Introduction
Rectal cancer is one of the most commonly diagnosed
malignancies and is one of the leading causes of cancer-associated
cases of mortality (1). For patients
with locally advanced rectal cancer, neoadjuvant chemoradiotherapy
(CRT) followed by radical surgery has been the standard treatment
for many years (2). However, the
response to CRT varies substantially among patients (3). A number of studies have revealed that
pathological complete response is associated with improved clinical
outcome (4,5). Biomarkers for early response prediction
prior to treatment could be helpful for identifying rectal cancer
patients who would not benefit from CRT. This may save potentially
nonresponsive patients from unnecessary treatment and possible side
effects, and allow alternative treatments, including surgery and
adjuvant therapy, to be administered without delay.
A number of studies have revealed a wide variety of
genetic and molecular biomarkers that have the potential to predict
the response to CRT, including epidermal growth factor receptor,
thymidylate synthase and Bax (5–7). However,
no previously identified biomarker has changed the approach to CRT
treatment or been used to perform clinical trials, due to
inadequate sensitivity and specificity. Improved predictive methods
are urgently required to discriminate patients who would or would
not clinically benefit from neoadjuvant CRT.
Recent technological advances in the genomics of
gene expression by DNA microarray have facilitated the simultaneous
analysis of a large numbers of genes; this supports the
identification of molecular markers with the potential to predict
response to CRT at an early time point (8). With the progress of technology, issues
have emerged due to differentially expressed (DE) gene lists for
the same tumor, identified from different microarray studies,
demonstrating very low reproducibility, which places a serious
impact on further research (9).
Considering the heterogeneity and complexity of carcinogenesis,
microarray-based biomarker discovery should be established by
network paradigms regarding gene interaction, which may eliminate
data inconsistencies.
The current study applied an integrative approach to
predict novel mRNAs that may be applied to predict chemosensitivity
and identify patients with rectal cancer as responders or
non-responders to neoadjuvant CRT. Integration of mRNA expression
data, mRNA-mRNA interaction data and other types of genomic
information may facilitate the identification of mRNA biomarker
panels for precision medicine in rectal cancer.
Materials and methods
Data collection
The gene expression dataset GSE35452 (10) was downloaded from the Gene Expression
Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). This dataset
contained 46 patients with rectal cancer who had been approved to
receive preoperative CRT. Biopsy specimens of rectal tumor were
collected during colonoscopic examination prior to preoperative
CRT, snap-frozen in liquid nitrogen and stored at −80°C until use.
Total RNA was isolated from frozen samples and gene expression
profiles were determined using Affymetrix Human Genome U133 Plus
2.0 arrays (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
study protocol was approved by the Ethics Committee of Teikyo
University (Tokyo, Japan). The current study was also approved by
the Ethics Committee of the Second Affiliated Hospital of Soochow
University (Suzhou, China).
Response to CRT
The semi-quantitative classification system defined
by the Japanese Society for Cancer of the Colon and Rectum was
applied to evaluate the response to CRT by histopathological
examination of surgically resected specimens (11). Based on the system, no tumor cell
necrosis or degeneration was defined as grade 0, tumor cell
necrosis or degeneration in less than two-thirds of the entire
lesion was defined as grade 1, prominent tumor cell necrosis or
degeneration in more than two-thirds of the entire lesion with
viable tumor cells remaining was defined as grade 2, and the
presence of no viable tumor cells was defined as grade 3. Tumors
were classified as ‘responders’ (histopathologic regression grade 2
or 3) and ‘nonresponders’ (histopathologic regression grade 0 or
1).
Gene expression profile analysis
The normalized data file of GSE35452 was downloaded
directly from the GEO database for further analysis. The DE genes
between the responders and non-responders to neoadjuvant CRT in
rectal cancer were identified by Student's t-test using the limma
package of R software (version 3.4.2, http://www.r-project.org/) (12). P<0.05 was considered to indicate a
statistically significant difference. Following identification of
the DE genes, bidirectional hierarchical clustering was conducted
with the expression values of the DE genes using the gplots package
of R software.
Functional enrichment analysis
Gene Ontology (GO) is a common method used for
annotating large numbers of genes at the functional level (13). Kyoto Encyclopedia of Genes and Genomes
(KEGG), a database containing known genes and their respective
biochemical functionalities, can be used for systematic analysis of
gene functions (14). The current
study conducted KEGG pathway and GO enrichment analyses of the
identified DE genes using the online version of Database for
Annotation, Visualization and Integrated Discovery (DAVID)
(https://david.ncifcrf.gov/). P<0.05
was considered to indicate a statistically significant enriched
term. In the present study, the functional enriched results were
screened for their relevance with CRT response by searching the
PubMed database (https://www.ncbi.nlm.nih.gov/pubmed).
Protein-protein interaction (PPI)
network construction and analysis
A PPI network of the DE genes was constructed using
the Search Tool for the Retrieval of Interacting Genes (STRING;
string-db.org) database, which is a unified
database of PPIs of 9,643,763 proteins from 2,031 organisms
(15). The PPI network was visualized
using Cytoscape 3.4.0 (http://www.cytoscape.org/). To evaluate the importance
of nodes in the PPI network, three common indices, including
betweenness centrality, closeness centrality and degree centrality,
were investigated. The Cytoscape plug-in, CytoNCA, was used to
calculate the three aforementioned indices (16). The Cytoscape plug-in, Molecular
Complex Detection (MCODE), was applied to identify the significant
modules of the PPI network (17).
Subsequently, GO analysis and KEGG pathway analysis was performed
using DAVID for the hub nodes and DE associated with the selected
modules (18). P<0.05 was
considered to indicate a statistically significant difference.
