Introduction
Lung adenocarcinoma (LUAD) is the most well-known
histological sub-type of non-small cell lung cancer (NSCLC), which
is the most significant cause of cancer-associated mortality
worldwide (1). Since LUAD tends to
form metastases at an early stage, the prognosis for patients with
LUAD is generally poor, with average 5-year survival rates of
<20% (2). Although recent advances
in molecular pathology and targeted therapies have improved
clinical therapies, the 5-year overall survival of patients with
LUAD remains low (3), requiring
continued research to identify novel biomarkers and effective
targeted molecular therapies.
Recent high-throughput transcriptome analyses have
demonstrated that >90% of the transcriptome is transcribed into
non-coding RNAs, among which microRNAs (miRNAs/miRs) and long
non-coding RNAs (lncRNAs) have been revealed to be involved in the
malignant behaviors of LUAD. By definition, lncRNAs are non-coding
RNAs that are >200 base pairs (4).
According to recent studies, lncRNAs are important in various
biological processes, including gene expression, cell proliferation
and differentiation, the immune response, apoptosis and human
diseases, including various types of cancer (5–8).
It has been reported that lncRNAs serve as competing
endogenous RNAs (ceRNAs) or miRNA sponges, inhibiting miRNA
response elements (MREs) from exchanging with mRNAs by competing
for common miRNAs during the tumorigenic process (9). The ceRNA hypothesis has been proposed as
a novel regulatory mechanism between non-coding RNAs and coding
RNAs (10), thereby contributing
toward the occurrence and progression of cancer (11,12). Based
on ceRNA theory, Akcakaya et al (13) demonstrated that miR-133b was
significantly decreased in CRC and associated with overall survival
and metastasis. Zhang et al (14) analyzed 372 lncRNAs from The Cancer
Genome Atlas (TCGA) and NCBI GEO omnibus (GSE65485), and identified
a complex cancer-specific ceRNA network in HCC. Zhang et al
(15) revealed MALAT1 to be the
direct target of miR-202 and reported that interfering with MALAT1
downregulates Gli2 through positive regulation of miR-202 to
suppress the development of gastric cancer.
As a novel post-transcriptional regulatory system,
ceRNAs provide a novel means of investigating the pathogenesis of
cancer and detecting novel signatures with high accuracy in
diagnosis. The development and survival of anomalous lncRNAs in
different types of cancer (including LUAD) have been examined,
revealing the potential of lncRNAs as prognostic cancer biomarkers
(16–20). Nevertheless, the study of ceRNAs in
LUAD is scarce, and there are few targets for tumor therapies.
There are little data on large-scale samples, and microarray
detection remains rare, and the association between cancer-specific
lncRNAs and clinical features is unknown.
The TCGA database is a public platform that can be
used to download the sequencing data of LUAD lncRNAs, miRNAs and
mRNAs. Following obtaining RNA sequence data, lncRNAs, miRNAs and
mRNAs with abnormal expression were identified, and an lncRNA/mRNA
expression network was constructed. These results increase
understanding of the molecular mechanisms and therapeutic targets
of lncRNAs in the treatment of LUAD.
Materials and methods
TCGA dataset and analysis of the
differentially expressed genes
LUAD individual RNA sequencing (RNA-Seq) data (level
3), computed on the Illumina HiSeq RNA-Seq platform, were obtained
from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/), which contains
512 LUAD and 58 adjacent non-tumor lung tissue samples, until
December 31, 2016. The raw RNA-Seq reads (lncRNA and mRNA) were
post-processed and normalized on the TCGA RNASeqV2 system. The
miRNA expression data (level 3) were collected by Illumina GA
microRNA sequencing platforms (Illumina, Inc., Hayward, CA, USA),
quantile-normalized prior to the analysis and expressed as reads
per million (RPM).
The exclusion criteria were as follows: i) The
histological diagnosis was not LUAD; ii) presence of a malignancy
other than LUAD; and iii) patient samples lacking complete data for
analysis.
A total of 493 patients with LUAD were included in
the present study. lncRNA expression profiles for normal lung
tissue samples were obtained from adjacent non-tumor lung tissues
(n=58). In addition, there were 170 LUAD patients with lymph node
metastases and 323 LUAD patients without lymph node metastases.
According to the Union for International Cancer Control (UICC),
there were 381 patients with stage I–II LUAD and 112 patients with
stage III–IV LUAD (21).
The expression data of lncRNAs are displayed as RPM
and the expression levels of each lncRNA were normalized by Deseq R
package for further analysis. The data were obtained from TCGA, a
community resource project that provides data for community
research. Following low-expression genes being filtered, the LUAD
RNA-Seq coverage data contained 20,321 mRNA and 181 lncRNAs
compared with the GENCODE database (http://www.gencodegenes.org/). Next, the
differentially expressed lncRNAs were counted using EdgeR and the
DEseq package of R (Padj <0.05 and absolute log2FC>1). The
final lncRNAs were selected using a combination of the two methods.
Student's t-test (SPSS 20.0, Inc., IBM Corp., Armonk, NY, USA) was
employed to analyze the difference in expression levels of these
lncRNAs between LUAD and non-cancerous lung tissues. The present
study meets the publication guidelines provided by TCGA (http://cancergenome.nih.gov/publications/publicationguidelines).
Functional enrichment analysis
The differentially expressed genes were analyzed by
Gene Ontology (GO) (22) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) (23) pathway enrichment using the cluster
Profiler package of R. P-values <0.05 and q values <0.05 were
considered to indicate statistically significant differences, and
were based on similar functional visualization and clustering.
Preparation of the prognostic
signature of differentially expressed lncRNAs for LUAD
The screening threshold criteria for differentially
expressed genes were as follows: |log2FC|>1 and FDR<0.05.
Additionally, the TCGA also collected clinical pathological
features, including survival information. Cases with insufficient
clinical data were omitted. Ultimately, the prognostic analysis
included 493 samples, with 6,280 genes representing 6,205 mRNAs and
75 lncRNAs. The endpoints of the patients with LUAD in terms of
overall survival (OS) were also studied. The mean follow-up period
for these patients with LUAD was 28 months.
The prognostic value of the genes and the
clinicopathological features were first evaluated by
single-variable Cox proportional regression analysis (P<0.05). A
multivariate Cox regression model with the survival and KMsurv
packs (24) further confirmed the
statistical indicators, including lncRNAs and clinicopathological
features.
Based on the linear combination of multiple genes in
the Cox regression model (β) and the linear combination of
regression coefficients, the prognostic risk score was established
based on a study undertaken by Tang et al (25). According to the definition of the
median prognostic index (PI), patients with LUAD were classified by
high and low scores, and the hazard ratio (HR) and 95% confidence
interval (CI) were calculated. The time-dependent receiver
operating characteristic (ROC) curve within 5 years was also
analyzed as the defining point with the R package ‘survival ROC’ to
evaluate the predictive accuracy of the prognostic model for the
results of time-dependent diseases. Kaplan-Meier survival curves
were drawn to assess the association between all parameters
(clinical examination and prognostic risk score) and the OS of
patients with LUAD (26).
Functional evaluation of lncRNAs with
WGCNA
In analyzing the incorporation of differentially
expressed lncRNAs into the network, WGCNA can describe the
correlation patterns in gene expression profiles (27). WGCNA R package was used to assess the
performance of an mRNA/lncRNA and its module membership, and the
correlation between each module and disease state was obtained
through single-factor variance analysis. All mRNAs associated with
an lncRNA in the module that were significantly associated with the
disease were considered potential targets for the lncRNA. The
function of an lncRNA was obtained by functional enrichment
analysis of the target. Following an evaluation of the weighted
correlations, the network feature was presented by Cytoscape 3.4.0.
(http://www.cytoscape.org/).
LncRNA function prediction
The coding gene in the same WGCNA network module is
a potential regulatory target gene for an lncRNA. The core
enrichment lncRNA in the WGCNA network module, which is
significantly associated with disease, is the risk lncRNA in the
present study. GO and KEGG enrichment analyses of the risk lncRNAs
and coding genes co-expressed in the same WGCNA module were
performed using the R clusterProfiler package to obtain the
functional information of disease risk lncRNAs.
Network analysis of ceRNA
lncRNAs can bind to miRNAs and affect ceRNA
function. MiRanda (http://miranda.org.uk/) is used to predict the
interaction between lncRNAs and miRNAs. miRanda is based mainly on
the combination of circRNA and miRNA free energy size: The smaller
the free energy, the stronger the binding capacity. Following
application of filters of a threshold maximum energy ≤-20 and score
>160, numerous microRNAs that bound to lncRNA were predicted,
but only microRNAs with the top 10 scores were studied.
Based on the TarBase (http://www.microrna.gr/tarbase), miRTarBase
(http://mirtarbase.mbc.nctu.edu.tw/php/index.php),
miRecards (http://c1.accurascience.com/miRecords/) and starBase
v2.0 (http://starbase.sysu.edu.cn/)
databases, information on the downstream target genes of microRNAs
in the ceRNA network was obtained. Cytoscape software was used to
construct microRNA-target and microRNA-lncRNA integration networks
that only showed the genes within the module being investigated.
The mRNAs in the network were analyzed by GO BP enrichment using
the clueGO plugins in Cytoscape software.
