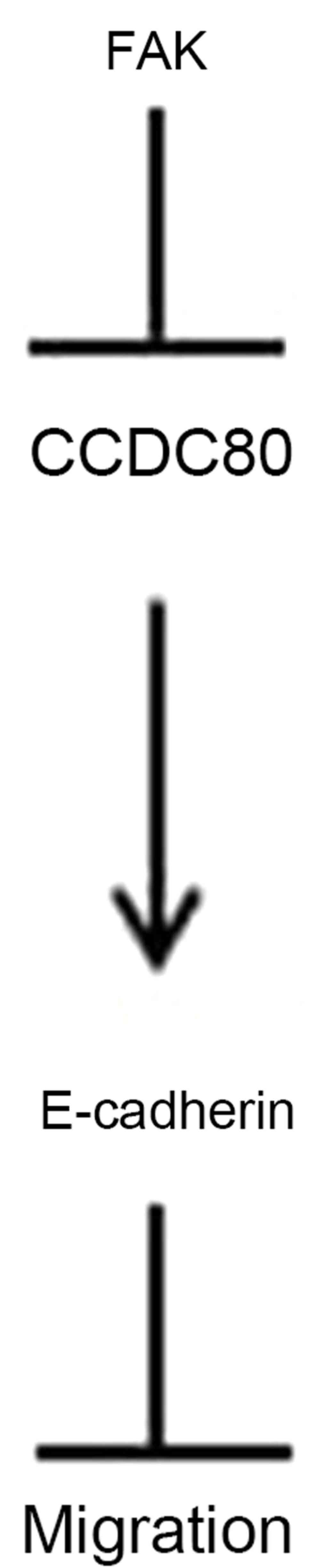
migration by FAK is potentially mediated by CCDC80 (Fig. 5).
| Table II.mRNA levels of FAK, CCDC80 and
E-Cadherin in the Riker melanoma dataset. |
Table II.
mRNA levels of FAK, CCDC80 and
E-Cadherin in the Riker melanoma dataset.
| Gene | Fold change (Melanoma
vs. normal) |
|---|
| FAK | 2.12 |
| CCDC80 | −3.23 |
| E-cadherin | −2.68 |
| Table III.mRNA levels of FAK, CCDC80 and
E-Cadherin in the Haqq melanoma dataset. |
Table III.
mRNA levels of FAK, CCDC80 and
E-Cadherin in the Haqq melanoma dataset.
| Gene | Fold change (Melanoma
vs. normal) |
|---|
| FAK | 1.36 |
| CCDC80 | −2.39 |
| E-cadherin | −5.32 |
| Table IV.mRNA levels of FAK, CCDC80 and
E-Cadherin in the Critchley-Thorne melanoma dataset. |
Table IV.
mRNA levels of FAK, CCDC80 and
E-Cadherin in the Critchley-Thorne melanoma dataset.
| Gene | Fold change (Melanoma
vs. normal) |
|---|
| FAK | −1.01 |
| CCDC80 |
1.05 |
| E-cadherin |
1.20 |
Discussion
Malignant melanoma is one of the most aggressive
types of malignancy (19–21), and the incidence of melanoma is
increasing more rapidly compared with any other type of cancer
globally (22). In the present study
it was demonstrated that FAK is overexpressed and may serve a key
role in melanoma progression, the role of FAK in B16F10 cell
migration was investigated and the underlying mechanism was
additionally explored. It was identified that the knockdown of FAK
promoted the expression levels of CCDC80 and E-cadherin, and
suppressed the migration of B16F10 cells. Concurrently, CCDC80
inhibited the migration of B16F10 cells, and increased the levels
of E-cadherin. In the present study, only the role of CCDC80 in
B16F10 cell migration was examined. As a potential target for tumor
therapy, the roles of CCDC80 in cell proliferation and apoptosis
require additional investigation.
However, it remains to be elucidated how FAK
regulates CCDC80 and E-cadherin in B16F10 cells. As demonstrated in
Fig. 1A, the RT-qPCR assay indicated
that the CCDC80 mRNA level was significantly increased in siFAK
cells, suggesting that FAK regulates CCDC80 expression at the
transcriptional level. It has been revealed that NCOA3 is
overexpressed in primary cutaneous melanoma, and that NCOA3 is a
negative regulator of CCDC80 (23).