Results
Detection of DE genes between CRT
responders and non-responders
Based on the aforementioned cut-off criteria
(P<0.05), a total of 445 upregulated DE genes and 465
downregulated DE genes were identified in tumor samples of CRT
responders and non-responders. Subsequently, hierarchical
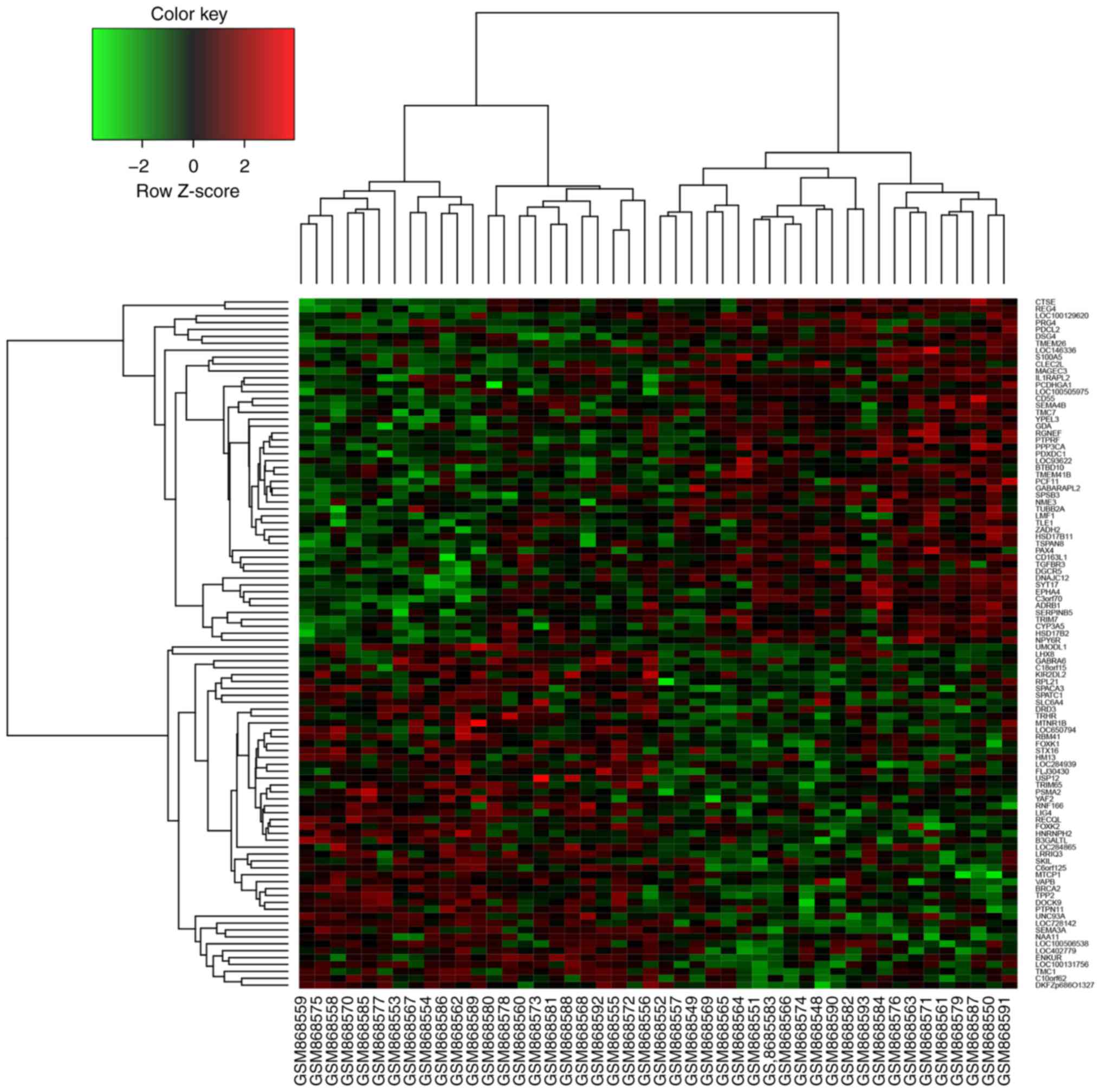
clustering analysis was performed with the 50 most upregulated and
50 most downregulated DE genes. As illustrated in Fig. 1, the DE genes could be used to clearly
distinguish responders from non-responders to preoperative CRT.
Functional annotation of the DE
genes
To analyze the altered biological function of the DE
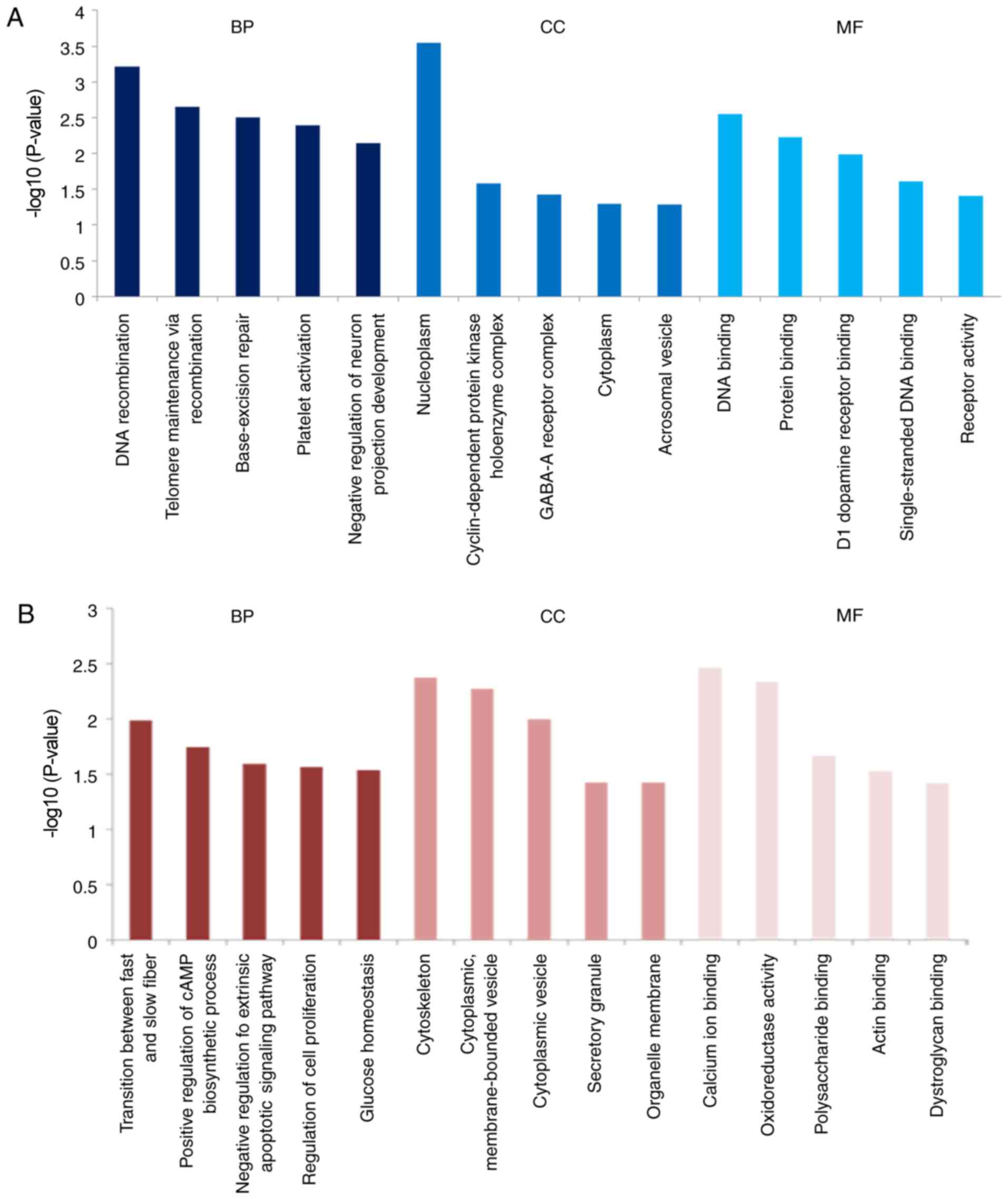
genes, GO enrichment analysis was performed using the DAVID online
tool. The enriched GO terms were divided into biological process
(BP), molecular function (MF) and cell component (CC) ontologies;
the five most significantly enriched terms for each ontology are
illustrated in Fig. 2. GO analysis
demonstrated that upregulated DE genes in CRT responders were
significantly enriched in several BP ontologies, including DNA
recombination, telomere maintenance via recombination and
base-excision repair. The downregulated DE genes in CRT responders
were largely involved in regulatory processes, including regulation
of the cAMP biosynthetic process, regulation of the extrinsic
apoptotic signaling pathway and regulation of cell proliferation.
For MF ontology, the upregulated DE genes in CRT responders were
mainly involved in DNA binding and protein binding, and the
downregulated DE genes were associated with calcium ion binding and
polysaccharide binding. In addition, GO terms in the CC ontology
revealed that the upregulated DE genes in CRT responders were
significantly enriched in the nucleoplasm, cytoplasm and nucleus,
and downregulated DE genes in CRT responders were significantly
enriched in the cytoskeleton, cytoplasmic vesicle and secretory
granule.
Pathway enrichment of the DE
genes
To further investigate the function of the DE genes,
KEGG pathway enrichment analysis was performed using DAVID. The
upregulated DE genes in CRT responders were enriched in excision
