Introduction
Initially discovered in avian genomes, MYC
proto-oncogene (MYC) is a key regulator of cell growth,
proliferation, metabolism, differentiation and apoptosis (1), and a potent activator of tumorigenesis.
MYC is dysregulated in various cancer types (1–3) and serves
a critical role in breast tumorigenesis and cancer progression
(4–6).
Dysregulation of MYC in breast cancer involves multiple mechanisms,
including gene amplification, transcriptional regulation, and mRNA
and protein stabilization. Previous studies demonstrated that the
MYC T58A point mutation impaired the apoptotic potential of MYC in
rodent cells (7–9), and a study reported that this mutation
reduced the occurrence of MYC-induced apoptosis in human mammary
epithelial cells (10). However, the
physiological functions of MYC and the consequences of its
dysfunction in breast cancer cells remain obscure.
Bim is a Bcl-2 homology 3 domain (BH3)-only protein
that is involved in stimulus-induced, cellular tumor antigen p53
(p53)-independent apoptosis (11,12). MYC
was reported to bind to the Bim promoter and promote Bim
transcription (13,14). Furthermore, dysregulated MYC
expression sensitized cells to apoptosis via a Bim-dependent
mechanism (11). Although mutant MYC
was unable to induce Bim expression, it activated the p53 pathway
to a similar extent as wild-type (WT) MYC (9). This result may help explain why tumors
with mutant MYC are less prone to apoptosis, and why mutant MYC is
more oncogenic compared with WT MYC (9). Previously, it was demonstrated that
tumor-derived MYC mutants are relevant to breast cancer pathology
as they are unable to upregulate Bim, which induces p53-independent
apoptosis (15). However, the
mechanisms underlying the observation that MYC mutants are unable
to upregulate Bim are unknown.
The principal model for the p53-independent
mechanism of apoptosis is that the tumor suppressor p14, which
binds to MYC directly and blocks the transcription of MYC canonical
target genes, also inhibits MYC-induced hyperproliferation and
transformation (16,17). Despite its inhibition of canonical MYC
activity, p14 has been reported to be essential for MYC to induce
p53-independent apoptosis in mouse embryonic fibroblasts (17,18). A
subsequent study demonstrated that E2F proteins serve a direct role
in the transcriptional regulation of p14 (19). Upregulated Bim expression levels in
prostate and breast cancer cells are dependent on E2F, and E2F
silencing leads to the loss of Bim expression (20). Thus, WT MYC appears to upregulate p14
in order to promote Bim-induced apoptosis. However, the mechanisms
by which MYC mutants regulate p14-induced p53-independent apoptosis
have not been fully clarified.
The cyclin-dependent kinase inhibitor protein p21
regulates cell cycle progression at the G1 phase, mediates cell
proliferation, differentiation, senescence and apoptosis, and may
influence transcription in a p53-dependent or independent manner
(21). Substantial data from
biochemical and genetic studies indicate that p21 acts as a master
effector of multiple tumor suppressor pathways to promote
antiproliferative activities that are independent of the classical
p53 tumor suppressor pathway. Furthermore, p21 suppresses the
induction of proapoptotic genes by MYC and E2F1 through direct
binding and inhibition of their transactivation functions (22). G1/S arrest induced by overexpression
of p21 was reported to involve suppression of Bim (23). Although a number of studies indicated
a proapoptotic role for p21, these studies only demonstrated that
apoptosis occurred concurrently with p21 induction, without
determining whether p21 was required for the induction of
apoptosis.
Apoptosis in response to dysregulated MYC is an
important failsafe mechanism that is essential in preventing the
proliferation of tumorigenic cells (24). Apoptosis induced by oncogenic MYC
occurs through p53-dependent and independent mechanisms that are
not well understood (9,25). The p53 gene is mutated in ~50% of
human tumors, and the loss of p53 antitumor activity is associated
with defects in cell cycle arrest and apoptosis (26). It has been proposed that the loss of
p53-initiated DNA repair processes underpins the high cancer
susceptibility observed in p53-deficient mice (27). DNA damage elicited by chemotherapeutic
drugs may induce apoptosis in the absence of p53 (28). It is apparent that various
p53-independent apoptotic mechanisms may be used to induce
apoptosis in p53-initiated cancer cells. It is therefore important
to further clarify the precise molecular mechanism of
p53-independent apoptosis. It appears likely that DNA lesions may
activate BH3-only proteins in a p53-independent manner (29). A recent study reported that the loss
of Bim strongly correlates with the loss of p53 in lymphomagenesis
(11). Thus, the effects of the MYC
point mutation T58A on the progression of breast cancer with p53
loss were analyzed, and the mechanism of p53-independent
MYC-induced apoptosis was investigated.
MYC dysregulation contributes to the initiation and
progression of breast cancer and is associated with poor outcomes,
particularly in the basal-like cancer subtype. Thus, targeting
MYC-regulated pathways may provide a promising therapeutic strategy
for breast cancer.
Materials and methods
Cells and reagents
Human breast cancer cells (HCC1937; China Center for
Type Culture Collection, Chinese Academy of Sciences, Shanghai,
China) were cultured in RPMI-1640 medium (HyClone; GE Healthcare
Life Sciences, Logan, UT, USA) and 10% fetal bovine serum (FBS;
Hyclone; GE Healthcare Life Sciences). The WT MYC-green fluorescent
protein (GFP)-lentivirus and T58A-GFP-lentivirus vectors were
constructed previously (30). The
p21-small interfering (si)RNA-red fluorescent protein
(RFP)-lentivirus and p14-siRNA-RFP-lentivirus vectors were
constructed by Shanghai Biological Engineering Co., Ltd. (Shanghai,
China). The terminal deoxynucleotidyl transferase-mediated dUTP
nick end labeling (TUNEL) assay kit was obtained from Beyotime
Institute of Biotechnology (Haimen, China). Antibodies against MYC,
p21, p14 and GAPDH were purchased from Abcam (Cambridge, UK).
Purified rabbit anti-Bim was from BD Pharmingen (BD Biosciences,
Franklin Lakes, NJ, USA).
T58A and WT MYC transfection and p21
and p14 interference of HCC1937 cells
Control lenti-GFP/neo virus, lenti-T58A-GFP/neo, and
lenti-WT MYC-GFP/neo particles were previously constructed and
packaged (30). The titer was
measured using the method described by Meng et al (30). HCC1937 cells were maintained in
RPMI-1640 medium supplemented with 10% FBS and plated into six-well
plates at 1×105 cells/well overnight. Cells at 50%
confluence were infected with lenti-GFP/neo, lenti-T58A-GFP/neo,
lenti-WT MYC-GFP/neo particles, and with p21- or
p14-siRNA-RFP-lentivirus vectors at a multiplicity of infection of
6. Cells were cultured in RPMI-1640 medium supplemented with 10%
FBS at 37°C in 5% CO2 for 2 days. Transfection
efficiencies were determined by assessing GFP expression using a
Leica DMI 4000B (Leica Microsystems GmbH, Wetzlar, Germany)
fluorescence microscope at magnification, ×100.