The correlations between LUAD-specific
lncRNAs and clinical features
According to the bioinformatics analysis and the
ceRNA network, prognostic lncRNAs were selected. Cox regression
analysis was also performed on the results for the clinical
factors, including sex, tumor pathological stage,
Tumor-Node-Metastasis (TNM) stage (21) and the number of pack-years.
Statistical analysis
Data are presented as the mean ± standard deviation
and Student's t-tests, two-sided log-rank test, single-factor cox
proportional risk regression, multi-factor cox proportional risk
regression, R package ‘survivalROC’ and ‘KMsurv’ were used to
perform statistical analysis. P<0.05 was considered to indicate
a statistically significant difference.
Results
Data source and pre-processing
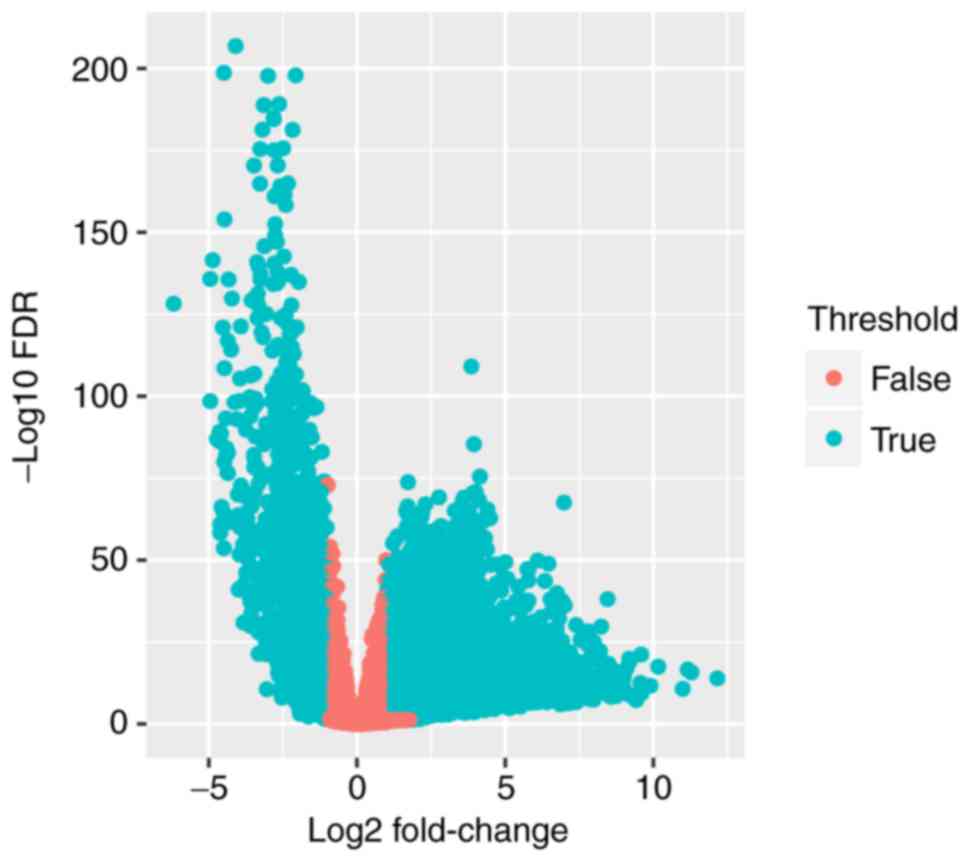
The expression data on 20,502 genes were extracted,
and R language (EdegR and DESeq) was used to calculate the
expression data. The differentially expressed genes (n=6280;
Table I) were selected to further
investigate their prognostic value (Fig.
1).
 | Table I.Differentially expressed genes. |
Table I.
Differentially expressed genes.
| Category | Upregulated | Downregulated | Total |
|---|
| lncRNA |
57 |
18 |
75 |
| mRNA | 4,311 | 1,894 | 6,205 |
| Total | 4,368 | 1,912 | 6,280 |
Prognostic effect of differentially
expressed genes
Following exclusion of those with insufficient
survival data, the prognosis of 493 patients was obtained.
Single-factor Cox proportional hazard regression analysis revealed
that 328 mRNAs and four lncRNAs had prognostic value in LUAD.
Subsequently, multi-factor Cox proportional hazard regression
analysis was used to evaluate these results, and 32 genes (29 mRNAs
and 3 lncRNAs; Table II) were
independent prognostic indicators of LUAD (HR>1; P<0.05). The
prognostic risk index PI was calculated for each of the 32
prognostic risk genes using a previously described formula, in
which the expression of all risk genes is multiplied by the HR
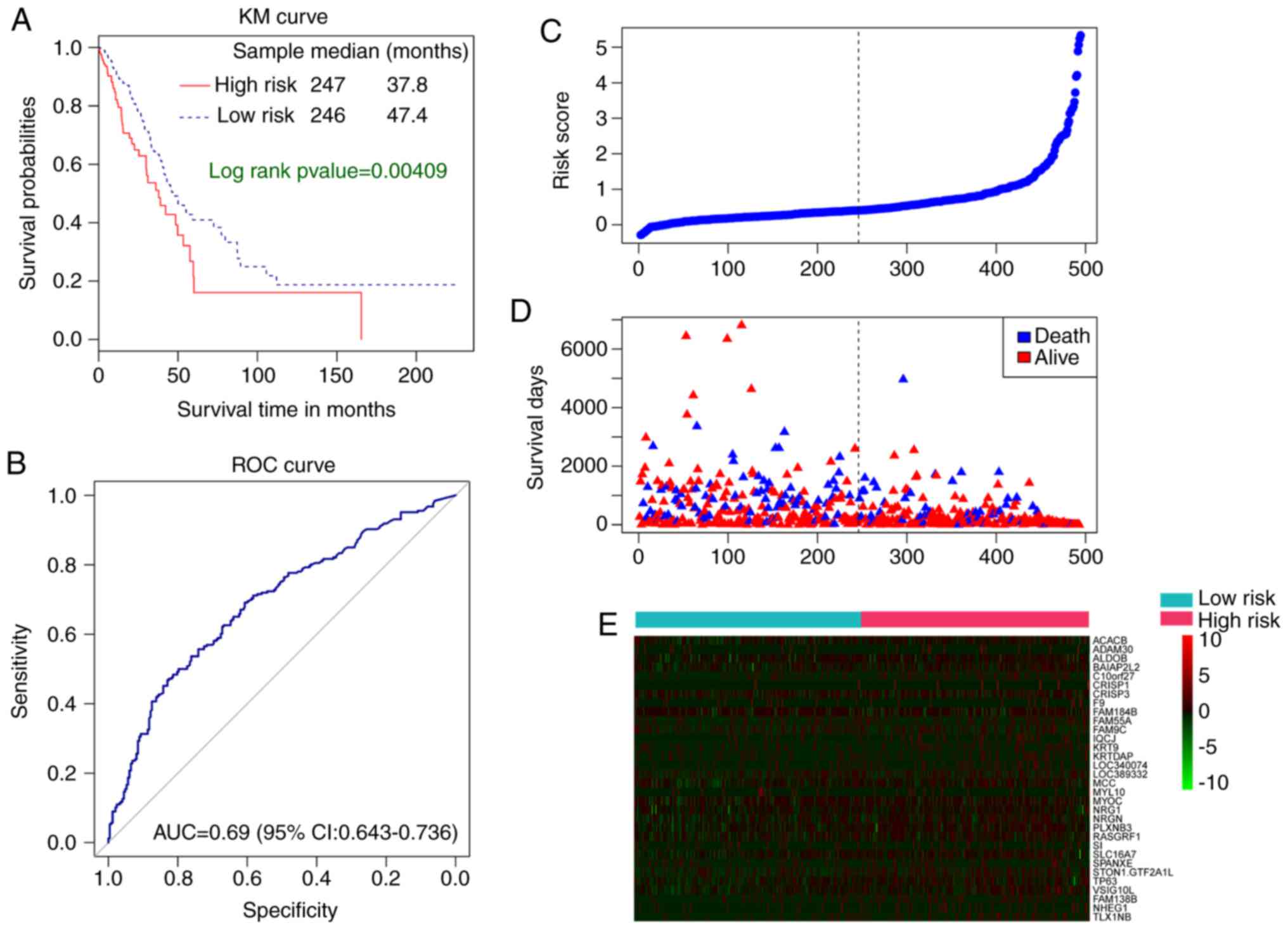
value and then summed (28,29). the Kaplan-Meier curve suggested that
the median survival time of patients in the high-risk group was
37.8 months, significantly less than that of the low-risk group
(47.4 months; P=0.00409; Fig. 2A). In
addition, the present study also assessed the difference in
expression levels of 32 genes between the high-risk and low-risk
groups. The prognostic risk index aided in predicting the 5-year
survival rate of patients with LUAD, with an AUC value of 0.69
(Fig. 2B). The median prognostic risk
index of ‘Risk score’ and ‘survival days’ is the separation for
low-risk and high-risk patients with LUAD. (Fig. 2C and D).
| Table II.32 genes serving as prognostic
indicators. |
Table II.