repair and homologous recombination pathways, while downregulated
DE genes in CRT responders were enriched in the Ras signaling
pathway, the forkhead box O (FoxO) signaling pathway, focal
adhesion and the Wnt signaling pathway (Table I).
 | Table I.Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis of differentially expressed
genes in chemoradiotherapy responders and non-responders. |
Table I.
Kyoto Encyclopedia of Genes and
Genomes pathway enrichment analysis of differentially expressed
genes in chemoradiotherapy responders and non-responders.
| A, Upregulated
genes |
|---|
|
|---|
| Pathway | Genes | P-value |
|---|
| Neuroactive
ligand-receptor interaction | 12 |
7.48×10−3 |
| Base excision
repair | 4 |
1.82×10−3 |
| Morphine
addiction | 6 |
1.94×10−2 |
| GABAergic
synapse | 5 |
5.70×10−2 |
| mRNA surveillance
pathway | 5 |
6.96×10−2 |
| Measles | 6 |
7.68×10−2 |
| Homologous
recombination | 3 |
8.71×10−2 |
|
| B, Downregulated
genes |
|
| Pathway | Genes | P-value |
|
| Linoleic acid
metabolism | 7 |
4.39×10−5 |
| Ras signaling
pathway | 12 |
3.23×10−3 |
| Steroid hormone
biosynthesis | 6 |
3.42×10−3 |
| Oxytocin signaling
pathway | 8 |
1.98×10−3 |
| Renin
secretion | 5 |
3.03×10−2 |
| FoxO signaling
pathway | 7 |
3.11×10−2 |
| Arrhythmogenic
right ventricular cardiomyopathy | 5 |
3.49×10−2 |
| Hypertrophic
cardiomyopathy | 5 |
5.51×10−2 |
| Vascular smooth
muscle contraction | 6 |
6.42×10−2 |
| Dilated
cardiomyopathy | 5 |
6.83×10−2 |
| Regulation of
lipolysis in adipocytes | 4 |
7.55×10−2 |
| Maturity onset
diabetes of the young | 3 |
7.84×10−2 |
| Fatty acid
elongation | 3 |
7.84×10−2 |
| Focal adhesion | 8 |
7.98×10−2 |
| Alpha-Linolenic
acid metabolism | 3 |
8.34×10−2 |
| Butanoate
metabolism | 3 |
8.92×10−2 |
| Osteoclast
differentiation | 6 |
9.26×10−2 |
| Glycerophospholipid
metabolism | 5 |
9.62×10−2 |
| Wnt signaling
pathway | 6 |
9.94×10−2 |
PPI network construction and
identification of key nodes
A PPI network of the DE genes was constructed using
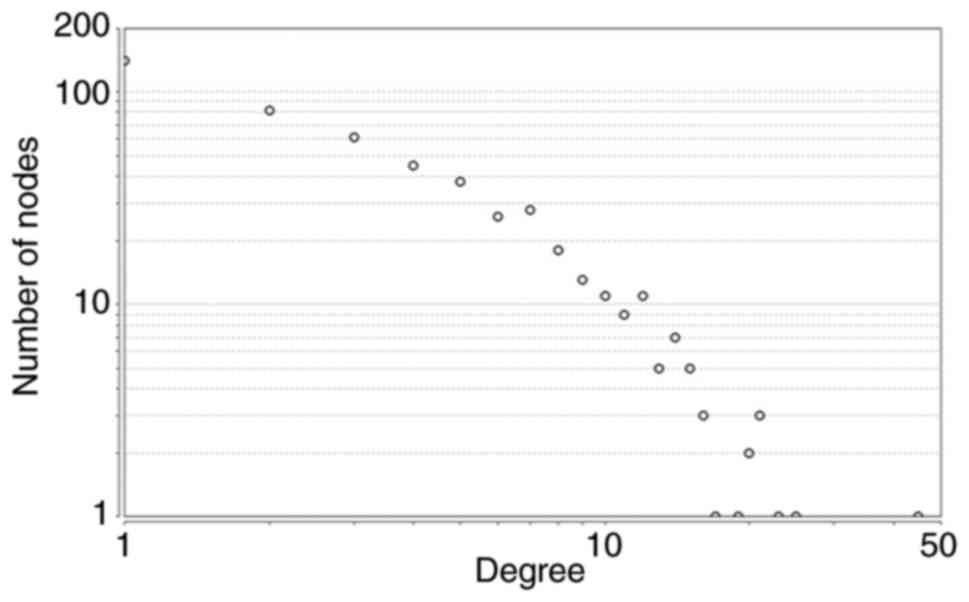
the STRING database. The PPI network contained 513 nodes with 910
DE genes (P<0.001). The degree distributions of the network
nodes are illustrated in Fig. 3.