NeoMYCin G418 (400 mg/l; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) was added to the medium to select the
stably transfected cells. NeoMYCin-resistant colonies were picked
up 2 weeks post-transfection. Stably transfected cells were
maintained in medium containing 400 mg/l neoMYCin.
RNA extraction and reverse
transcription-semi-quantitative polymerase chain reaction
(RT-sqPCR) analysis
Total RNA was isolated from the cells using TRIzol
reagent (Thermo Fisher Scientific, Inc.). The first strand of cDNA
was synthesized by reverse transcription of 2 µg total RNA using an
Advantage RT-for-PCR kit (Takara Biotechnology Co., Ltd., Dalian,
China). The temperature protocol for reverse transcription was as
follows: 72°C for 2 min, 42°C for 1 h and 94°C for 5 min. A 0.6 µl
aliquot of the RT reaction mixture was used for subsequent sqPCR
analysis. The sequences of the forward and reverse primer pairs
(designed and synthesized by Shanghai Sangon Biological Engineering
Technology Co., Ltd., Shanghai, China) were as follows: MYC sense,
5′-GATTCTCTGCTCTCCTCGAC-3′ and antisense,
5′-TCCAGACTCTGACCTTTTGC-3′; p21 sense, 5′-ACTGTGATGCGCTAATGGC-3′
and antisense, 5′-ATGGTCTTCCTCTGCTGTCC-3′; p14 sense,
5′-CACCGGAATCCTGGACCAG-3′ and antisense,
5′-GCAGTTCGAATCTGCACCGT-3′; Bim sense, 5′-AGATCCCCGCTTTTCATCTT-3′
and antisense, 5′-AGGACTTGGGGTTTGTGTTG-3′; and GAPDH sense,
5′-CTGCACCACCAACTGCTTAG-3′ and antisense,
5′-TGAAGTCAGAGGAGACCACC-3′. Each 20 µl volume of PCR mixture
contained 10 µl premixed Taq polymerase (Takara Biotechnology Co.,
Ltd.), 0.5 µl forward primer, 0.5 µl reverse primer and 8.4 µl
double-distilled water. The PCR thermocycling conditions were as
follows: 94°C for 3 min, followed by 30 cycles of 94°C for 45 sec,
60°C for 45 sec and 70°C for 2 min, and a final extension step at
72°C for 7 min. A 2-µl aliquot of PCR product was analyzed by
electrophoresis on a 2% agarose gel containing ethidium bromide,
visualized under UV light, and quantified using ImageJ (version
2.1.4.7; http://imagej.nih.gov/ij/).
Western blot analysis
Cells were harvested using radioimmunoprecipitation
assay lysis buffer (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
The protein concentration of each cell extract was measured using a
bovine serum albumin protein assay kit (Bio-Rad Laboratories Inc.,
Hercules, CA, USA). Proteins were separated by SDS-PAGE on a 10%
gel (30 µg/lane). Proteins were transferred to Hybond-P
polyvinylidene difluoride membranes (GE Healthcare Life Sciences),
which were blocked with 5% skimmed milk in Tris-buffered saline
containing 0.1% Tween-20 for 60 min at room temperature. The
membranes were incubated overnight at 4°C with FLAG-conjugated
primary antibodies, including anti-p21, anti-p14, anti-MYC and
anti-β-actin (cat. nos. sc-53393, sc-71808, sc-70469 and sc-58673,
respectively; all 1:200 dilution; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), and anti-Bim (cat. no. 559685; 1:200 dilution; BD
Pharmingen), followed by incubation at room temperature for 1 h
with an anti-FLAG horseradish peroxidase (HRP)-conjugated
monoclonal antibody (cat. no. A8592; 1:5,000 dilution;
Sigma-Aldrich; Merck KGaA) in 20 mM TBS with Tween-20.
Immunoreactions were visualized using an enhanced chemiluminescence
kit (GE Healthcare, Chicago, IL, USA), in accordance with the
manufacturer's protocol. The intensity of each band relative to
β-actin was determined quantitatively using ImageQuant TL software
(version 7.0; GE Healthcare Life Sciences). Data are reported as
the mean ± standard deviation of three replicates for each
experiment.
Colony formation assay
HCC1937 cells were seeded in 6-well plates. Cells at
50% confluence were subsequently transfected with WT or mutant
(T58A) MYC in triplicate and cultured for 12 days, followed by
fixation with methanol and staining with 0.4% crystal violet at
room temperature for 10 min. Colonies containing ≥10 cells were
counted under an inverted microscope at magnification, ×100. The
colony formation ratio (%) was determined as the number of cell
clones divided by 500 and multiplied by 100.
MTT assay
HCC1937 cells were transfected in 6-well plates and
cultured for 48 h. Cells with the indicated treatments were
harvested and transferred to 96-well plates. Following 24, 48 and
72 h incubation, the medium was removed and replaced with 100 µl
fresh culture medium. MTT (5 mg/ml; 20 µg) was added to each well,
followed by incubation at 37°C for 4 h. Dimethyl sulfoxide (100 µg;
Promega Corporation, Madison, WI, USA) was added to each well,
followed by thorough mixing for 15 sec. Absorbance at 490 nm was
measured using an automated plate reader. Each sample was analyzed
in triplicate, and each experiment was repeated three times. Cell
growth curves were calculated using mean values for each group.
TUNEL assay
Glass coverslips (~30-mm diameter) were placed in
the wells of a 6-well plate and cultured in RPMI-1640 medium.
HCC1937 cells in the logarithmic growth phase were seeded in the
plates at 1×105 cells/well. When the cells reached 50%
confluence, they were transfected and cultured for 48 h. Cell
apoptosis was measured by the TUNEL assay according to the
manufacturer's protocol. The cells were fixed in freshly prepared
4% methanol-free paraformaldehyde solution in PBS (pH 7.4) for 25
min at 4°C. Following fixation, the cells were permeabilized in
0.2% Triton X-100 solution in PBS for 5 min, washed with PBS and
covered with 0.3% H2O2 in PBS for 20 min, at
room temperature. The labeling reaction was performed using TUNEL
reaction-mixture label solution incubated for 1 h at 37°C. The
samples were incubated with streptavidin-HRP for 30 min at room
temperature. Following 3 PBS washes, the samples were stained with
diaminobenzidine developing solution for 10 min at room
temperature, and washed again 3 times. A total of 5 equal-sized
fields were randomly chosen and analyzed under a Leica DMI 4000B
light microscope (Leica Microsystems GmbH). Density was evaluated
in each positively stained field, yielding the density of dead
cells (cell death index).