When the expression of NCOA3 was silenced in MCF-7 cells, there was
a significant increase in the expression level of CCDC80 (12). Additionally, it was suggested that ERK
may modulate NCOA3 (24). An
increased phosphorylation of ERK was accompanied by an increased
expression of NCOA3 in MCF7-YB-1 cells (25). Preliminary data indicated that the
protein level of p-ERK was decreased in siFAK cells (data not
shown). In addition, the results from the gene chip assay (Table V) and RT-qPCR assay (data not shown)
revealed that the mRNA level of NCOA3 was downregulated in siFAK
cells. The data suggest that FAK may regulate CCDC80 via the
ERK/NCOA3 signaling pathway.
| Table V.Gene chip analysis of FAK and NCOA3
gene expression. |
Table V.
Gene chip analysis of FAK and NCOA3
gene expression.
| Gene | Fold change (siFAK
vs. siNC) |
|---|
| FAK | −2.12 |
| NCOA3 | −1.09 |
There are also certain indications of how CCDC80
regulates E-cadherin expression. As revealed in Fig. 3A, the overexpression of CCDC80
increased the mRNA level of E-cadherin, indicating that CCDC80
regulates E-cadherin expression at the transcriptional level. A
previous study demonstrated that the silencing of CCDC80 decreases
the mRNA level of peroxisome proliferator-activated receptor γ
(PPARγ), suggesting that PPARγ is a downstream target of CCDC80
(14). Preliminary data from the
present study indicated that FAK knockdown promoted PPARγ
expression, and that PPARγ knockdown decreased E-cadherin
expression (data not shown). Therefore, it was hypothesized that
CCDC80 promotes E-cadherin expression via PPARγ. However,
additional studies are required to provide complete
characterization of this interaction.
In conclusion, the data of the present study suggest
that FAK potentially regulates B16F10 cell migration by the
CCDC80/E-cadherin pathway. These results may assist in melanoma
treatment by the identification of a novel biomarker for
diagnosis.
Acknowledgements
The authors would like to thank Mrs. Min Lu and Mr
Tongyang Zhu (The State Key Laboratory of Pharmaceutical
Biotechnology, School of Life Sciences, Nanjing University,
Nanjing, China) for their assistance with experimental apparatus
and materials.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos.: 81421091,
30425009, 30330530, 31200572, 81121062 and 31071196), the Natural
Science Foundation of Jiangsu province (grant no.: BE2013630) and
Bureau of Science and Technology of Changzhou (grant no.:
CE20135013).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZCH undertook study design; GP and YL were
responsible for study design and conducting the experiments. WL
designed the primers, GP wrote the manuscript and LJ contributed
substantially to the analysis and interpretation of data for the
work and revised the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
FAK
|
focal adhesion kinase
|
|
CCDC80
|
coiled-coil domain containing 80
|
|
NC
|
normal control
|
|
NCOA3
|
nuclear receptor coactivator-3
|
|
RT-qPCR
|
quantitative real time polymerase
chain reaction
|
|
siFAK cell line
|
stable interference of FAK in B16F10
cells
|
|
siNC cell line
|
negative control for siFAK cell
line
|
References
|
1
|
Bandarchi B, Jabbari CA, Vedadi A and
Navab R: Molecular biology of normal melanocytes and melanoma
cells. J Clin Pathol. 66:644–648. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cary LA and Guan JL: Focal adhesion kinase
in integrin-mediated signaling. Front Biosci. 4:D102–D113. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schlaepfer DD, Hauck CR and Sieg DJ:
Signaling through focal adhesion kinase. Prog Biophys Mol Biol.
71:435–478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee BY, Timpson P, Horvath LG and Daly RJ:
FAK signaling in human cancer as a target for therapeutics.
Pharmacol Ther. 146:132–149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McLean GW, Carragher NO, Avizienyte E,
Evans J, Brunton VG and Frame MC: The role of focal-adhesion kinase
in cancer-a new therapeutic opportunity. Nat Rev Cancer. 5:505–515.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sulzmaier FJ, Jean C and Schlaepfer DD:
FAK in cancer: Mechanistic findings and clinical applications. Nat
Rev Cancer. 14:598–610. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brusegan C, Pistocchi A, Frassine A, Della
Noce I, Schepis F and Cotelli F: Ccdc80-l1 Is involved in axon
pathfinding of zebrafish motoneurons. PLoS One. 7:e318512012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferraro A, Schepis F, Leone V, Federico A,
Borbone E, Pallante P, Berlingieri MT, Chiappetta G, Monaco M,
Palmieri D, et al: Tumor suppressor role of the CL2/DRO1/CCDC80
gene in thyroid carcinogenesis. J Clin Endocrinol Metab.