Degree centrality, betweenness centrality and closeness centrality
were evaluated in the constructed network to evaluate the
relationship between nodes and identify the hub genes within the
network. The 10 genes with the highest degree centrality,
betweenness centrality and closeness centrality are listed in
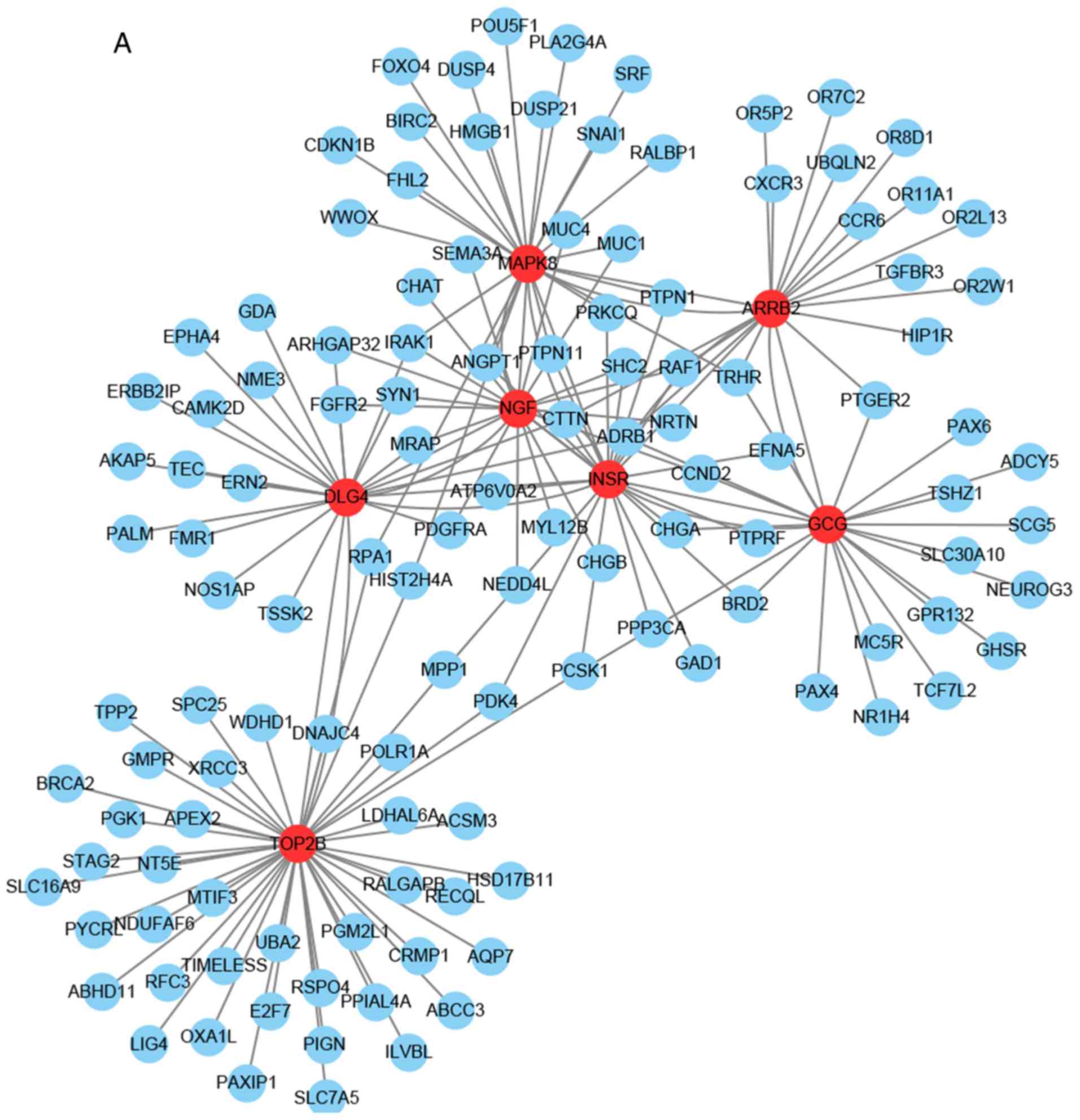
Tables II–IV. Seven genes (TOP2B, MAPK8, DLG4, GCG,
NGF, INSR and ARRB2) were identified as hub genes as they were
identified by all three of the aforementioned methods. The
sub-network was reconstructed with the selected hub nodes and their
first neighbor genes, presented in Fig.
4A. The biological function of the selected hub genes were
assessed and it was revealed that these genes serve a role in the
Ras signaling pathway (MAPK8, INSR and NGF), the mitogen-activated
protein kinase (MAPK) signaling pathway (ARRB2, MAPK8 and NGF) and
the FoxO signaling pathway (MAPK8 and INSR) (Fig. 4B). Furthermore, the hub genes were
identified to be involved in regulatory processes (Fig. 4C), including positive regulation of
protein kinase B signaling (ARRB2 and INSR), positive regulation of
the extracellular signal-regulated kinase (ERK) 1 and ERK2 cascades
(GCG and ARRB2), positive regulation of gene expression (MAPK8 and
NGF) and negative regulation of apoptotic processes (GCG and
MAPK8).
| Table II.Top 10 genes identified by
betweenness centrality in the protein-protein interaction
network. |
Table II.