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) flow cytometry
Following 48 h of culture, cells in the indicated
treatment groups were resuspended in binding buffer at a density of
1×106 cells/ml. A 100-µl sample of cells was mixed with
5 µg FITC-Annexin V (Sigma-Aldrich; Merck KGaA) and PI (20 µg/ml,
10 µl; Sigma-Aldrich; Merck KGaA). The cells were incubated for 20
min in the dark at room temperature, and 1×104 cells
were analyzed from each sample by flow cytometry (FACSCaliber).
Cell apoptosis was assayed using Cell Quest software (version 5.1;
BD Biosciences). Cells that were positive for Annexin V (Annexin
V+) and negative for PI (PI−) were scored as
early apoptotic cells. Cells that were Annexin V+ and
PI+ were scored as late apoptotic cells. In this way,
necrotic cells were excluded (Annexin V+ and
PI−/PI+ cells).
Statistical analysis
All statistical analyses were performed using SPSS
17.0 software (SPSS Inc., Chicago, IL, USA). Each result is
presented as the mean ± standard deviation of three replicate
assays. One-way analysis of variance was used to analyze
differences between groups. The LSD post hoc test was used to
determine pairwise differences between means. P<0.05 was
considered to indicate a statistically significant difference.
Results
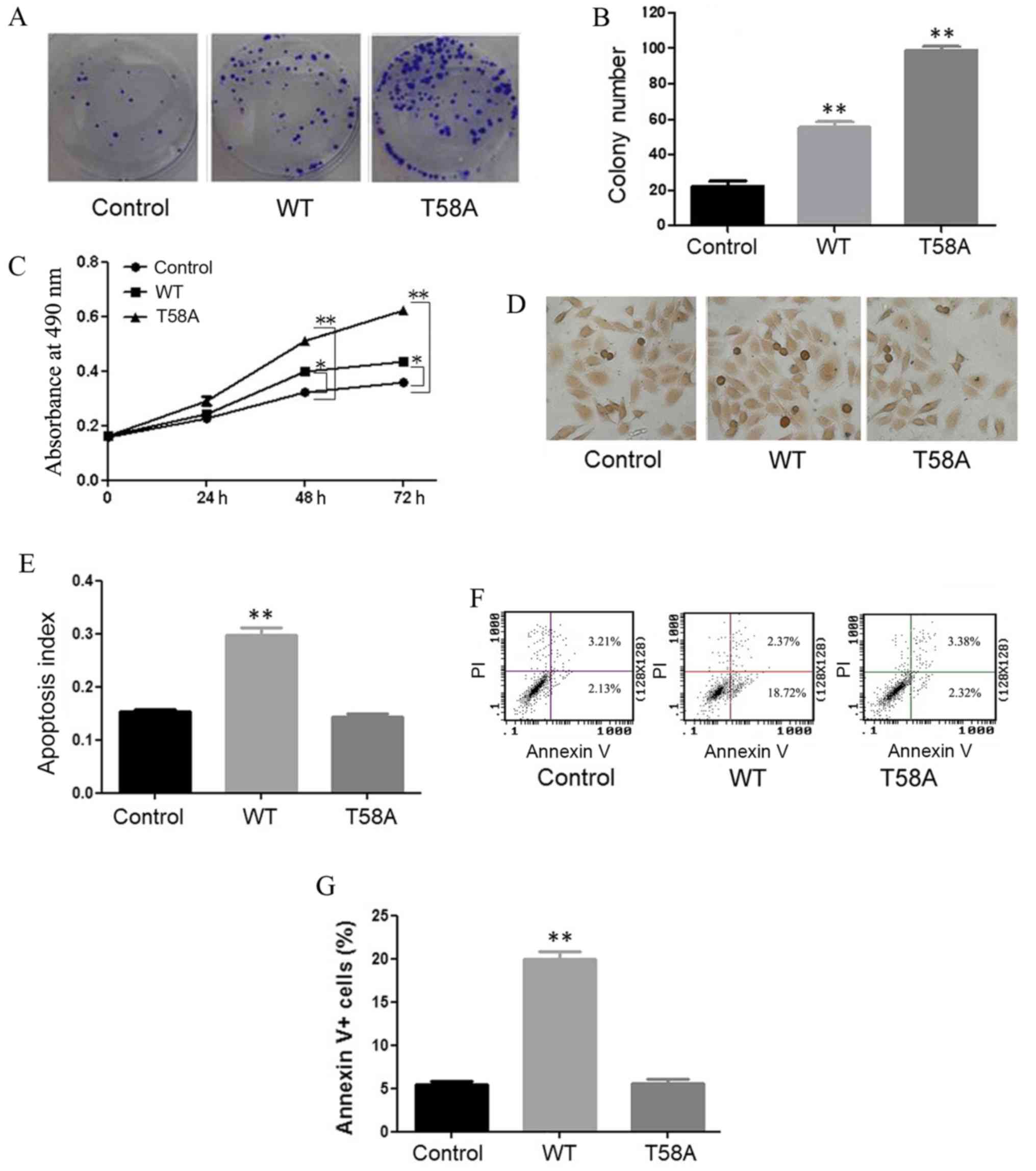
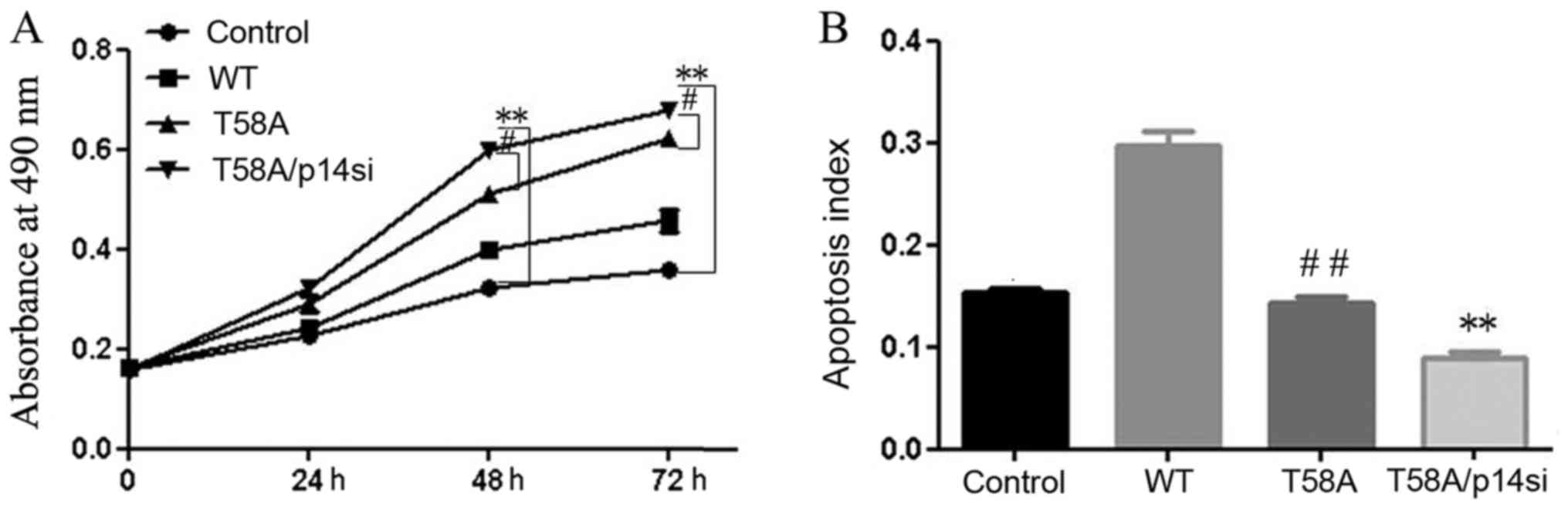
MYC mutant does not induce apoptosis
in HCC1937 cells
HCC1937 cells were successfully transfected with WT
and mutant MYC. Colony formation (Fig. 1A
and B) and MTT (Fig. 1C) assays
demonstrated that cell growth was induced by WT and mutant MYC
compared with the control. Mutant MYC resulted in greater induction
of cell proliferation compared with WT MYC.