32 genes serving as prognostic
indicators.
| lncRNA |
|
| mRNA |
|
|
|---|
| FAM138B | CRISP1 | MYL10 | NRG1 | FAM9C | MYOC |
|
| TP63 | CRISP3 | BC092501 | KRTDAP | SLC16A7 |
| TLX1NB | KRT9 | STON1.GTF2A1L | SI | VSIG10L | IQCJ |
|
| ADAM30 | SPANXE | ACACB | FAM55A | F9 |
| NHEG1 | PLXNB3 | ALDOB | BC040891 | NRGN | MCC |
|
| BAIAP2L2 | FAM184B | C10orf27 | RASGRF1 |
|
WGCNA module division and lncRNA
target gene prediction
The WGCNA network module was constructed for all
differentially expressed mRNAs and lncRNAs, setting the number of
genes in each module to be no less than 20. A total of 19 network
modules were divided, and the correlation between the module and
LUAD was analyzed by one-way analysis of variance. It was revealed
that 11 modules were significantly correlated significantly with
the disease. The present study focused on the network modules of
the lncRNAs FAM138B (downregulated in LUAD), NHEG1 (downregulated
in LUAD) and TLX1NB (upregulated in LUAD). FAM138B is located in
the turquoise module containing 3,257 genes, 311 of which were
co-expressed with FAM138B. We hypothesized that these 311 genes are
potential FAM138B target genes. NHEG1 is located in the blue module
containing 900 genes, 582 of which were co-expressed with NHEG1.
Therefore, the 582 genes are potential NHEG1 target genes. TLX1NB
lies in the turquoise module containing 3,257 genes, 2,262 of which
are co-expressed with TLX1NB. We believe that these 2262 genes are
potential TLX1NB target genes.
Functional analysis of the lncRNA
target gene
From the WGCNA analysis of the FAM138B, NHEG1 and
TLX1NB lncRNAs, the target genes of each lncRNA were obtained. The
target genes of the lncRNAs were analyzed by GO and KEGG enrichment
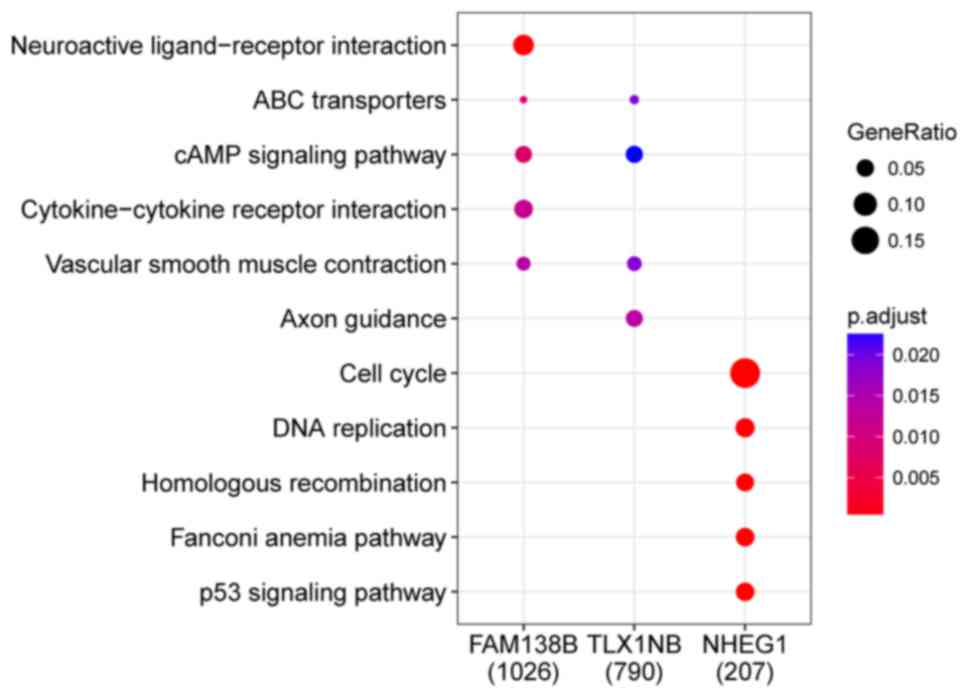
using the cluster Profiler package of R. The top three upregulated
pathways of FAM138B and TLX1NB were ABC transporters, cAMP
signaling and vascular smooth muscle contraction. The top five
upregulated pathways of NHEG1 were cell cycle, DNA replication,
homologous recombination, Fanconi anemia and p53 signaling
(Fig. 3).
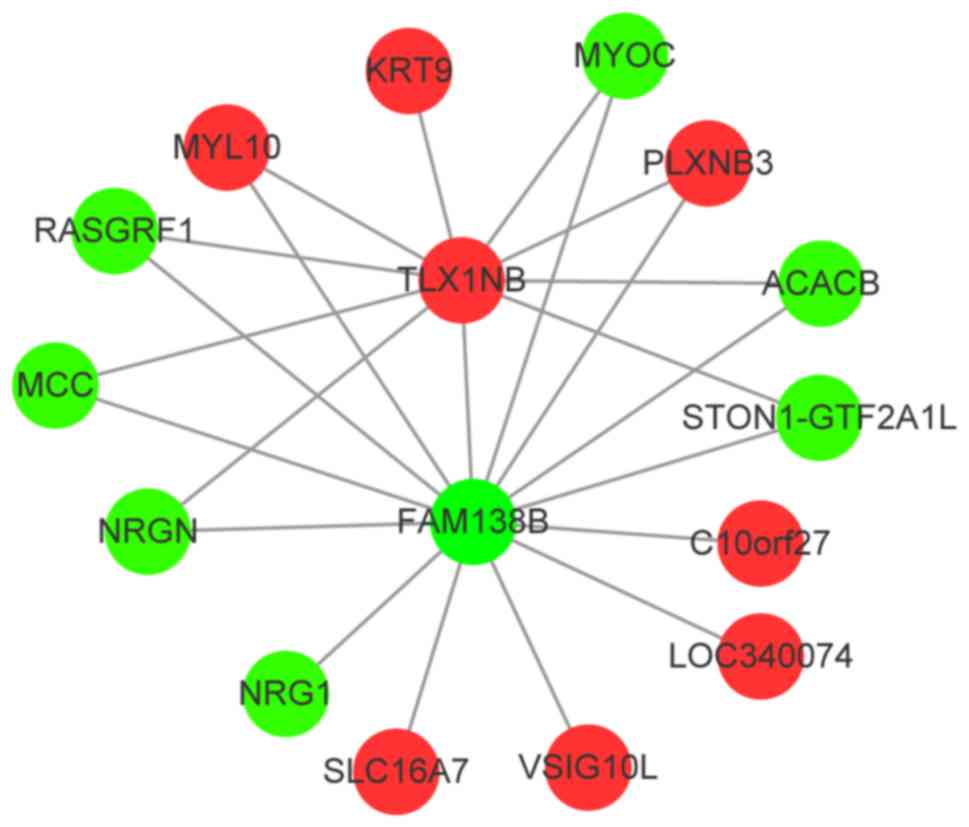
There were 14 prognostic risk genes among the target
genes of FAM138B and 10 genes among the target genes of TLX1NB
(Table III). The two lncRNAs
FAM138B and TLX1NB shared MYOC, PLXNB3 and ACACB among the eight
prognostic risk genes as target genes (Fig. 4).
| Table III.lncRNA FAM138B and TLX1NB target
gene. |
Table III.
lncRNA FAM138B and TLX1NB target
gene.
| LncRNA | Target gene | log2FC | FDR |
|---|
| FAM138B | MYOC | −4.357717939 | 0.0000000294 |
|
| PLXNB3 | 3.878550766 | 0.0000000191 |
|
| ACACB | −1.379164532 | 0.0000000121 |
|
| MCC | −1.27636241 | 0.0000000488 |
|
| RASGRF1 | −2.055290402 | 0.0000000424 |
|
| VSIG10L | 2.102105505 | 0.0000000327 |
|
| SLC16A7 | 1.372035277 | 0.0000000011 |
|
| NRGN | −1.203111742 | 0.00000000132 |
|
| NRG1 | −1.17178188 | 0.00000029 |
|
| TLX1NB | 2.102744205 | 0.0000335 |
|
| LOC340074 | 1.475322606 | 0.000465275 |
|
| STON1-GTF2A1L | −1.249288199 | 0.000666914 |
|
| MYL10 | 1.376749843 | 0.02548923 |
|
| C10orf27 | 1.038217387 | 0.03122004 |
| TLX1NB | MYOC | −4.35771794 | 0.000000000272 |
|
| PLXNB3 | 3.87855077 | 0.000000000943 |
|
| ACACB | −1.37916453 | 0.000000000669 |
|
| MCC | −1.27636241 | 0.000000000432 |
|
| RASGRF1 | −2.0552904 | 0.000000000571 |
|
| NRGN | −1.20311174 | 0.000000000444 |
|
| KRT9 | 2.93858028 | 0.00000755 |
|
| FAM138B | −1.11756478 | 0.00016011 |
|
| STON1-GTF2A1L | −1.2492882 | 0.000392098 |
|
| MYL10 | 1.37674984 | 0.018236455 |
Construction of lncRNA-miRNA
network
Miranda predicted microRNAs that were bound to the
three lncRNAs, and 712 miRNAs were obtained at the thresholds.