Top 10 genes identified by
betweenness centrality in the protein-protein interaction
network.
| Gene name | Betweenness
centrality |
|---|
| TOP2B | 50299.086 |
| MAPK8 | 27945.004 |
| DLG4 | 19934.455 |
| PPP3CA | 16902.625 |
| INSR | 15489.692 |
| GCG | 14696.811 |
| ARRB2 | 14580.526 |
| NGF | 12627.297 |
| POLR1A | 12130.832 |
| UBA2 | 11705.651 |
| Table IV.Top 10 genes identified by degree
centrality in the protein-protein interaction network. |
Table IV.
Top 10 genes identified by degree
centrality in the protein-protein interaction network.
| Gene name | Degree
centrality |
|---|
| TOP2B | 45 |
| MAPK8 | 25 |
| DLG4 | 23 |
| ADCY5 | 21 |
| GCG | 21 |
| NGF | 21 |
| PPP3CA | 20 |
| INSR | 20 |
| ARRB2 | 19 |
| NEDD4L | 17 |
Module analysis of PPI network
The most significant modules in the PPI network were
identified using MCODE in Cytoscape software and presented in
Fig. 4D. According to KEGG pathway
enrichment analysis, the DE genes in the significant modules were
predominantly involved in several CRT response associated pathways
including the chemokine signaling pathway (CCR6, ADCY5, CXCR3,
CXCL11 and CCL27), cytokine-cytokine receptor interaction (CCR6,
CXCR3, CXCL11 and CCL27) and the cAMP signaling pathway (HTR1B,
ADCY5 and GABBR1) (Fig. 4E).
Identification of CRT
response-associated functions and pathways
Based on the aforementioned results, the enriched
functional categories and pathways were screened for their
associations with CRT response by manually searching citations in
PubMed (https://www.ncbi.nlm.nih.gov/pubmed; Table V).
| Table V.Pathways associated with the
chemoradiotherapy response. |
Table V.
Pathways associated with the
chemoradiotherapy response.
| Pathway | Enriched genes | Representative
PubMed references |
|---|
| Base excision
repair | Upregulated DE
genes | 21420246,
20832016 |
| Homologous
recombination | Upregulated DE
genes | 23746696,
25017126 |
| Ras signaling
pathway | Downregulated DE
genes and hub genes | 20619549,
15549191 |
| FoxO signaling
pathway | Downregulated DE
genes | 17183370,
20734804 |
| Focal adhesion | Downregulated DE
genes and hub genes | 14703945,
24930861 |
| Wnt signaling
pathway | Downregulated DE
genes | 27363012,
26072169 |
| MAPK signaling
pathway | Hub genes | 12947395,
26926139 |
| Chemokine signaling
pathway | Module genes | 21292447,
28957436 |
| Cytokine-cytokine
receptor interaction | Module genes | 28763256,
27745982 |
| cAMP signaling
pathway | Module genes | 24568192,
26989332 |
For the upregulated DE genes, the most highly
enriched GO terms at the BP level were associated with DNA
recombination, telomere maintenance via recombination and
base-excision repair. For downregulated DE genes, the more highly
enriched GO terms at the BP level were related to regulatory
processes, including cAMP biosynthetic process, extrinsic apoptotic
signaling pathway and cell proliferation. Notably, these enriched
terms have been demonstrated to be closely related to radiation
response (19,20). The enriched GO terms in MF mainly
included the function of binding, including DNA binding, protein
binding (upregulated DE genes), calcium ion binding and
polysaccharide binding (downregulated DE genes). The binding of
important molecules, including proteins and DNA may highly
influence the ionizing radiation (21). Most GO CC items converged on the
hallmarks of a cell including the nucleoplasm, cytoplasm and
nucleus for the upregulated DE genes and the cytoskeleton,
cytoplasmic vesicle and secretory granule for downregulated DE
genes. These basic structural hallmarks of a cell have a major
influence on radiation sensitivity through affecting the integrity
of cell structure and exerting impact on cell growth,
differentiation, proliferation, apoptosis and metastasis (22).
With regard to the KEGG pathway enrichment results,
the upregulated DE genes in CRT responders were mainly involved in
pathways including base excision repair and homologous
recombination, while downregulated DE genes in CRT responders were
enriched in pathways including the Ras signaling pathway, FoxO
signaling pathway, focal adhesion and Wnt signaling pathway. These
pathways were also indicated to be highly associated with radiation
response (23–26). The seven hub nodes (TOP2B, MAPK8,
DLG4, GCG, NGF, INSR and ARRB2) were associated with the Ras
signaling pathway, MAPK signaling pathway and FoxO signaling
pathway. Furthermore, the hub genes were identified to participate
in specific processes, including regulation of protein kinase B
signaling, ERK1 and ERK2 cascade, gene expression and apoptosis
through the BP ontology. These enriched pathways and ontology terms
were confirmed to be responsible for the radiation response
according to the literature search (27,28).
The DE genes involved in the selected module were
mainly associated with the chemokine signaling pathway,
cytokine-cytokine receptor interaction and cAMP signaling pathway.
These pathways also play important roles in radiation response
(19,29,30).
Discussion
Increasing importance is being placed on improving
personalized precision treatment methods for cancer. Developing
methods for response prediction may help to distinguish responders
from non-responders to preoperative CRT. In the current study, a
bioinformatics approach was conducted to predict the potential
crucial genes and pathways related to CRT response. A total of 445
upregulated DE genes and 465 downregulated DE genes were identified
between CRT responders and non-responders. Subsequently, the
functions of the DE genes were investigated using GO annotation and
pathway enrichment analyses. Finally, the interactions between DE
genes were explored by creating a PPI network. As a result,
specific key genes and pathways were identified which may play
vital roles in CRT response and could become potential biomarkers
for the optimization of CRT.