TUNEL assays were performed to detect in situ
apoptosis (Fig. 1D and E), with dark
brown staining of nuclei indicating apoptotic cells. Compared with
control cells, WT cells displayed a higher apoptosis rate
(P<0.01). No significant difference in apoptosis was observed
between mutant MYC and control cells (P=0.30). Flow cytometry was
performed in order to confirm the roles of mutant and WT MYC in
apoptosis (Fig. 1F and G). The same
results were obtained in the two experiments, indicating that
mutant MYC induced proliferation and did not promote apoptosis in
HCC1937 cells.
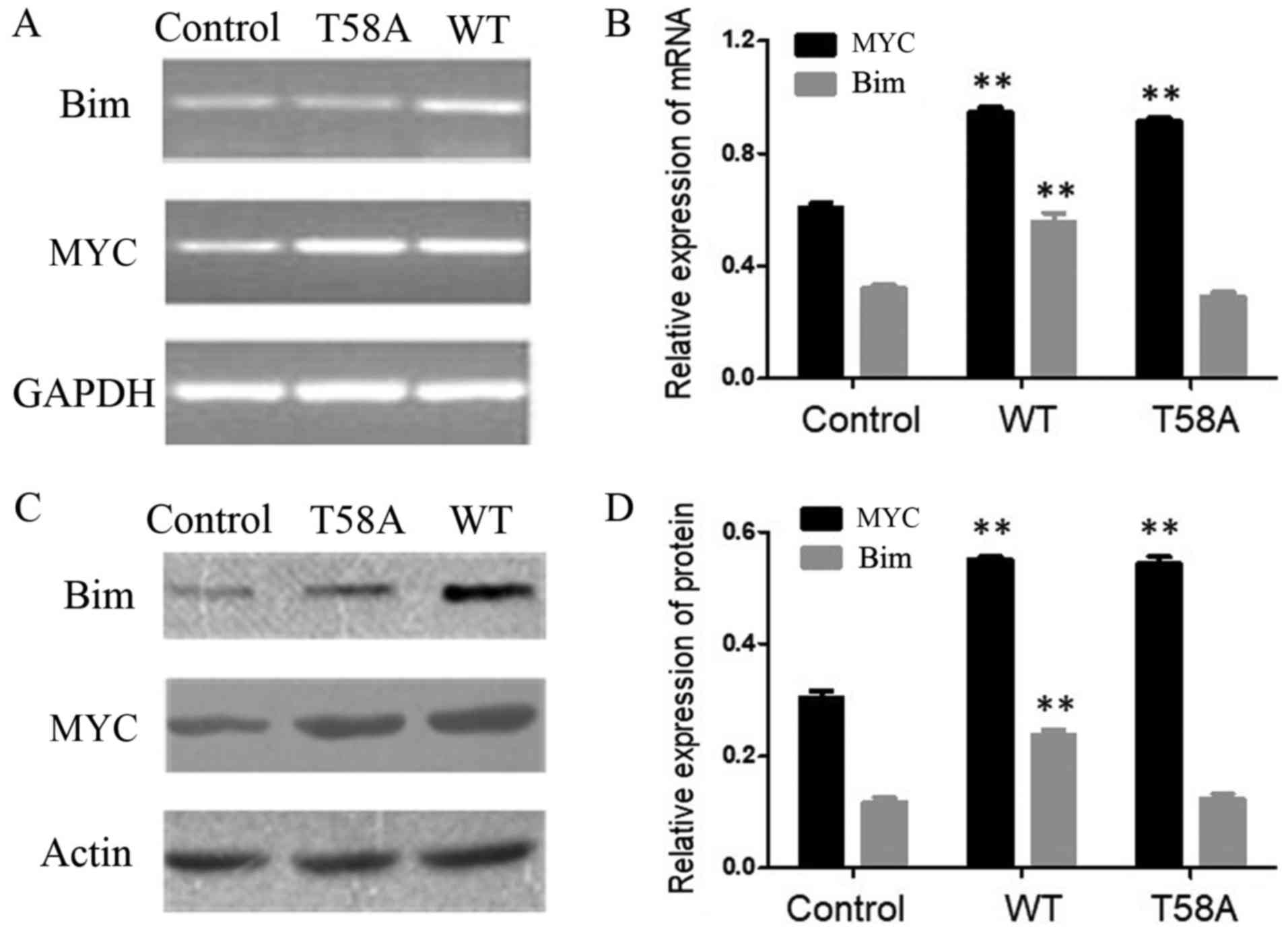
Impairment of Bim induced by mutant
MYC
MYC was demonstrated to bind to Bim, which is
involved in stimulus-induced p53-independent apoptosis (11–14). The
expression levels of Bim in cells transfected with WT and mutant
MYC were investigated (Fig. 2) to
further examine why mutant MYC did not induce apoptosis. Compared
with control cells, the Bim expression level was increased in WT
cells (P<0.01). Bim expression was comparable in control
and mutant MYC cells (P=0.92). These results indicated that the
inability of mutant MYC to upregulate Bim is associated with its
inability to induce apoptosis.