According to the predicted scores, the present study focused on the
27 miRNAs with the 10 highest scores (including duplicate scores)
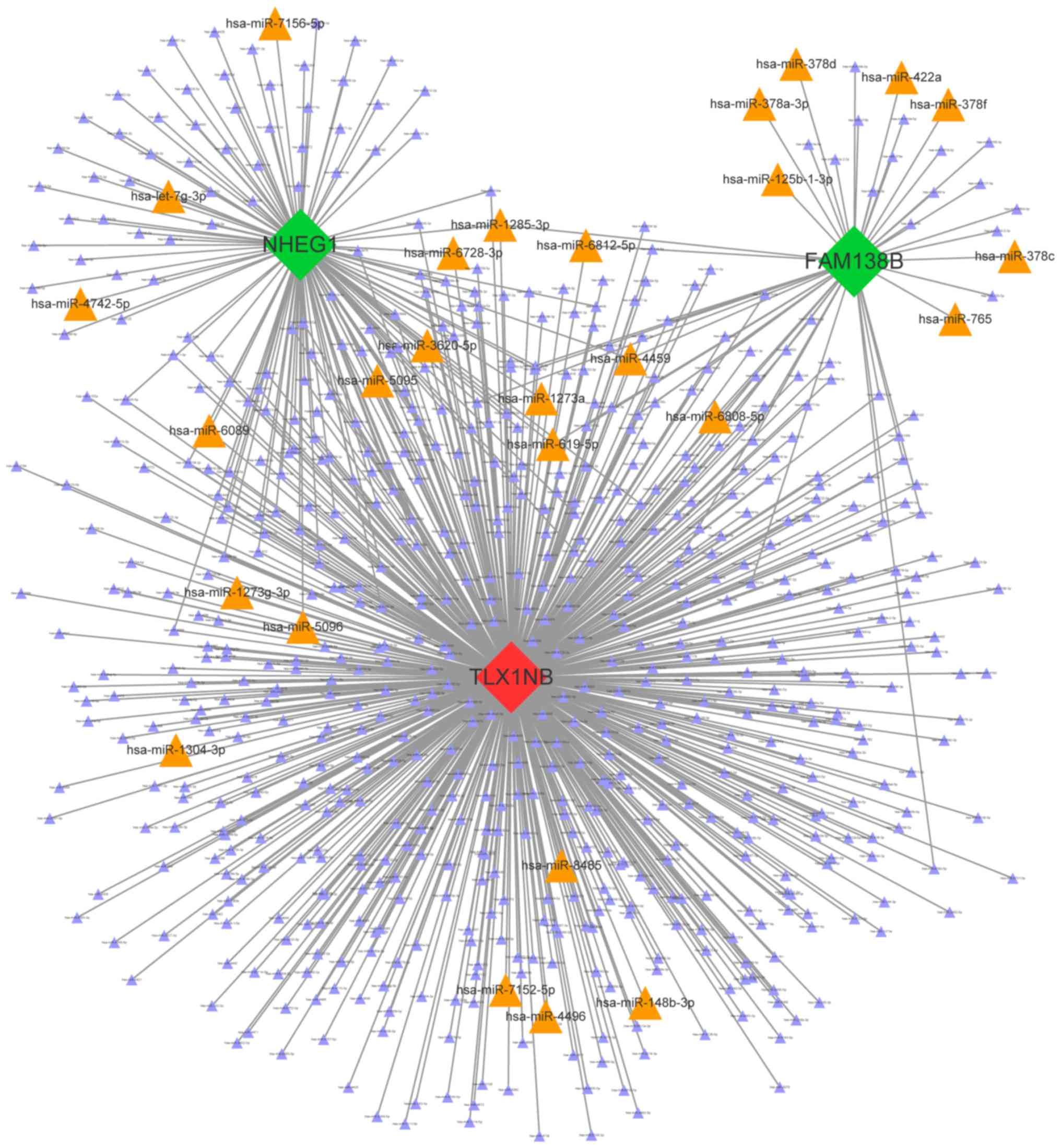
that bound to each lncRNA. The miRNA and lncRNA interaction
network, which contained 715 nodes and 815 edges, was constructed
(Fig. 5).
miRNA target gene prediction
Based on the TarBase, miRTarBase, miRecards and
starBase v2.0 databases, the target genes of the aforementioned 27
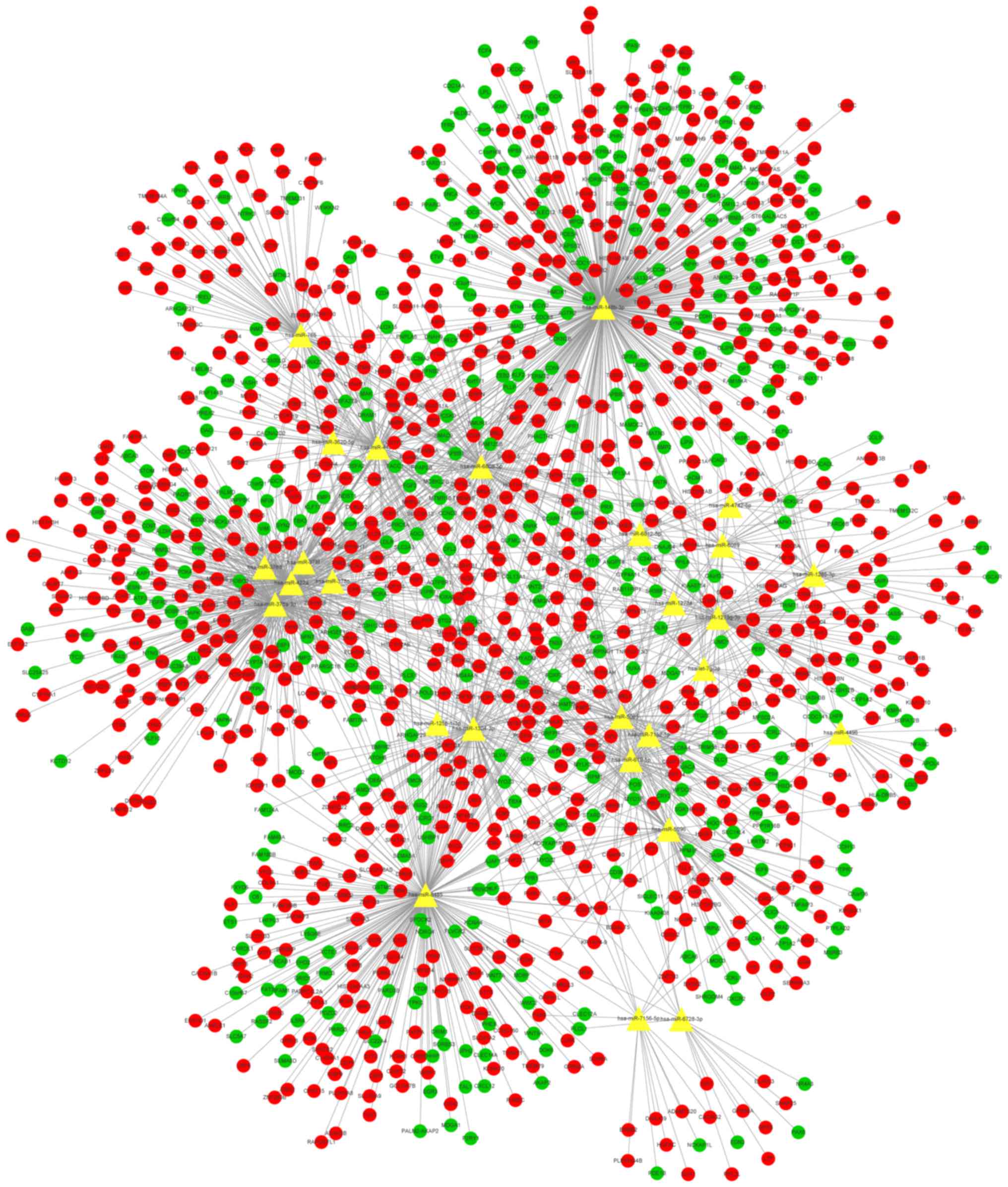
key miRNAs were obtained, and the microRNA-mRNA network was
constructed. The network contained 1,961 sides, 27 microRNAs and
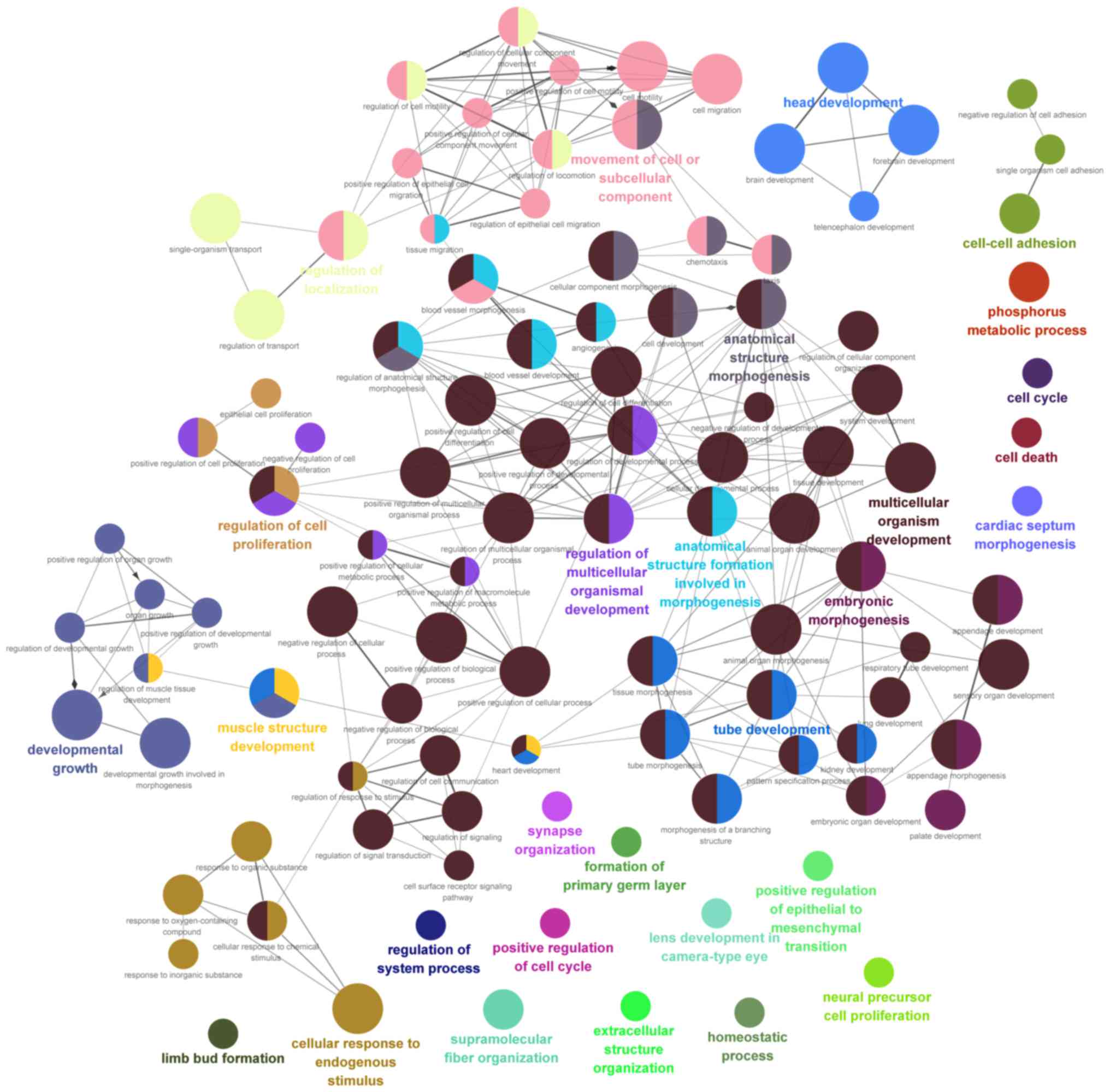
1,229 mRNAs (816 upregulated and 413 downregulated; Fig. 6). Furthermore, GO BP enrichment
analysis of the mRNAs in the network was performed (Fig. 7).
Construction of lncRNA-miRNA-mRNA
ceRNA network
The miRNA-mRNA and lncRNA-miRNA pairs were used to
build the lncRNA-miRNA-mRNA ceRNA network. The 29 previous
prognostic risk mRNAs and their microRNAs were separately
extracted, and a regulatory network was established. The network
also added lncRNAs, which regulate potential miRNAs, and
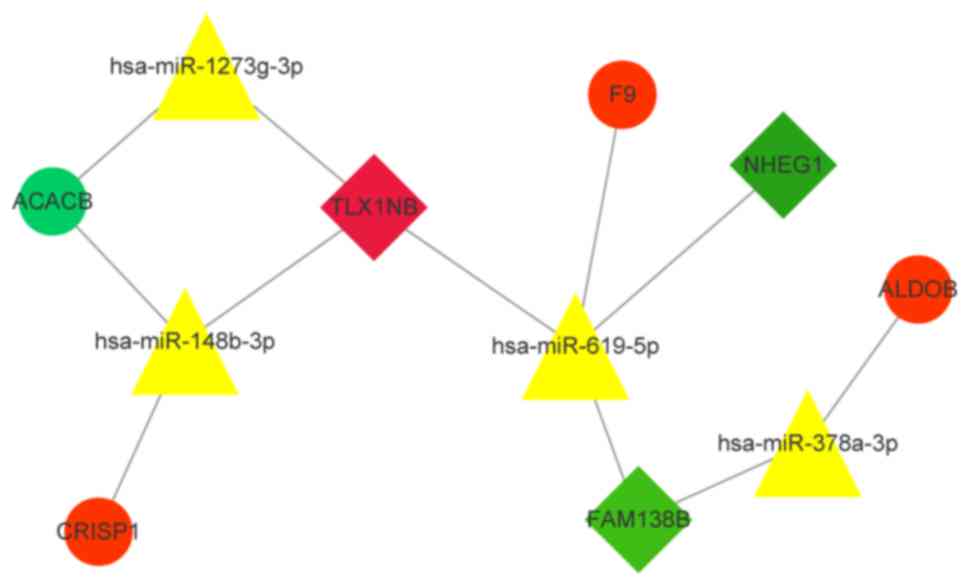
constructed an important lncRNA-miRNA-mRNA network (Fig. 8). As demonstrated, hsa-miR-619-5p has
a regulatory association with three prognostic risk factors. Three
upregulated mRNAs and one downregulated mRNA were potentially
affected by the lncRNA-ceRNA network.
Correlations between LUAD-specific
lncRNAs and clinical features
According to the single-factor and multi-factor Cox
proportional risk regression analysis, three specific
lncRNAs-FAM138B, NHEG1 and TLX1NB-were selected. Cox regression
analysis for clinical factors, including sex, tumor pathological
stage, TNM stage and number of pack-years, was then performed. The
expression levels of these three lncRNAs were significantly altered
by ‘T’ stage and primary tumor size (P<0.05; Table IV).
| Table IV.The correlations between lung
adenocarcinoma specific lncRNAs and clinical features. |
Table IV.
The correlations between lung
adenocarcinoma specific lncRNAs and clinical features.
| Id | Coef | Secoef | HR | Lower.95.CI | Upper.95.CI | P-value |
|---|
| Sex | 0.00980122 | 0.08968855 | 1.0098494 | 0.84705885 | 1.2039256 | 0.99727594 |
| Smoke | 0.00000723 | 0.00211715 | 1.0000072 | 0.99586625 | 1 | 0.99727594 |
| Stage | 0.48304592 | 0.47140751 | 1.6210043 | 0.64345974 | 4.08363557 | 0.30551012 |
| Metastasis | −0.1477427 | 0.24226305 | 0.8626531 | 0.53656395 | 1.38691821 | 0.93402361 |
| Node | −0.0575892 | 0.11703873 | 0.9440377 | 0.75052609 | 1.18744332 | 0.93402361 |
| Tumor | 1.29785383 | 0.19225169 | 3.6614302 | 2.51192012 | 5.34 |