At the BP level, many enriched GO terms were
significantly associated with DNA recombination, telomere
maintenance via recombination, base-excision repair, cAMP
biosynthetic process, the extrinsic apoptotic signaling pathway and
cell proliferation. DNA recombination is known to play a central
role in the DNA damage and repair response, which is a vital
determinant of both tumor development and tumor outcome following
radiation therapy (31). Telomere
maintenance via recombination is closely associated with telomerase
activity, which is associated with radiosensitivity (32). The use of telomerase inhibitors during
CRT may sensitize cancer cells to chemotherapy and radiotherapy
(33). Furthermore, the cAMP
biosynthetic process, the extrinsic apoptotic signaling pathway and
cell proliferation also contribute to the radiotherapy response
(19). The intrinsic apoptotic
pathway is sufficient to impart radiation resistance (34), while inhibition of cell proliferation
may enhance the radiosensitivity of cancer cells (35).
At the MF level, the enriched GO terms were mainly
associated with binding functions, including DNA binding, protein
binding, calcium ion binding and polysaccharide binding. These
functions influence ionizing radiation through the binding of
important molecules, including proteins and DNA (21). At the CC level, most GO terms involved
crucial components of a cell, including the nucleoplasm, cytoplasm,
nucleus, cytoskeleton, cytoplasmic vesicle and secretory granule.
These structural components of a cell have a major influence on
radiosensitivity (22).
KEGG pathway enrichment analysis identified several
pathways associated with the upregulated DE genes in CRT
responders, including base excision repair and homologous
recombination. Meanwhile, the downregulated DE genes in CRT
responders were associated with several pathways, including the Ras
signaling pathway, FoxO signaling pathway, focal adhesion and Wnt
signaling pathway. The base excision repair pathway is a vital
determinant of ionizing radiation sensitivity and has previously
been demonstrated to be associated with the cellular response to
radiotherapy in many studies (36,37).
Targeting base excision repair may provide a potential strategy to
optimize CRT (38). Previous studies
have also suggested that homologous recombination repair serves an
important role in the process of sensitizing cells to ionizing
radiation (39). Targeting the
homologous recombination pathway may be a potential translational
approach for increasing sensitivity to ionizing radiation (40). Furthermore, Ras is a significant
contributor to radiation resistance (41). Radiation sensitization could be
achieved in vitro and in vivo by inhibiting the
activation of Ras (23).
Additionally, FoxO serves an important role in mediating apoptosis
and regulating cell death and survival in response to radiation
(24). Focal adhesion is involved in
the regulation of cellular response to ionizing radiation and may
be associated with increased radioresistance (25). Targeting focal adhesion kinase could
provide a potential therapeutic approach for improving radiotherapy
sensitivity (42). A recent study has
identified that the Wnt/β-catenin signaling pathway serves an
important role in the development of radioresistance and the
inhibition of Wnt/β-catenin signaling may have significant
radiosensitizing effects (26). The
current study has revealed pathways that may contribute to the CRT
response of rectal cancer and may be potential predictive
biomarkers. The pathways identified provide a new insight into the
role of the DE genes in radiation response that could help
introduce therapeutic methods for radiosensitization of tumor
cells.
Through PPI network construction, a series of hub
genes were identified based on three different network analysis
methods, including TOP2B, MAPK8, DLG4, GCG, NGF, INSR and ARRB2. It
was revealed that these hub genes participate in several processes
at the BP level, including the regulation of protein kinase B
signaling, the ERK1 and ERK2 cascade, gene expression and apoptotic
process. Protein kinase B serves a critical role in promoting the
radiation-induced apoptotic response and mediating cell survival
during oxidative stress, the regulation of which may have an effect
on response to radiotherapy (27).
The ERK1 and ERK2 cascade, gene expression and apoptotic process
have also been demonstrated to exhibit synergistic roles in
regulating and controlling the response to radiotherapy for rectal
cancer (20,28). Pathway enrichment analysis of the
selected hub genes was also performed to further evaluate their
function. Several significant pathways were identified, including
the Ras signaling pathway, MAPK signaling pathway and FoxO
signaling pathway. The MAPK signaling pathway is a vital pathway
involved in radioresistance of tumor cells, therefore inhibiting
the MAPK pathway may be beneficial for improving the response to
radiotherapy for rectal cancer (43).
The aforementioned results suggest the selected hub genes are
closely associated with CRT response in rectal cancer and may be
used to predict the CRT response or provide potential treatment
targets for radiation sensitization.
Module analysis of the PPI network revealed that the
most significant module was associated with the chemokine signaling
pathway, cytokine-cytokine receptor interaction and cAMP signaling
pathway. The chemokine signaling pathway has previously been
reported to be responsible for mediating radiation sensitivity in
tumor cells by serving a crucial role in the early phase of the
radiation reaction (29). Cytokines,
useful markers involved in cancer initiation, progression and
pathogenesis, have previously been demonstrated to be closely
associated with modulating personalized radiotherapy (30). The cAMP signaling pathway serves a
role in the regulation of numerous cellular responses, including
gene expression, growth, differentiation, proliferation, apoptosis
and metastasis (19). A previous
study has proposed that cAMP signaling contributes to the response
to ionizing radiation by inhibiting DNA-damage induced apoptosis
(19). Module analysis demonstrated
that the most significant module of the PPI network was involved in
CRT-associated pathways, further confirming that the identified DE
genes were highly associated with the CRT response.
Previously, considerable effort has been made to
predict and explain prognosis variability in response to CRT.
However, no predictive molecular biomarkers for response to CRT
have been sufficiently robust to reach the clinic. Tumors are
heterogeneous and varying genes are expressed by different patients
with the same cancer type. Therefore, CRT response prediction in
rectal cancer cannot be determined by a single or limited number of
molecules. Rectal cancer is a highly heterogeneous disease,
characterized by many coordinated molecular changes; therefore
potential biomarkers should be explored in a systematic and dynamic
manner (44,45). Gene expression profiling performed by
microarray technology has successfully provided many promising
biomarkers for response prediction in rectal cancer. However, the
development of microarray technology has revealed that low
reproducibility is common in many high-throughput post genomic
areas, including proteomics and metabolomics (46). A network-based method can provide a
systemic approach for integrating diverse information into a
systematical framework and improve reproducibility (47). In the current study, seven gene
signatures were identified from the PPI network of DE genes by
using three different network analysis methods. This improves the
robustness of the study, as an integrative approach allows a choice
of the best biomarkers compared with a single analysis method. The
identified genes and their associated pathways could potentially be
developed as biomarkers for predicting CRT response. Notably, the
identified genes and pathways provide potential therapeutic
strategies for the radiosensitization of tumor cells.