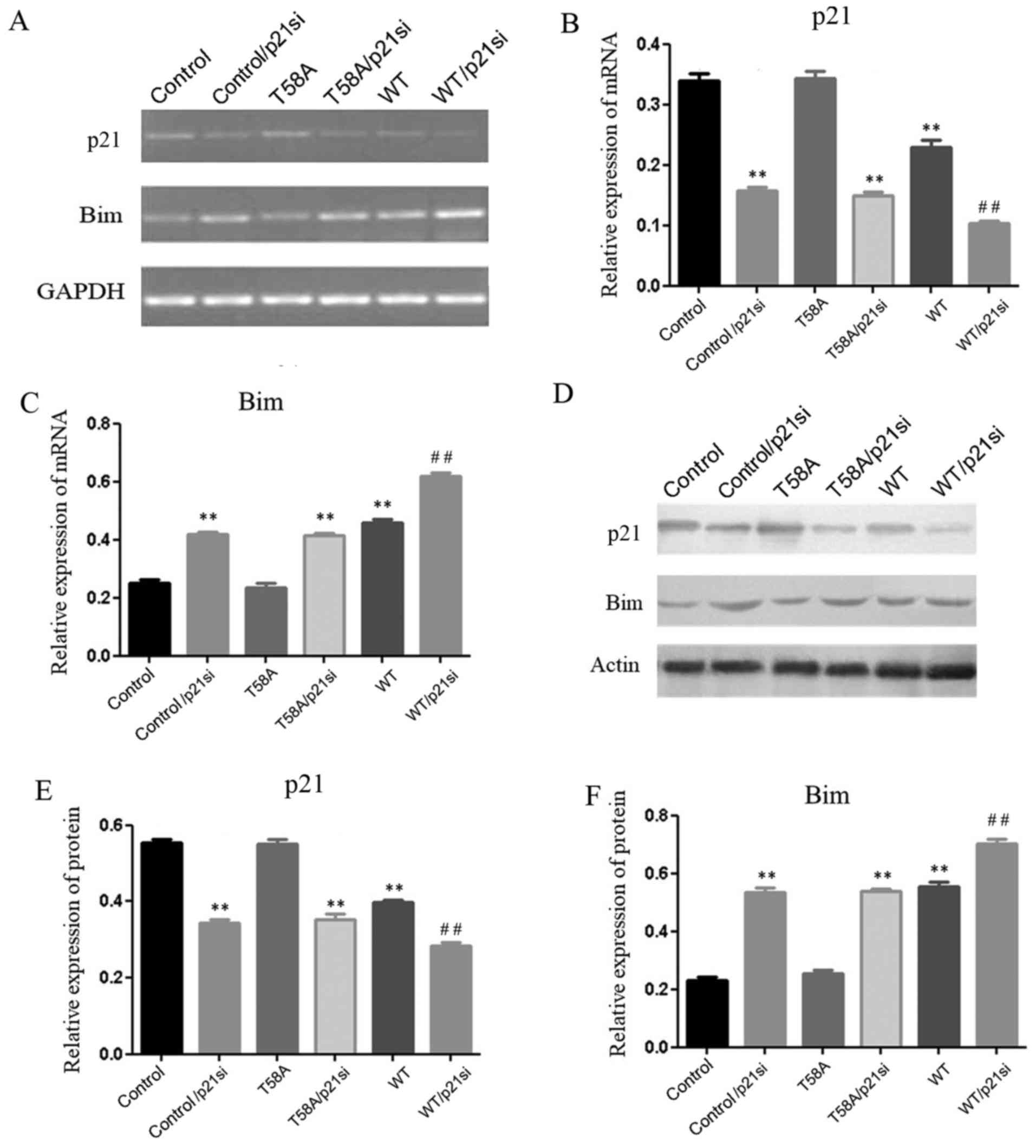
Mutant MYC is unable to suppress p21
or to induce Bim
The present study aimed to assess why mutant MYC is
unable to upregulate the expression of Bim (Fig. 3). Earlier studies supported the view
that MYC represses the expression of p21 (31), while p21 represses the expression of
Bim (23), thereby maintaining the
balance between cellular proliferation and apoptosis. In the
present study, in HCC1937 cells with p21 knockdown (P<0.01;
Fig. 3B and E), the expression of Bim
was increased (P<0.01; Fig. 3C and
F). It was also demonstrated that p21 expression was
significantly diminished (P<0.01; Fig.
3B and E), and Bim expression was significantly increased
(P<0.01; Fig. 3C and F), in WT
MYC-transfected cells compared with controls. In addition, when p21
was knocked down (P<0.01; Fig. 3B and
E), p21 expression was further diminished and Bim expression
was further increased in WT MYC/p21-siRNA transfected cells
compared with p21-siRNA transfected cells (P<0.01; Fig. 3). These results suggested that p21 and
Bim may be involved in a negative regulatory mechanism with
MYC.
HCC1937 cells were transfected with mutant MYC and
the expression levels of Bim and p21 were determined, using
RT-sqPCR and western blot analyses (Fig.
3). Bim and p21 are associated with apoptosis and
proliferation. The p21 and Bim expression levels were similar in
mutant MYC and control cells (P=0.85, 0.82, 0.51 and 0.27,
respectively), indicating that mutant MYC was unable to suppress
p21 or induce Bim. To confirm this regulatory mechanism, HCC1937
cells were transfected with T58A and the p21 siRNA construct
(T58A/p21si). The expression level of p21 was demonstrated to be
decreased (P<0.01; Fig. 3B and
E), and the expression level of Bim was significantly increased
(P<0.01; Fig. 3C and F).