0.0000000000147 |
GEO validation
The prognostic value of these three new lncRNAs was
then assessed using GEO data. However, there were not enough
survival data available in the GEO datasets. Therefore, the
prognostic value of these lncRNA signatures requires further
verification using other approaches in a larger number of
samples.
Discussion
Lung cancer has the highest incidence and mortality
rate of any disease worldwide (30),
and lung glandular cell cancer is one of the most common types of
LUAD (31). Surgery is considered the
most effective method of curing LUAD. However, the majority of
patients with LUAD in hospital have missed the opportunity for
surgery (32). Chemotherapy and
radiotherapy are the typical second-line treatments for LUAD, but
their effects are not satisfactory (33). In previous years, the diagnosis of
lung cancer, surgical techniques and novel drug treatments have
improved, but the 5-year response rate for lung cancer continues to
be maintained at 10% (34), mainly
because lung cancer cannot be diagnosed in the early stages.
Although numerous tests can support lung cancer examination and
staging, including tissue biopsy and thoracoscopy, these methods
are time-consuming and will delay early diagnosis and treatment
(35). Therefore, to improve this
situation, more attention is being paid to the occurrence and
development of lung cancer-related genes and their regulatory
mechanisms.
With the development of high-throughput sequencing
technology and bioinformatics, lncRNAs have been revealed to be
involved in the development of numerous diseases, particularly
tumors (36–39). However, the study of lncRNA expression
profiles in LUAD has gradually increased. Using TNM I stage lung
adenocarcinoma samples in the TCGA database, Tian et al
(40) demonstrated that FENDRR and
LINC00312 had diagnostic value in patients with LUAD. A recent
study has demonstrated that lncRNA can act as a ceRNA that adsorbs
onto miRNAs to regulate mRNA expression (41). The ceRNA hypothesis is a novel
regulatory mechanism that serves a role through miRNA competition
(10,42). However, the study of ceRNAs in LUAD is
not extensive. Li et al (43)
revealed MIAT, MEG3 and LINC00115 to be the basic therapeutic
targets of LUAD as ceRNAs that regulate miRNA-mRNA pairs. Sui et
al (44) demonstrated that
AFAP1-AS1 is associated with tumor pathological stage and that
LINC00472 is associated with lymph node metastasis. Liu et
al (45) presented lncRNA
landscapes and revealed highly altered, differentially expressed
lncRNAs in LUSC and LUAD. Two lncRNAs (IGF2BP2-AS1 and DGCR5) were
correlated with an improved OS in LUSC, and three (MIR31HG,
CDKN2A-AS1 and LINC01600) predicted a poor OS in LUAD.