The ideal method for screening rectal cancer CRT
response-related genes is to perform microarray analysis using two
groups of rectal cancer tissue samples from patients with the same
treatment strategy, one from CRT sensitive tumor tissues and the
other from insensitive tumor tissues. However, such tissue samples
are difficult to obtain. In addition, the current study could not
perform further exploration as detailed information regarding the
primary histological type of cancer, staging and histological
grading, and socio-demographic data were not available from the
expression profile GSE35452 database. In the future, biological
experiments and large-scale investigations should be conducted to
confirm the predictive power of the approach taken in the current
study and to provide further understanding regarding the mechanisms
underlying therapeutic outcome differences. Additionally,
edge-variation in the network should be considered to further
improve therapeutic response prediction.
In conclusion, the current study provided an
integrative bioinformatics analysis of a gene expression profile to
identify key genes and their involved pathways associated with CRT
response. The current study also provided insights into the
mechanisms underlying the therapeutic outcome difference, providing
valuable information for both basic research and clinical
optimization of CRT. However, further biological experiments are
required to confirm and endorse the results presented in the
current study.
Acknowledgements
Not applicable.
Funding
The present study was funded by Jiangsu Medical
Innovation Team (grant no. CXDT-37), Suzhou Clinical Medical Center
Construction Project (grant no. SZZXJ201503), Suzhou Science and
Technology Development Program (grant no. SZS201509) and the Second
Affiliated Hospital of Soochow University Preponderant Clinic
Discipline Group Project funding (grant no. XKQ2015005).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
QP, KL and YS conceived the idea for the study and
wrote the manuscript. QP, KL, YS, PZ and SF collected the data and
analyzed the data. LZ, YTS and YZ conceived the idea for the study
and revised the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ,
Meester RGS, Barzi A and Jemal A: Colorectal cancer statistics,
2017. CA Cancer J Clin. 67:177–193. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Erlandsson J, Holm T, Pettersson D,
Berglund Å, Cedermark B, Radu C, Johansson H, Machado M, Hjern F,
Hallböök O, et al: Optimal fractionation of preoperative
radiotherapy and timing to surgery for rectal cancer (Stockholm
III): A multicentre, randomised, non-blinded, phase 3,
non-inferiority trial. Lancet Oncol. 18:336–346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim KH, Shin SJ, Cho MS, Ahn JB, Jung M,
Kim TI, Park YS, Kim H, Kim NK and Koom WS: A phase II study of
preoperative mFOLFOX6 with short-course radiotherapy in patients
with locally advanced rectal cancer and liver-only metastasis.
Radiother Oncol. 118:369–374. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huh JW, Kim HR and Kim YJ: Clinical
prediction of pathological complete response after preoperative
chemoradiotherapy for rectal cancer. Dis Colon Rectum. 56:698–703.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burbach JP, den Harder AM, Intven M, van
Vulpen M, Verkooijen HM and Reerink O: Impact of radiotherapy boost
on pathological complete response in patients with locally advanced
rectal cancer: A systematic review and meta-analysis. Radiother
Oncol. 113:1–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akiyoshi T, Kobunai T and Watanabe T:
Predicting the response to preoperative radiation or chemoradiation
by a microarray analysis of the gene expression profiles in rectal
cancer. Surg Today. 42:713–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kamensek U, Sersa G and Cemazar M:
Evaluation of p21 promoter for interleukin 12 radiation induced
transcriptional targeting in a mouse tumor model. Mol Cancer.
12:1362013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Botling J, Edlund K, Lohr M, Hellwig B,
Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Pontén F, et
al: Biomarker discovery in non-small cell lung cancer: Integrating
gene expression profiling, meta-analysis, and tissue microarray
validation. Clin Cancer Res. 19:194–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang M, Yao C, Guo Z, Zou J, Zhang L,
Xiao H, Wang D, Yang D, Gong X, Zhu J, et al: Apparently low
reproducibility of true differential expression discoveries in
microarray studies. Bioinformatics. 24:2057–2063. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watanabe T, Kobunai T, Akiyoshi T, Matsuda
K, Ishihara S and Nozawa K: Prediction of response to preoperative
chemoradiotherapy in rectal cancer by using reverse transcriptase
polymerase chain reaction analysis of four genes. Dis Colon Rectum.
57:23–31. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trakarnsanga A, Gönen M, Shia J, Nash GM,
Temple LK, Guillem JG, Paty PB, Goodman KA, Wu A, Gollub M, et al:
Comparison of tumor regression grade systems for locally advanced
rectal cancer after multimodality treatment. J Natl Cancer Inst.
106:dju2482014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gene Ontology Consortium: The Gene
Ontology (GO) project in 2006. Nucleic Acids Res. 34:(Database
Issue). D322–D326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database Issue). D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang Y, Li M, Wang J, Pan Y and Wu FX:
CytoNCA: A cytoscape plugin for centrality analysis and evaluation
of protein interaction networks. Biosystems. 127:67–72. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su G, Morris JH, Demchak B and Bader GD:
Biological network exploration with Cytoscape 3. Curr Protoc
Bioinformatics. 47:8.13.11-24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cho EA, Kim EJ, Kwak SJ and Juhnn YS: cAMP
signaling inhibits radiation-induced ATM phosphorylation leading to
the augmentation of apoptosis in human lung cancer cells. Mol
Cancer. 13:362014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Furlong H, Mothersill C, Lyng FM and Howe
O: Apoptosis is signalled early by low doses of ionising radiation
in a radiation-induced bystander effect. Mutat Res. 741–742. 35–43.
2013.
|
|
21
|
Badawi A, Hehlgans S, Pfeilschifter J,
Rödel F and Eberhardt W: Silencing of the mRNA-binding protein HuR
increases the sensitivity of colorectal cancer cells to ionizing
radiation through upregulation of caspase-2. Cancer Lett.