This further demonstrated that mutant MYC was unable to suppress
p21 or induce Bim. T58A MYC-transfected cells displayed marked
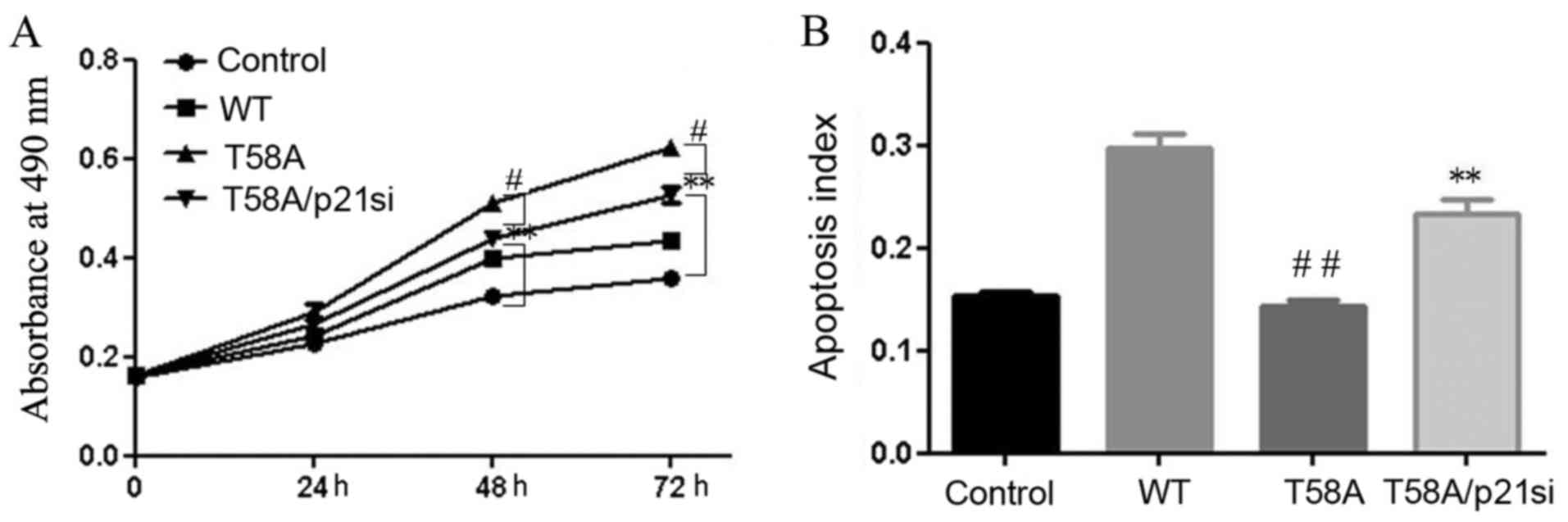
proliferation, yet no induction of apoptosis (P<0.01; Fig. 1). In addition, these effects of mutant
MYC on proliferation and apoptosis were weakened when cells were
transfected with T58A and p21 siRNA together (P<0.05; Fig. 4). These results indicated that the
breast cancer-derived MYC mutation T58A does not suppress p21 or
induce Bim, which may be the reason why it does not induce
apoptosis.
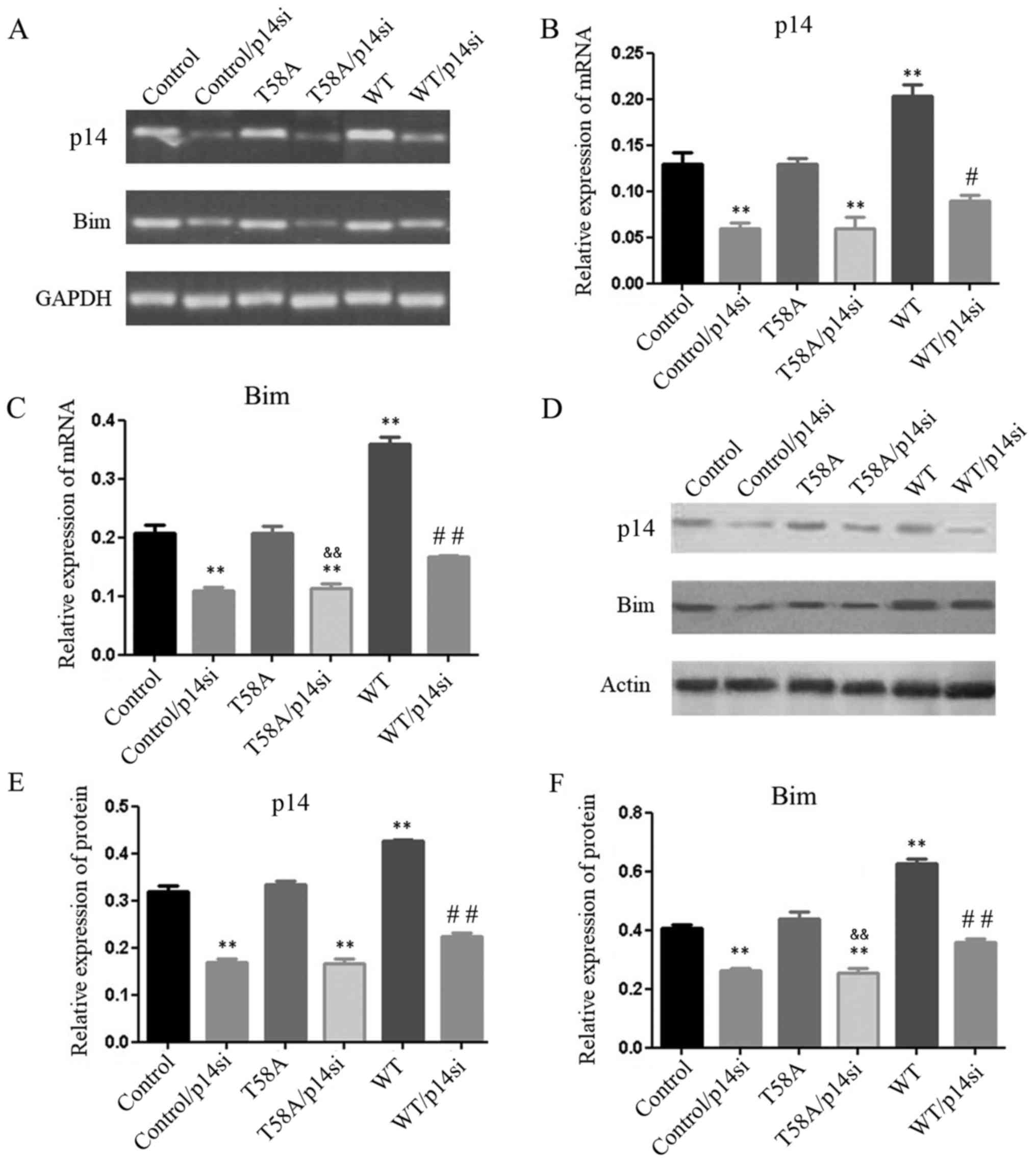
Mutant MYC is unable to induce p14 or
Bim
The tumor suppressor p14 is able to inhibit
MYC-induced hyperproliferation and induce p53-independent apoptosis
(18). In the present study it was
reported that in HCC1937 cells transfected with p14 siRNA
(P<0.05; Fig. 5), Bim expression
was decreased (P<0.05; Fig. 5C and
F). WT cells displayed significantly increased expression
levels of p14 and Bim (P<0.05; Fig.
5); however, when p14 was knocked down, p14 and Bim expression
levels were further diminished in WT MYC/p14-siRNA transfected
cells compared with p14-siRNA transfected cells (P<0.05,
P<0.01; Fig. 5). Thus, p14 and Bim
appear to form a positive regulatory mechanism with MYC.