The public lncRNA data were mined in the latest
study (46,47) from TCGA, which identified risk lncRNAs
and mRNAs to construct a ceRNA network. The present study first
obtained tumor tissue and their corresponding normal tissue lncRNAs
and mRNAs, and subsequently, those RNAs that tended to have
prognostic value were selected by univariate and multivariate
analysis. Next, 29 mRNAs and three lncRNAs were yielded to
construct the PI value (25).
Notably, the PI value proved to be an independent prognostic
indicator of LUAD. It was revealed that the PI, constructed from 32
differential genes, had good prognostic value in LUAD. The present
study used TCGA data that were generated by RNA-Seq and used to
assess the survival of a large number of patients with LUAD in 493
cases.
A total of 29 mRNAs and three lncRNAs were involved
in LUAD risk genes. The present study investigated the molecular
mechanisms of these lncRNAs. However, none of these lncRNAs was
identified in reference records, and little was known regarding the
functional mechanisms of these three lncRNAs. Therefore, a WGCNA
study on lncRNA-associated genes was conducted, introducing the 14
candidates that could be the most relevant genes in LUAD into the
network. The present study focused on the three risk lncRNAs, and
genes within these modules that had a weighted association. These
genes were the lncRNA target genes. Subsequently, the
bioinformatics of abnormally expressed mRNAs was studied, and the
most highly enriched KEGG pathway annotations could be the
functions of lncRNAs. Based on the KEGG pathway analysis, several
cancer-associated pathways were identified, including p53 signaling
and the cell cycle.
Lu et al (48)
assessed the expression and function of the lncRNA SOX21 antisense
RNA 1 (sox21-as1) in 68 pairs of LUAD tissues and the corresponding
non-tumor tissues, and demonstrated that sox21-as1 is associated
with the development of LUAD. Furthermore, the knockdown of
sox21-as1 inhibited the proliferation of LUAD cells in vitro
and in vivo, inducing cell cycle stagnation and apoptosis.
Wu et al (49) integrated two
datasets to search for novel target genes and pathways to explain
the pathogenicity of LUAD, and discovered that PPM1D and GADD45B
could modulate the progression of LUAD through p53 signaling.
Furthermore, the present study investigated the
target genes of three lncRNAs. Fourteen of the FAM138B target genes
were prognostic risk genes, as were 10 TLX1NB target genes-8 of
which were shared. In the absence of calcium, NRGN, which combines
calmodulin with synaptic protein kinase substrates, is reduced in
monkeys and rats following exposure to welding smoke (50). MCC is a candidate colorectal tumor
suppressor gene that is thought to negatively modulate cell cycle
progression (51) and encodes guanine
nucleotide exchange factor (GEF). RASGRF1 stimulates the
dissociation of GDP from RAS. This gene is expressed in various
tissues other than the brain, including the lung and pancreas, and
several tumor cell lines (52). KRT9
serves an important role in mature palm and plantar skin tissue and
in the morphological processes that form these tissues, and is
upregulated (53,54) in platycodin D (PD)-treated HepG2
cells. PLXNB3, a part of the coding family of plexiform proteins,
acts as a receptor for signal 5A and serves a role in invasive
growth, axon guidance and cell migration. Plexin and B3 serve vital
roles in the invasion and metastasis of gastric cancer (55). ACACB may be involved in the regulation
of fatty acid oxidation. It inhibits the synthesis of fatty acids
and tumor development in non-small cell lung cancer in preclinical
models (56).
lncRNAs can function as ceRNAs by binding miRNAs,
regulating the expression of genes. LUAD-associated lncRNA-related
ceRNA networks provide vital hints in detecting crucial RNAs of
ceRNA-mediated gene regulation networks during the initiation and
growth of LUAD. There is increasing evidence that lncRNAs are a key
part of ceRNA networks by regulating the transcription of other
RNAs (5,57–61). For
example, HOTAIR can use miR-331-3p as endogenous lodging to
eliminate the inhibitory activity of the miRNA induced by HER2
3′-UTR and increase its post-transcriptional levels (62). Therefore, as potential connections to
lncRNA, miRNA and mRNA may be involved in the initiation, as well
as the development, of LUAD. The present study predicted a network
involving three types of risk lncRNAs and 712 possible target
miRNAs. To facilitate the next step, 27 miRNAs with a score of
TOP10 were selected. Based on the TarBase, miRTarBase, miRecards
and starBase v2.0 databases, 1,229 objective genes of these 27
miRNAs were obtained, and GO enrichment analysis was performed on
these target genes. The GO enrichment results included positive
regulation of the epithelial-to-mesenchymal transition, regulation
of cell proliferation, positive regulation of the cell cycle and
cell death, and multicellular organism development.
Among the 1,229 genes, there were four prognostic
risk genes. ALDOB is associated with the GO term phosphorus
metabolic process. The main function is to catalyze the reversible
transformation of fructose-1, 6-diphosphite to dihydroxyacetone and
glyceraldehyde 3-phosphate (63).
Abnormal changes in ALDOB are associated with a number of diseases,
including hereditary fructose intolerance, hepatitis, cirrhosis and
cancer (64). Using data mined from
the Gene Expression Omnibus for the colorectal cancer transcriptome
(GSE35452) database, Tian et al (65) reported that ALDOB is associated with
glycolysis and is the most significantly increased transcript (GO:
0006096). It may serve a significant role in colorectal cancer
progression and the response to neoadjuvant concurrent
chemoradiotherapy, and as a novel prognostic biomarker. ACACB
positively regulates organ growth. It catalyzes the ATP-dependent
carboxylation of acetyl-CoA to malonyl-CoA (66). Recent studies have reported that ACACB
is increased in a variety of human cancer types, most likely to
promote fat generation and meet the requirement for rapid growth
and proliferation (67–69). Jones et al (70) demonstrated that knockout of ACACB
changes the lipid content in glioblastoma multiform U87 cells,
increases caspase activity and inhibits the growth of fat to
generate tumor treatment effects. The F9 gene encodes vitamin
K-dependent coagulation factor IX, which circulates in the blood as
an inactive zymogen (71).
Alterations in this gene cause blood coagulation factor IX
deficiency, which is a recessive X-linked disorder, also called
hemophilia B or Christmas disease. The presentation of hemophilia B
is consistent with easy bruising, urinary tract bleeding and
nosebleeds. We hypothesized that tumor-associated mutations in F9
may exacerbate bleeding symptoms due to necrosis of the tumor.
CRISP1, also known as astrocyte elevated gene-1 (AEG-1), is an
oncogene that is overexpressed in a wide range of cancer types and
serves a key role in regulating carcinogenesis (72). AEG-1 serves an important role in
regulating hepatocellular carcinoma (HCC). AEG-1-overexpression at
the mRNA and protein expression levels has been observed in a high
percentage (>90%) of patients with HCC. A progressive increase
in the levels of AEG-1 has been observed in patients with HCC,
correlating directly with disease stage, a higher rate of
recurrence and a poorer overall survival (73,74).
The present study predicted a network consisting of
lncRNAs, miRNAs and target genes, and these target genes are also
cancer-related genes regulated by lncRNAs though the ceRNA network.
The lncRNA FAM138B regulates ALDOB through hsa-miR-378a-3p and
regulates F9 through hsa-miR-619-5p; the lncRNA NHEG1 also
regulates F9 through hsa-miR-619-5p. The lncRNA TLX1NB regulates
CRISP1 through hsa-miR-148b-3p and regulates ACACB through
hsa-miR-1273g-3p. Therefore, hsa-miR-619-5p is at the center of
three prognostic risk lncRNA ceRNA regulation networks and may be a
key miRNA.
Therefore, three lncRNAs may be candidate prognostic
biomarkers for LUAD patients with significant clinical
significance. In summary, the present study examined lncRNAs in 493
patients with LUAD from TCGA and constructed three ceRNA networks
as novel prognostic indicators of LUAD. However, further
experiments are required to verify the clinical and biological
functions of these three lncRNAs.
Acknowledgements
The authors would like to thank Dr Sean Kim (Yale
University) for editing the English text of the first draft of this
manuscript.
Funding
The present study was supported by funds from the
Gansu Provincial Health Bureau Scientific Research Project (grant
no. GSWSKY2018-09).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PR and LW contributed toward the conception and
design of the study; BL analyzed and interpreted the data; and LW
and XL designed the experiments, drafted, and revised the
manuscript..
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
LUAD
|
lung adenocarcinoma
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
lncRNA
|
long non-coding RNA
|
|
NSCLC
|
non-small cell lung cancer
|
|
GO
|
gene ontology
|
|
ROC
|
receiver operating characteristic
|
|
TCGA
|
The Cancer Genome Atlas
|
|
AUC
|
area under the curve
|
|
ceRNA
|
competing endogenous RNAs
|
|
WGCNA
|
weighted gene co-expression network
analysis
|
|
MREs
|
miRNA response elements
|
|
CI
|
confidence intervals
|
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin JJ, Cardarella S, Lydon CA, Dahlberg
SE, Jackman DM, Jänne PA and Johnson BE: Five-Year survival in
EGFR-Mutant metastatic lung adenocarcinoma treated with EGFR-TKIs.