393:103–112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Terry SY and Vallis KA: Relationship
between chromatin structure and sensitivity to molecularly targeted
auger electron radiation therapy. Int J Radiat Oncol Biol Phys.
83:1298–1305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brunner TB, Hahn SM, McKenna WG and
Bernhard EJ: Radiation sensitization by inhibition of activated
Ras. Strahlentherapie Onkologie. 180:731–740. 2004. View Article : Google Scholar
|
|
24
|
Luo X, Puig O, Hyun J, Bohmann D and
Jasper H: Foxo and Fos regulate the decision between cell death and
survival in response to UV irradiation. EMBO J. 26:380–390. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Beinke C, Van Beuningen D and Cordes N:
Ionizing radiation modules of the expression and tyrosine
phosphorylation of the focal adhesion-associated proteins focal
adhesion kinase (FAK) and its substrates p130cas and paxillin in
A549 human lung carcinoma cells in vitro. Int J Radiat Biol.
79:721–731. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yin L, Gao Y, Zhang X, Wang J, Ding D,
Zhang Y, Zhang J and Chen H: Niclosamide sensitizes triple-negative
breast cancer cells to ionizing radiation in association with the
inhibition of Wnt/β-catenin signaling. Oncotarget. 7:42126–42138.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang M, Gou X and Wang L: Protein kinase B
promotes radiation-induced regulatory T cell survival in bladder
carcinoma. Scand J Immunol. 76:70–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rahmanian N, Hosseinimehr SJ and Khalaj A:
The paradox role of caspase cascade in ionizing radiation therapy.
J Biomed Sci. 23:882016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Müller K and Meineke V: Radiation-induced
mast cell mediators differentially modulate chemokine release from
dermal fibroblasts. J Dermatol Sci. 61:199–205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bravatà V, Minafra L, Forte GI, Cammarata
FP, Russo G, Di Maggio FM, Augello G, Lio D and Gilardi MC:
Cytokine profile of breast cell lines after different radiation
doses. Int J Radiat Biol. 93:1217–1226. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Goldstein M and Kastan MB: The DNA damage
response: Implications for tumor responses to radiation and
chemotherapy. Annu Rev Med. 66:129–143. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shim G, Ricoul M, Hempel WM, Azzam EI and
Sabatier L: Crosstalk between telomere maintenance and radiation
effects: A key player in the process of radiation-induced
carcinogenesis. Mutat Res Rev Mutat Res. S1383–5742, (14)00002-7.
2014.PubMed/NCBI
|
|
33
|
Wesbuer S, Lanvers-Kaminsky C,
Duran-Seuberth I, Bölling T, Schäfer KL, Braun Y, Willich N and
Greve B: Association of telomerase activity with radio- and
chemosensitivity of neuroblastomas. Radiat Oncol. 5:662010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Crowther AJ, Ocasio JK, Fang F, Meidinger
J, Wu J, Deal AM, Chang SX, Yuan H, Schmid R, Davis I and Gershon
TR: Radiation sensitivity in a preclinical mouse model of
medulloblastoma relies on the function of the intrinsic apoptotic
pathway. Cancer Res. 76:3211–3223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fang Y, DeMarco VG and Nicholl MB:
Resveratrol enhances radiation sensitivity in prostate cancer by
inhibiting cell proliferation and promoting cell senescence and
apoptosis. Cancer Sci. 103:1090–1098. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yin M, Liao Z, Liu Z, Wang LE, Gomez D,
Komaki R and Wei Q: Functional polymorphisms of base excision
repair genes XRCC1 and APEX1 predict risk of radiation pneumonitis
in patients with non-small cell lung cancer treated with definitive
radiation therapy. Int J Radiat Oncol Biol Phys. 81:e67–e73. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Toprani SM and Das B: Radio-adaptive
response of base excision repair genes and proteins in human
peripheral blood mononuclear cells exposed to gamma radiation.
Mutagenesis. 30:663–676. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vens C and Begg AC: Targeting base
excision repair as a sensitization strategy in radiotherapy. Semin
Radiat Oncol. 20:241–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Somaiah N, Yarnold J, Lagerqvist A,
Rothkamm K and Helleday T: Homologous recombination mediates
cellular resistance and fraction size sensitivity to radiation
therapy. Radiother Oncol. 108:155–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lim YC, Roberts TL, Day BW, Stringer BW,
Kozlov S, Fazry S, Bruce ZC, Ensbey KS, Walker DG, Boyd AW and
Lavin MF: Increased sensitivity to ionizing radiation by targeting
the homologous recombination pathway in glioma initiating cells.
Mol Oncol. 8:1603–1615. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kidd AR III, Snider JL, Martin TD,
Graboski SF, Der CJ and Cox AD: Ras-related small GTPases RalA and
RalB regulate cellular survival after ionizing radiation. Int J
Radiat Oncol Biol Phys. 78:205–212. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu FF, Tao TQ, Wang XR, Li YZ, Song DD,
Liu M and Liu XH: Cytosolic calreticulin inhibits microwave
radiation-induced microvascular endothelial cell injury through the
integrin-focal adhesion kinase pathway. Microcirculation.
21:717–729. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dent P, Yacoub A, Fisher PB, Hagan MP and
Grant S: MAPK pathways in radiation responses. Oncogene.
22:5885–5896. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peng Q, Zhu J, Shen P, Yao W, Lei Y, Zou
L, Xu Y, Shen Y and Zhu Y: Screening candidate microRNA-mRNA
regulatory pairs for predicting the response to chemoradiotherapy
in rectal cancer by a bioinformatics approach. Sci Rep.
7:113122017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ren X, Wang Y, Chen L, Zhang XS and Jin Q:
ellipsoidFN: A tool for identifying a heterogeneous set of cancer
biomarkers based on gene expressions. Nucleic Acids Res.
41:e532013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ransohoff DF: Lessons from controversy:
Ovarian cancer screening and serum proteomics. J Natl Cancer Inst.
97:315–319. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zeng T, Sun SY, Wang Y, Zhu H and Chen L:
Network biomarkers reveal dysfunctional gene regulations during
disease progression. FEBS J. 280:5682–5695. 2013. View Article : Google Scholar : PubMed/NCBI
|