To examine how mutant MYC regulated p14 and Bim, the
expression levels of Bim and p14 were analyzed, using RT-sqPCR and
western blot analyses in mutant MYC cells (Fig. 5). The expression levels of p14 and Bim
were similar in mutant MYC and control cells (P=1.0, 0.41, 0.95 and
0.25, respectively). HCC1937 cells were transfected with T58A and
the p14 siRNA construct (T58A/p14si). When p14 was blocked
(P<0.05; Fig. 5B and E),
T58A/p14si cells displayed significantly lower expression levels of
Bim (P<0.01; Fig. 5C and F)
compared with cells transfected with T58A alone. T58A
MYC-transfected cells demonstrated notable proliferation and
decreased apoptosis (P<0.01; Fig.
1). In addition, these effects of mutant MYC on proliferation
and apoptosis were strengthened when cells were transfected with
T58A and the p14 siRNA together (P<0.05; Fig. 6). These results suggested that mutant
MYC does not induce p14 or Bim, in contrast with previous reports.
Hemann et al (9) reported that
mutant MYC induced p14 and not Bim. This finding may have been due
to the expression of p53. The results of the present study
demonstrated that the breast cancer-derived MYC mutation T58A does
not induce p14 or Bim, which may be another reason why it does not
induce apoptosis.
Discussion
The results of the present study revealed novel
insights into the biology of the MYC oncoprotein in breast cancer
cells. In breast cancer with loss of p53, WT MYC and T58A mutant
MYC are able to promote cell proliferation. In addition, WT MYC may
also induce cell apoptosis, whereas T58A mutant MYC does not induce
cell apoptosis. These results demonstrated that MYC, and not the
MYC mutant, may be able to induce apoptosis in a p53-independent
manner.
Hemann et al (9) previously reported that mutant MYC was
unable to induce Bim expression, yet still activated the p53
pathway to a similar extent compared with WT MYC in Burkitt
lymphoma. This may explain why tumors with mutant MYC are less
prone to apoptosis (9). The results
of the present study indicated that the tumor-derived MYC mutant
T58A is relevant in breast cancer pathology, as it is unable to
upregulate Bim or to induce apoptosis to protect against
unrestrained proliferation in breast cancer.
Until now, MYC was thought to regulate Bim through
the MYC-p14-Bim and MYC-p21-Bim networks, which interact with each
other via mechanisms that are poorly understood. However, the
reason why the MYC mutant is unable to promote apoptosis is
unclear. Earlier studies supported the view that MYC represses the
expression of p21, and that p21 represses the expression of Bim, to
regulate cellular proliferation and the tumor surveillance
response. In the present study, when HCC1937 cells were transfected
with WT MYC, the expression level of p21 was significantly
diminished and the expression level of Bim was significantly
increased. Thus, p21 and Bim seem to form a negative regulatory
pathway with MYC. Furthermore, mutant MYC did not suppress p21,
induce Bim, or induce apoptosis to prevent unrestrained
proliferation. When p21 was blocked, the expression of Bim was
significantly increased and the effects of mutant MYC on
proliferation and apoptosis were weakened. From these results, it
was concluded that breast cancer-derived T58A MYC does not induce
apoptosis, which is associated with its failure to suppress p21 and
activate Bim.
p14 is a tumor suppressor protein, which is
regulated by the MYC protein and is able to induce apoptosis
(16). Further analysis has revealed
that p14 may be combined with E2F to serve a role in tumor
detection (19). The E2F family is a
class of transcription factors that accumulate in Bim enhancers to
regulate apoptosis induction. It seems that normal MYC increases
p14, which interacts with Bim to induce apoptosis. The present
study reported that the expression levels of p14 and Bim were
significantly increased in WT MYC-transfected HCC1937 cells. These
results confirm that p14 and Bim may form a positive regulatory
association with MYC. It was also reported that mutant MYC was
unable to upregulate p14 or Bim. When p14 was blocked, the
expression level of Bim was significantly decreased due to
transfected T58A. In addition, these effects of mutant MYC on
proliferation and apoptosis were strengthened. Therefore, it was
concluded that the breast cancer-derived MYC mutation T58A does not
induce apoptosis, which is associated with its failure to activate
p14 or Bim.
MYC-initiated alterations in gene expression have a
variety of effects resulting in the formation of breast cancer. A
previous study reported that the T58A mutation in MYC may enhance
the formation of tumors (32). In
recent work, the T58A mutations in MYC reduced the dependence on
KRAS proto-oncogene mutations for tumorigenesis (33). In the present study, the T58A mutation
was MYC is not able to regulate p14/p21 in MYC-induced apoptosis in
p53-deficient breast cancer. However, the role of the T58A mutation
in MYC-induced apoptosis is unclear. Further studies on the
mechanism of the T58A MYC mutation in the development of breast
cancer are required.
Substantial evidence has established that p53 is a
key tumor suppressor, apoptosis-inducer and prognostic marker in
cancer (34–36). The p53 status of a tumor may have a
strong influence on tumor sensitivity to commonly used anticancer
drugs and radiotherapy, given that the majority of anticancer
treatments trigger DNA damage-induced apoptosis. The mechanism by
which MYC activates Bim may provide important insights into novel
tumor-specific treatment strategies, as activated Bim induces a
p53-independent form of apoptosis that is induced by oncogenes
rather than by DNA damage. Considering that p53 expression is lost
in one-half of all human tumors (34), the unique MYC-p14-Bim and MYC-p21-Bim
apoptosis pathways may have potential in the development of future
therapeutic agents as an alternative to p53 reactivation.
Acknowledgements
The authors would like to thank Dr Xuefeng Zhang,
Medical School of Qingdao University, Qingdao, China, for providing
statistical advice, Professor Xiangping Liu, The Affiliated
Hospital of Qingdao University (Qingdao, China), for technical
support, and colleagues in the laboratory for helpful
discussions.
Funding
The present study was funded by a grant from the
Natural Science Foundation of Shandong Province (grant nos.
y2007C134 and ZR2017PH032), the Higher Educational Science and
Technology Program of Shandong Province (grant no. J17B092) and the
National Natural Science Foundation of China (grant no. 81302290
and 81700029).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DJJ designed and performed experiments, and wrote
the manuscript. YS and FL designed experiments and revised the
manuscript. DJ helped to culture cells. WC and XW made substantial
contributions to the conception and design of the study, and
revised the manuscript. ZL and ZY helped to analyze the
experimental data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fallah Y, Brundage J, Allegakoen P and
Shajahan-Haq AN: MYC-Driven pathways in breast cancer subtypes.