J Thorac Oncol. 11:556–565. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qi L, Li Y, Qin Y, Shi G, Li T, Wang J,
Chen L, Gu Y, Zhao W and Guo Z: An individualised signature for
predicting response with concordant survival benefit for lung
adenocarcinoma patients receiving platinum-based chemotherapy. Br J
Cancer. 115:1513–1519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee JT: Epigenetic regulation by long
noncoding RNAs. Science. 338:1435–1439. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou M, Wang X, Shi H, Cheng L, Wang Z,
Zhao H, Yang L and Sun J: Characterization of long non-coding
RNA-associated ceRNA network to reveal potential prognostic lncRNA
biomarkers in human ovarian cancer. Oncotarget. 7:12598–12611.
2016.PubMed/NCBI
|
|
6
|
Wang PL, Liu B, Xia Y, Pan CF, Ma T and
Chen YJ: Long non-coding RNA-Low expression in tumor inhibits the
invasion and metastasis of esophageal squamous cell carcinoma by
regulating p53 expression. Mol Med Rep. 13:3074–3082. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shang C, Guo Y, Zhang J and Huang B:
Silence of long noncoding RNA UCA1 inhibits malignant proliferation
and chemotherapy resistance to adriamycin in gastric cancer. Cancer
Chemother Pharmacol. 77:1061–1067. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Zhang Y, Yang T, Zhao W, Wang N,
Li P, Zeng X and Zhang W: Long non-coding RNA MALAT1 for promoting
metastasis and proliferation by acting as a ceRNA of miR-144-3p in
osteosarcoma cells. Oncotarget. 8:59417–59434. 2017.PubMed/NCBI
|
|
9
|
Wang P, Zhi H, Zhang Y, Liu Y, Zhang J,
Gao Y, Guo M, Ning S and Li X: miRSponge: A manually curated
database for experimentally supported miRNA sponges and ceRNAs.
Database (Oxford). 2015(pii): bav0982015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: the Rosetta stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan JY, Sirey T, Honti F, Graham B,
Piovesan A, Merkenschlager M, Webber C, Ponting CP and Marques AC:
Extensive microRNA-mediated crosstalk between lncRNAs and mRNAs in
mouse embryonic stem cells. Genome Res. 25:655–666. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Karreth FA and Pandolfi PP: ceRNA
cross-talk in cancer: When ce-bling rivalries go awry. Cancer
Discov. 3:1113–1121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akcakaya P, Ekelund S, Kolosenko I,
Caramuta S, Ozata DM, Xie H, Lindforss U, Olivecrona H and Lui WO:
miR-185 and miR-133b deregulation is associated with overall
survival and metastasis in colorectal cancer. Int J Oncol.
39:311–318. 2011.PubMed/NCBI
|
|
14
|
Zhang J, Fan D, Jian Z, Chen GG and Lai
PB: Cancer specific long noncoding RNAs show differential
expression patterns and competing endogenous RNA potential in
hepatocellular carcinoma. PLoS One. 10:e01410422015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Chen Z, Li MJ, Guo HY and Jing
NC: Long non-coding RNA metastasis-associated lung adenocarcinoma
transcript 1 regulates the expression of Gli2 by miR-202 to
strengthen gastric cancer progression. Biomed Pharmacother.
85:264–271. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu T, Zhang X, Yang YM, Du LT and Wang
CX: Increased expression of the long noncoding RNA CRNDE-h
indicates a poor prognosis in colorectal cancer, and is positively
correlated with IRX5 mRNA expression. Onco Targets Ther.
9:1437–1448. 2016.PubMed/NCBI
|
|
17
|
Han D, Gao X, Wang M, Qiao Y, Xu Y, Yang
J, Dong N, He J, Sun Q, Lv G, et al: Long noncoding RNA H19
indicates a poor prognosis of colorectal cancer and promotes tumor
growth by recruiting and binding to eIF4A3. Oncotarget.
7:22159–22173. 2016.PubMed/NCBI
|
|
18
|
Qiu JJ and Yan JB: Long non-coding RNA
LINC01296 is a potential prognostic biomarker in patients with
colorectal cancer. Tumour Biol. 36:7175–7183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yin DD, Liu ZJ, Zhang E, Kong R, Zhang ZH
and Guo RH: Decreased expression of long noncoding RNA MEG3 affects
cell proliferation and predicts a poor prognosis in patients with
colorectal cancer. Tumour Biol. 36:4851–4859. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Y, Zhang M, Liang L, Li J and Chen YX:
Over-expression of lncRNA DANCR is associated with advanced tumor
progression and poor prognosis in patients with colorectal cancer.
Int J Clin Exp Pathol. 8:11480–11484. 2015.PubMed/NCBI
|
|
21
|
Edge SB and Compton CC: The American Joint
Committee on Cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gene Ontology Consortium, . The gene
ontology: Enhancements for 2011. Nucleic Acids Res. 40(Database
Issue): D559–D564. 2012.PubMed/NCBI
|
|
23
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–103. 101–103. 119–128. 244–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Foulger RE, Osumi-Sutherland D, McIntosh
BK, Hulo C, Masson P, Poux S, Le Mercier P and Lomax J:
Representing virus-host interactions and other multi-organism
processes in the Gene Ontology. BMC Microbiol. 15:1462015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang RX, Chen WJ, He RQ, Zeng JH, Liang L,
Li SK, Ma J, Luo DZ and Chen G: Identification of a RNA-Seq based
prognostic signature with five lncRNAs for lung squamous cell
carcinoma. Oncotarget. 8:50761–50773. 2017.PubMed/NCBI
|
|
26
|
Zhou M, Sun Y, Sun Y, Xu W, Zhang Z, Zhao
H, Zhong Z and Sun J: Comprehensive analysis of lncRNA expression
profiles reveals a novel lncRNA signature to discriminate
nonequivalent outcomes in patients with ovarian cancer. Oncotarget.
7:32433–32448. 2016.PubMed/NCBI
|
|
27
|
Presson AP, Sobel EM, Papp JC, Suarez CJ,
Whistler T, Rajeevan MS, Vernon SD and Horvath S: Integrated
weighted gene co-expression network analysis with an application to
chronic fatigue syndrome. BMC Syst Biol. 2:952008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lossos IS, Czerwinski DK, Alizadeh AA,
Wechser MA, Tibshirani R, Botstein D and Levy R: Prediction of
survival in diffuse large-B-cell lymphoma based on the expression
of six genes. N Engl J Med. 350:1828–1837. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alizadeh AA, Gentles AJ, Alencar AJ, Liu
CL, Kohrt HE, Houot R, Goldstein MJ, Zhao S, Natkunam Y, Advani RH,
et al: Prediction of survival in diffuse large B-cell lymphoma
based on the expression of 2 genes reflecting tumor and
microenvironment. Blood. 118:1350–1358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meza R, Meernik C, Jeon J and Cote ML:
Lung cancer incidence trends by gender, race and histology in the
United States, 1973–2010. PLoS One. 10:e01213232015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kerr KM: Pulmonary adenocarcinomas:
Classification and reporting. Histopathology. 54:12–27. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aokage K, Yoshida J, Hishida T, Tsuboi M,
Saji H, Okada M, Suzuki K, Watanabe S and Asamura H: Limited
resection for early-stage non-small cell lung cancer as
function-preserving radical surgery: A review. Jpn J Clin Oncol.
47:7–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Provencio M, Isla D, Sanchez A and Cantos
B: Inoperable stage III non-small cell lung cancer: Current
treatment and role of vinorelbine. J Thorac Dis. 3:197–204.
2011.PubMed/NCBI
|
|
34
|
Ortea I, Rodriguez-Ariza A, Chicano-Galvez
E, Arenas Vacas MS and Jurado Gamez B: Discovery of potential
protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage
fluid by SWATH MS data-independent acquisition and targeted data
extraction. J Proteomics. 138:106–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Navani N, Nankivell M, Lawrence DR, Lock
S, Makker H, Baldwin DR, Stephens RJ, Parmar MK, Spiro SG, Morris
S, et al: Lung cancer diagnosis and staging with endobronchial
ultrasound-guided transbronchial needle aspiration compared with
conventional approaches: An open-label, pragmatic, randomised
controlled trial. Lancet Respir Med. 3:282–289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Y, Yang Y, Li L, Liu Y, Geng P, Li G
and Song H: LncRNA SNHG1 enhances cell proliferation, migration and
invasion in cervical cancer. Biochem Cell Biol. 96:38–43. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao M, Sun D, Li X, Xu Y, Zhang H, Qin Y
and Xia M: Overexpression of long noncoding RNA PEG10 promotes
proliferation, invasion and metastasis of hypopharyngeal squamous
cell carcinoma. Oncol Lett. 14:2919–2925. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bao H, Guo CG, Qiu PC, Zhang XL, Dong Q
and Wang YK: Long non-coding RNA Igf2as controls hepatocellular
carcinoma progression through the ERK/MAPK signaling pathway. Oncol
Lett. 14:2831–2837. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lai Y, Xu P, Liu J, Li Q, Ren D, Zhang J
and Wang J: Decreased expression of the long non-coding RNA MLLT4
antisense RNA 1 is a potential biomarker and an indicator of a poor
prognosis for gastric cancer. Oncol Lett. 14:2629–2634. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tian Z, Wen S, Zhang Y, Shi X, Zhu Y, Xu
Y, Lv H and Wang G: Identification of dysregulated long non-coding
RNAs/microRNAs/mRNAs in TNM I stage lung adenocarcinoma.