98:2834–2843. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Visconti R, Schepis F, Iuliano R,
Pierantoni GM, Zhang L, Carlomagno F, Battaglia C, Martelli ML,
Trapasso F, Santoro M and Fusco A: Cloning and molecular
characterization of a novel gene strongly induced by the adenovirus
E1A gene in rat thyroid cells. Oncogene. 22:1087–1097. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bommer GT, Jäger C, Dürr EM, Baehs S,
Eichhorst ST, Brabletz T, Hu G, Fröhlich T, Arnold G and Kress DC:
DRO1, a gene down-regulated by oncogenes, mediates growth
inhibition in colon and pancreatic cancer cells. J Biol Chem.
280:7962–7975. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Herbst A, Bayer C, Wypior C and Kolligs
FT: DRO1 sensitizes colorectal cancer cells to receptor-mediated
apoptosis. Oncol Lett. 2:981–984. 2011.PubMed/NCBI
|
|
12
|
Ferragud J, Avivar-Valderas A, Pla A, De
Las Rivas J and Font de Mora J: Transcriptional repression of the
tumor suppressor DRO1 by AIB1. FEBS Lett. 585:3041–3046. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lan Y and Hua ZC: Construction of stable
focal adhesion kinase knockdown cell line and preliminary study of
its properties. Yao Xue Xue Bao. 47:1128–1133. 2012.(In Chinese).
PubMed/NCBI
|
|
15
|
Singhai R, Patil VW, Jaiswal SR, Patil SD,
Tayade MB and Patil AV: E-Cadherin as a diagnostic biomarker in
breast cancer. N Am J Med Sci. 3:227–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan P, Manna A, Saha S, Mohanty S,
Mukherjee S, Mazumdar M, Guha D and Das T: Aspirin inhibits
epithelial-to-mesenchymal transition and migration of oncogenic
K-ras-expressing non-small cell lung carcinoma cells by
down-regulating E-cadherin repressor Slug. BMC Cancer. 16:392016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Venza M, Visalli M, Catalano T, Biondo C,
Beninati C, Teti D and Venza I: DNA methylation-induced E-cadherin
silencing is correlated with the clinicopathological features of
melanoma. Oncol Rep. 35:2451–2460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cui T, Srivastava AK, Han C, Yang L, Zhao
R, Zou N, Qu M, Duan W, Zhang X and Wang QE: XPC inhibits NSCLC
cell proliferation and migration by enhancing E-Cadherin
expression. Oncotarget. 6:10060–10072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Russo AE, Torrisi E, Bevelacqua Y,
Perrotta R, Libra M, McCubrey JA, Spandidos DA, Stivala F and
Malaponte G: Melanoma: Molecular pathogenesis and emerging target
therapies (Review). Int J Oncol. 34:1481–1489. 2009.PubMed/NCBI
|
|
20
|
Shoo BA and Kashani-Sabet M: Melanoma
arising in African-, Asian-, Latino- and Native-American
populations. Semin Cutan Med Surg. 28:96–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rigel DS and Carucci JA: Malignant
melanoma: Prevention, early detection, and treatment in the 21st
century. CA Cancer J Clin. 50:215–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rangel J, Torabian S, Shaikh L, Nosrati M,
Baehner FL, Haqq C, Leong SP, Miller JR III, Sagebiel RW and
Kashani-Sabet M: Prognostic significance of nuclear receptor
coactivator-3 overexpression in primary cutaneous melanoma. J Clin
Oncol. 24:4565–4569. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Font de Mora J and Brown M: AIB1 is a
conduit for kinase-mediated growth factor signaling to the estrogen
receptor. Mol Cell Biol. 20:5041–5047. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ito T, Kamijo S, Izumi H, Kohno K, Amano J
and Ito K: Alteration of Y-box binding protein-1 expression
modifies the response to endocrine therapy in estrogen
receptor-positive breast cancer. Breast Cancer Res Treat.
133:145–159. 2012. View Article : Google Scholar : PubMed/NCBI
|