Biomolecules. 7:E532017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eilers M and Eisenman RN: MYC's broad
reach. Genes Dev. 22:2755–2766. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y and Olopade OI: MYC in breast tumor
progression. Expert Rev Anticancer Ther. 8:1689–1698. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hynes NE and Stoelzle T: Key signalling
nodes in mammary gland development and cancer: MYC. Breast Cancer
Res. 11:2102009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Efstratiadis A, Szabolcs M and Klinakis A:
Notch, MYC and breast cancer. Cell Cycle. 6:418–429. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang DW, Claassen GF, Hann SR and Cole
MD: The MYC transactivation domain is a direct modulator of
apoptotic versus proliferative signals. Mol Cell Biol.
20:4309–4319. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Conzen SD, Gottlob K, Kandel ES, Khanduri
P, Wagner AJ, O'Leary M and Hay N: Induction of cell cycle
progression and acceleration of apoptosis are two separable
functions of MYC: Transrepression correlates with acceleration of
apoptosis. Mol Cell Biol. 20:6008–6018. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hemann MT, Bric A, Teruya-Feldstein J,
Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP and
Lowe SW: Evasion of the p53 tumour surveillance network by
tumour-derived MYC mutants. Nature. 436:807–811. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thibodeaux CA, Liu X, Disbrow GL, Zhang Y,
Rone JD, Haddad BR and Schlegel R: Immortalization and
transformation of human mammary epithelial cells by a tumor-derived
MYC mutant. Breast Cancer Res Treat. 116:281–294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delbridge AR, Pang SH, Vandenberg CJ,
Grabow S, Aubrey BJ, Tai L, Herold MJ and Strasser A: RAG-induced
DNA lesions activate proapoptotic BIM to suppress lymphomagenesis
in p53-deficient mice. J Exp Med. 213:2039–2048. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kovi RC, Paliwal S, Pande S and Grossman
SR: An ARF/CtBP2 complex regulates BH3-only gene expression and
p53-independent apoptosis. Cell Death Differ. 17:513–521. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Muthalagu N, Junttila MR, Wiese KE, Wolf
E, Morton J, Bauer B, Evan GI, Eilers M and Murphy DJ: BIM is the
primary mediator of MYC-induced apoptosis in multiple solid
tissues. Cell Rep. 8:1347–1353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Campone M, Noël B, Couriaud C, Grau M,
Guillemin Y, Gautier F, Gouraud W, Charbonnel C, Campion L,
Jézéquel P, et al: MYC dependent expression of pro-apoptotic Bim
renders HER2-overexpressing breast cancer cells dependent on
antiapoptotic Mcl-1. Mol Cancer. 10:1102011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Li F, Meng C, Jiang D and Liu S:
Effects of mutant and wild-type c-myc on Bim expression in
non-p53-dependent pathway. Med J Qilu. 27:395–397. 2012.
|
|
16
|
Datta A, Nag A, Pan W, Hay N, Gartel AL,
Colamonici O, Mori Y and Raychaudhuri P: MYC-ARF (alternate reading
frame) interaction inhibits the functions of MYC. J Biol Chem.
279:36698–36707. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qi Y, Gregory MA, Li Z, Brousal JP, West K
and Hann SR: p19ARF directly and differentially controls the
functions of MYC independently of p53. Nature. 431:712–717. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gregory MA, Qi Y and Hann SR: The ARF
tumor suppressor: Keeping MYC on a leash. Cell Cycle. 4:249–252.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aslanian A, Iaquinta PJ, Verona R and Lees
JA: Repression of the Arf tumor suppressor by E2F3 is required for
normal cell cycle kinetics. Genes Dev. 18:1413–1422. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gogada R, Yadav N, Liu J, Tang S, Zhang D,
Schneider A, Seshadri A, Sun L, Aldaz CM, Tang DG and Chandra D:
Bim, a proapoptotic protein, up-regulated via transcription factor
E2F1-dependent mechanism, functions as a prosurvival molecule in
cancer. J Biol Chem. 288:368–381. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abbas T and Dutta A: p21 in cancer:
Intricate networks and multiple activities. Nat Rev Cancer.
9:400–414. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dotto GP: p21WAF1/Cip1: More than a break
to the cell cycle? Biochim Biophys Acta. 1471:M43–M56.
2000.PubMed/NCBI
|
|
23
|
Collins NL, Reginato MJ, Paulus JK, Sgroi
DC, Labaer J and Brugge JS: G1/S Cell Cycle arrest provides anoikis
resistance through erk-mediated bim suppression. Mol Cell Biol.
25:5282–5291. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sherr CJ: The INK4a/ARF network in tumour
suppression. Nat Rev Mol Cell Biol. 2:731–737. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eischen CM, Roussel MF, Korsmeyer SJ and
Cleveland JL: Bax loss impairs MYCinduced apoptosis and circumvents
the selection of p53 mutations during MYCmediated lymphomagenesis.
Mol Cell Biol. 21:7653–7662. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Soussi T and Wiman KG: Shaping genetic
alterations in human cancer: The p53 mutation paradigm. Cancer
Cell. 12:303–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Valente LJ, Gray DH, Michalak EM,
Pinon-Hofbauer J, Egle A, Scott CL, Janic A and Strasser A: p53
efficiently suppresses tumor development in the complete absence of
its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and
Noxa. Cell Rep. 3:1339–1345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Strasser A, Harris AW, Jacks T and Cory S:
DNA damage can induce apoptosis in proliferating lymphoid cells via
p53-independent mechanisms inhibitable by Bcl-2. Cell. 79:329–339.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang DC and Strasser A: BH3-Only
proteins-essential initiators of apoptotic cell death. Cell.
103:839–842. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meng CH, Li FN and Zhang DL: Construction
and significance of lentiviral vector with c-myc. Chin J Exp Surg.
4:543–546. 2011.
|
|
31
|
Wanzel M, Herold S and Eilers M:
Transcriptional repression by Myc. Trends Cell Biol. 13:146–150.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Cunningham M, Zhang X, Tokarz S,
Laraway B, Troxell M and Sears RC: Phosphorylation regulates
c-Myc's oncogenic activity in the mammary gland. Cancer Res.
71:925–936. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hollern DP, Yuwanita I and Andrechek ER: A
mouse model with T58A mutations in Myc reduces the dependence on
KRasmutations and has similarities to claudin-low human breast
cancer. Oncogene. 32:1296–1304. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Farnebo M, Bykov VJ and Wiman KG: The p53
tumor suppressor: A master regulator of diverse cellular processes
and therapeutic target in cancer. Biochem Biophys Res Commun.
396:85–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mihara M, Erster S, Zaika A, Petrenko O,
Chittenden T, Pancoska P and Moll UM: p53 has a direct apoptogenic
role at the mitochondria. Mol Cell. 11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee JY, Cho KS, Diaz RR, Choi YD and Choi
HY: p53 expression as a prognostic factor in upper urinary tract
urothelial carcinoma: A systematic review and meta-analysis. Urol
Int. 94:50–57. 2015. View Article : Google Scholar : PubMed/NCBI
|