Oncotarget. 8:51703–51718. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen G, Peng L, Zhu Z, Du C, Shen Z, Zang
R, Su Y, Xia Y and Tang W: LncRNA AFAP1-AS functions as a competing
endogenous RNA to regulate RAP1B expression by sponging miR-181a in
the HSCR. Int J Med Sci. 14:1022–1030. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li DS, Ainiwaer JL, Sheyhiding I, Zhang Z
and Zhang LW: Identification of key long non-coding RNAs as
competing endogenous RNAs for miRNA-mRNA in lung adenocarcinoma.
Eur Rev Med Pharmacol Sci. 20:2285–2295. 2016.PubMed/NCBI
|
|
44
|
Sui J, Li YH, Zhang YQ, Li CY, Shen X, Yao
WZ, Peng H, Hong WW, Yin LH, Pu YP and Liang GY: Integrated
analysis of long non-coding RNA-associated ceRNA network reveals
potential lncRNA biomarkers in human lung adenocarcinoma. Int J
Oncol. 49:2023–2036. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu B, Chen Y and Yang J: LncRNAs are
altered in lung squamous cell carcinoma and lung adenocarcinoma.
Oncotarget. 8:24275–24291. 2017.PubMed/NCBI
|
|
46
|
Zhang Y, Wen DY, Zhang R, Huang JC, Lin P,
Ren FH, Wang X, He Y, Yang H, Chen G and Luo DZ: A preliminary
investigation of PVT1 on the effect and mechanisms of
hepatocellular carcinoma: Evidence from clinical data, a
meta-analysis of 840 cases, and in vivo validation. Cell Physiol
Biochem. 47:2216–2232. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jiang R, Zhao C, Gao B, Xu J, Song W and
Shi P: Mixomics analysis of breast cancer: Long non-coding RNA
linc01561 acts as ceRNA involved in the progression of breast
cancer. Int J Biochem Cell Biol. 102:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lu X, Huang C, He X, Liu X, Ji J, Zhang E,
Wang W and Guo R: A novel long Non-coding RNA, SOX21-AS1, indicates
a poor prognosis and promotes lung adenocarcinoma proliferation.
Cell Physiol Biochem. 42:1857–1869. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu X, Zang W, Cui S and Wang M:
Bioinformatics analysis of two microarray gene-expression data sets
to select lung adenocarcinoma marker genes. Eur Rev Med Pharmacol
Sci. 16:1582–1587. 2012.PubMed/NCBI
|
|
50
|
Heo JD, Oh JH, Lee K, Kim CY, Song CW,
Yoon S, Han JS and Yu IJ: Gene expression profiling in the lung
tissue of cynomolgus monkeys in response to repeated exposure to
welding fumes. Arch Toxicol. 84:191–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fukuyama R, Niculaita R, Ng KP, Obusez E,
Sanchez J, Kalady M, Aung PP, Casey G and Sizemore N: Mutated in
colorectal cancer, a putative tumor suppressor for serrated
colorectal cancer, selectively represses beta-catenin-dependent
transcription. Oncogene. 27:6044–6055. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guerrero C, Rojas JM, Chedid M, Esteban
LM, Zimonjic DB, Popescu NC, Font de Mora J and Santos E:
Expression of alternative forms of Ras exchange factors GRF and
SOS1 in different human tissues and cell lines. Oncogene.
12:1097–1107. 1996.PubMed/NCBI
|
|
53
|
Langbein L, Heid HW, Moll I and Franke WW:
Molecular characterization of the body site-specific human
epidermal cytokeratin 9: cDNA cloning, amino acid sequence, and
tissue specificity of gene expression. Differentiation. 55:57–71.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lu JJ, Lu DZ, Chen YF, Dong YT, Zhang JR,
Li T, Tang ZH and Yang Z: Proteomic analysis of hepatocellular
carcinoma HepG2 cells treated with platycodin D. Chin J Nat Med.
13:673–679. 2015.PubMed/NCBI
|
|
55
|
Pan GQ, Ren HZ, Zhang SF, Wang XM and Wen
JF: Expression of semaphorin 5A and its receptor plexin B3
contributes to invasion and metastasis of gastric carcinoma. World
J Gastroenterol. 15:2800–2804. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Svensson RU, Parker SJ, Eichner LJ, Kolar
MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A,
Vera L, et al: Inhibition of acetyl-CoA carboxylase suppresses
fatty acid synthesis and tumor growth of non-small-cell lung cancer
in preclinical models. Nat Med. 22:1108–1119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li CY, Liang GY, Yao WZ, Sui J, Shen X,
Zhang YQ, Peng H, Hong WW, Ye YC, Zhang ZY, et al: Integrated
analysis of long non-coding RNA competing interactions reveals the
potential role in progression of human gastric cancer. Int J Oncol.
48:1965–1976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Guo LL, Song CH, Wang P, Dai LP, Zhang JY
and Wang KJ: Competing endogenous RNA networks and gastric cancer.
World J Gastroenterol. 21:11680–11687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hu Y, Tian H, Xu J and Fang JY: Roles of
competing endogenous RNAs in gastric cancer. Brief Funct Genomics.
15:266–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wu Q, Guo L, Jiang F, Li L, Li Z and Chen
F: Analysis of the miRNA-mRNA-lncRNA networks in ER+ and ER-breast
cancer cell lines. J Cell Mol Med. 19:2874–2887. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Song X, Cao G, Jing L, Lin S, Wang X,
Zhang J, Wang M, Liu W and Lv C: Analysing the relationship between
lncRNA and protein-coding gene and the role of lncRNA as ceRNA in
pulmonary fibrosis. J Cell Mol Med. 18:991–1003. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB,
Yin DD, Kong R, Xia R, Lu KH, Li JH, De W, et al: Lnc RNA HOTAIR
functions as a competing endogenous RNA to regulate HER2 expression
by sponging miR-331-3p in gastric cancer. Mol Cancer. 13:922014.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Penhoet E, Rajkumar T and Rutter WJ:
Multiple forms of fructose diphosphate aldolase in mammalian
tissues. Proc Natl Acad Sci USA. 56:pp. 1275–1282. 1966; View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Asaka M, Kimura T, Meguro T, Kato M, Kudo
M, Miyazaki T and Alpert E: Alteration of aldolase isozymes in
serum and tissues of patients with cancer and other diseases. J
Clin Lab Anal. 8:144–148. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tian YF, Hsieh PL, Lin CY, Sun DP, Sheu
MJ, Yang CC, Lin LC, He HL, Solórzano J, Li CF and Chang IW: High
expression of Aldolase B confers a poor prognosis for rectal cancer
patients receiving neoadjuvant chemoradiotherapy. J Cancer.
8:1197–1204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wakil SJ and Abu-Elheiga LA: Fatty acid
metabolism: Target for metabolic syndrome. J Lipid Res. 50
Suppl:S138–S143. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Milgraum LZ, Witters LA, Pasternack GR and
Kuhajda FP: Enzymes of the fatty acid synthesis pathway are highly
expressed in in situ breast carcinoma. Clin Cancer Res.
3:2115–2120. 1997.PubMed/NCBI
|
|
68
|
Yahagi N, Shimano H, Hasegawa K, Ohashi K,
Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, et
al: Co-ordinate activation of lipogenic enzymes in hepatocellular
carcinoma. Eur J Cancer. 41:1316–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Swinnen JV, Brusselmans K and Verhoeven G:
Increased lipogenesis in cancer cells: New players, novel targets.
Curr Opin Clin Nutr Metab Care. 9:358–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Jones JE, Esler WP, Patel R, Lanba A, Vera
NB, Pfefferkorn JA and Vernochet C: Inhibition of Acetyl-CoA
carboxylase 1 (ACC1) and 2 (ACC2) reduces proliferation and De Novo
lipogenesis of EGFRvIII Human Glioblastoma cells. PLoS One.
12:e01695662017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Simioni P, Tormene D, Tognin G, Gavasso S,
Bulato C, Iacobelli NP, Finn JD, Spiezia L, Radu C and Arruda VR:
X-linked thrombophilia with a mutant factor IX (factor IX Padua). N
Engl J Med. 361:1671–1675. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Sarkar D and Fisher PB: AEG-1/MTDH/LYRIC:
Clinical significance. Adv Cancer Res. 120:39–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yoo BK, Emdad L, Su ZZ, Villanueva A,
Chiang DY, Mukhopadhyay ND, Mills AS, Waxman S, Fisher RA, Llovet
JM, et al: Astrocyte elevated gene-1 regulates hepatocellular
carcinoma development and progression. J Clin Invest. 119:465–477.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhu K, Dai Z, Pan Q, Wang Z, Yang GH, Yu
L, Ding ZB, Shi GM, Ke AW, Yang XR, et al: Metadherin promotes
hepatocellular carcinoma metastasis through induction of
epithelial-mesenchymal transition. Clin Cancer Res. 17:7294–7302.
2011. View Article : Google Scholar : PubMed/NCBI
|